Luminescent iminophosphorane gold, palladium and platinum complexes as potential anticancer agents†‡
Malgorzata
Frik
a,
Josefina
Jiménez
b,
Vadim
Vasilevski
a,
Monica
Carreira
a,
Andreia
de Almeida
c,
Elena
Gascón
b,
Farrah
Benoit
a,
Mercedes
Sanaú
d,
Angela
Casini
*c and
María
Contel
*a
aDepartment of Chemistry, Brooklyn College and The Graduate Center, The City University of New York, Brooklyn, NY 11210, USA. E-mail: mariacontel@brooklyn.cuny.edu
bDepartamento de Química Inorgánica, Facultad de Ciencias-Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, Pedro Cerbuna 12, 50009 Zaragoza, Spain
cDepartment of Pharmacokinetics, Toxicology and Targeting, Research Institute of Pharmacy, University of Groningen, 9713 AV Groningen, The Netherlands. E-mail: a.casini@rug.nl
dDepartamento de Química Inorgánica, Universidad de Valencia, Burjassot, Valencia 46100, Spain
First published on 7th March 2014
Abstract
A series of coordination gold(III), palladium(II), and platinum(II) complexes with a luminescent iminophosphorane ligand derived from 8-aminoquinoline [Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C9H6N] (1) have been synthesized and structurally characterized. The coordination palladium(II) and platinum(II) compounds can evolve further, under appropriate conditions, to give stable cyclometalated endo species [M{κ3-C,N,N-C6H4(PPh2N-8-C9H6N)}Cl] (M = Pd, Pt) by C–H activation of the phenyl group of the PPh3 fragment. Iminophosphorane 1 and the new metallic complexes are luminescent in DMSO or DMSO–H2O (1
N–C9H6N] (1) have been synthesized and structurally characterized. The coordination palladium(II) and platinum(II) compounds can evolve further, under appropriate conditions, to give stable cyclometalated endo species [M{κ3-C,N,N-C6H4(PPh2N-8-C9H6N)}Cl] (M = Pd, Pt) by C–H activation of the phenyl group of the PPh3 fragment. Iminophosphorane 1 and the new metallic complexes are luminescent in DMSO or DMSO–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture) solutions at RT. The compounds have been evaluated for their antiproliferative properties in a human ovarian cancer cell line (A2780S), in human lung cancer cells (A549) and in a non-tumorigenic human embryonic kidney cell line (HEK-293T). Most compounds have been more toxic to the ovarian cancer cell line than to the non-tumorigenic cell line. The new complexes interact with human serum albumin (HSA) faster than cisplatin. Studies of the interactions of the compounds with DNA indicate that, in some cases, they exert anticancer effects in vitro based on different mechanisms of action with respect to cisplatin.
:1 mixture) solutions at RT. The compounds have been evaluated for their antiproliferative properties in a human ovarian cancer cell line (A2780S), in human lung cancer cells (A549) and in a non-tumorigenic human embryonic kidney cell line (HEK-293T). Most compounds have been more toxic to the ovarian cancer cell line than to the non-tumorigenic cell line. The new complexes interact with human serum albumin (HSA) faster than cisplatin. Studies of the interactions of the compounds with DNA indicate that, in some cases, they exert anticancer effects in vitro based on different mechanisms of action with respect to cisplatin.
Introduction
Cisplatin and the follow-on drugs carboplatin (paraplatin™) and oxaliplatin (eloxatin™) have been the treatment of choice for ovarian, testicular, head and neck, and non-small cell lung cancer for the past 40 years.1 However, their effectiveness is still hindered by clinical problems, including acquired or intrinsic resistance, a limited spectrum of activity, and high toxicity leading to side effects.1,2 During the past two decades promising anticancer activities of a variety of other metal complexes have been reported.3–8 Recent progress in this field has brought a better understanding of the mode of action of some of these derivatives.9 In particular, a number of gold compounds have overcome cisplatin resistance to specific cancer cells,10 which makes them attractive potential therapeutics. In addition, it has been found that DNA is not the primary target for most gold compounds, reinforcing the idea that their mode of action is different with respect to cisplatin.10,11 This has prompted the search for alternative biomolecular targets not only for gold but for other non-platinum complexes10–13 and a revision of the mode of action of anticancer platinum compounds themselves.14 Thus, multiple biological pathways have been proposed for non-platinum derivatives including the inhibition of mitochondrial enzymes and of the proteasome for gold compounds.15–19We have reported that non-toxic iminophosphorane or iminophosphane (IM) compounds serve as stabilizing (C,N- or N,N) chelating ligands in the preparation of anticancer organometallic and coordination d8 metal complexes (Chart 1).20–24 Organogold(III) complexes containing iminophosphorane (C,N-IM) ligands of the type PPh3NPh (such as a in Chart 1) displayed a high cytotoxicity in vitro against human ovarian cancer and leukemia cell lines (in some cases with IC50 values in the nanomolar range)20,21 while being less toxic to normal T-lymphocytes by a non-cisplatin mode of action (involving mitochondrial production of reactive oxygen species).21 We have also reported on the cytotoxicity of coordination and organometallic Pt(II) and Pd(II) complexes with water-soluble iminophosphorane ligands.22,23 Interestingly, we observed that some of these Pt and Pd compounds (b–d) were more cytotoxic than cisplatin to leukemia cell lines (both T-Jurkat and cisplatin-resistant Jurkat sh-Bak) by a mode of action different from that of cisplatin.22,23
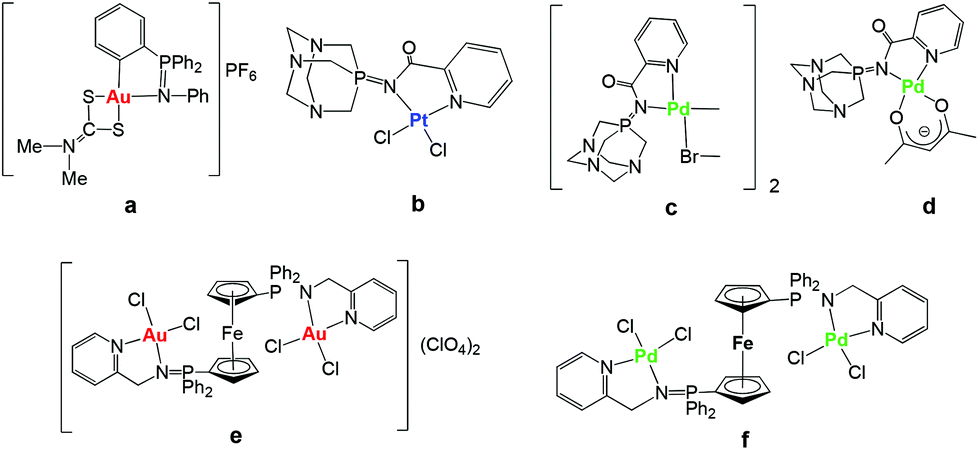 | ||
| Chart 1 Selected iminophosphorane (IM) d8 transition metal complexes with significant anticancer properties prepared in our group.20–24 | ||
More recently we have reported on the preparation of di- and tri-metallic Au(III) and Pd(II) coordination complexes containing iminophosphorane ligands derived from ferrocenyl-phosphanes (like e and f in Chart 1).24 The trimetallic complexes [({Cp-P(Ph2)N-CH2-2-NC5H4}MCl2)2Fe]A2 (M = Au, A = ClO4−e M = Pd, A = none f) were more cytotoxic to cancer cells than their corresponding monometallic fragments. Moreover, these complexes were significantly more cytotoxic than cisplatin in the resistant human ovarian A2780R and in the human breast MCF7 cancer cell lines. Studies of the interactions of the trimetallic compounds with DNA and the zinc-finger protein PARP-1 indicate that they exert anticancer effects in vitro based on different mechanisms of action with respect to cisplatin.24
Within this frame, we aimed to explore the biological properties of related IM complexes containing d8 transition metal centers incorporating a luminescent molecule. The potential of metal-based theranostics, biologically active luminescent metal complexes that can target and probe a specific biomolecule, in the treatment of human diseases has been recently highlighted.25 In this context, we decided to explore the possibility of exploiting the native luminescence of metal complexes to determine its intracellular distribution by using fluorescence microscopy.26,27 Thus, we report here on the synthesis, characterization and luminescence studies of gold, palladium and platinum derivatives containing the previously reported28 iminophosphorane ligand derived from 8-aminoquinoline [Ph3PN-C9H6N] (1). The complexes have been evaluated for their antiproliferative properties in a human ovarian cancer cell line (A2780S), in human lung cancer cells (A549) and in a non-tumorigenic human embryonic kidney cell line (HEK-293T). Most compounds have been more effective on the ovarian cancer cell line than on the non-tumorigenic cell line. The compounds have also been tested for their possible interactions with plasmid (pBR322) DNA used as a model nucleic acid, and for their reactivity with the transport protein human serum albumin (HSA).
Results and discussion
Synthesis and characterization
Ligand [Ph3PN-C9H6N] (1) was prepared by a modification of a previously27 reported procedure. 8-Azidoquinoline was obtained by reaction of 8-aminoquinoline with trifyl azide28 and subsequently reacted with PPh3 by the Staudinger method.29
The addition of this ligand to an in situ prepared CH3CN solution of [AuCl2(CH3CN)2]A (A = ClO4−, PF6−), or CH2Cl2 solutions/suspensions of PdCl2(COD) and PtCl2 afforded coordination complexes of gold(III) (2, 3), palladium(II) (4) and platinum(II) (5) in moderate to high yields (Scheme 1).
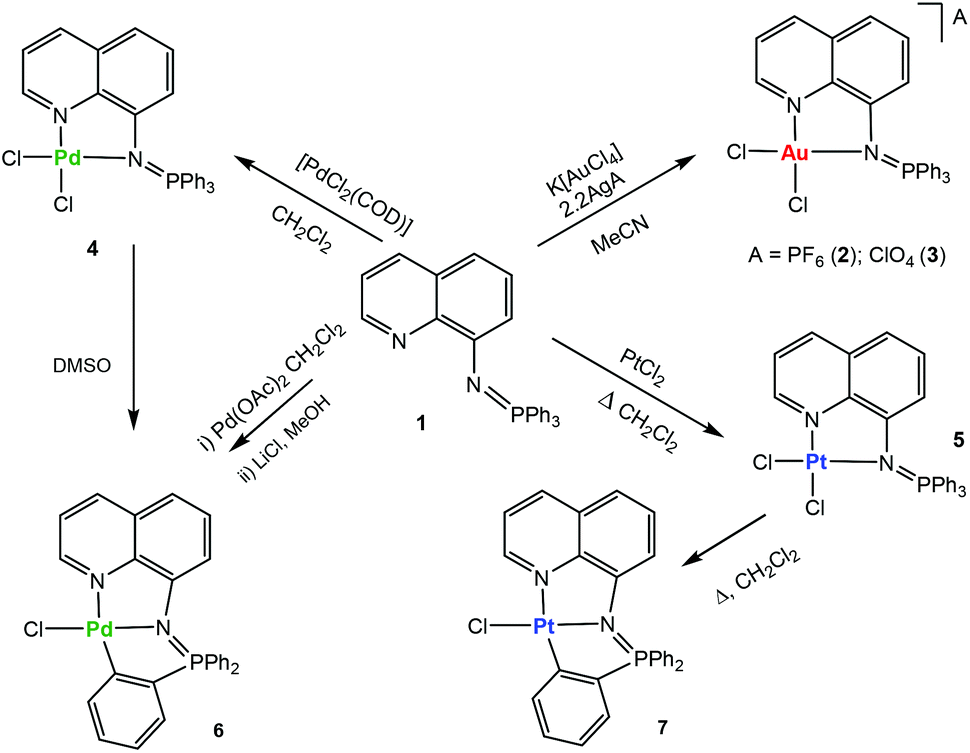 | ||
| Scheme 1 Preparation of the new luminescent metallic complexes 2–7. | ||
The coordination complexes have been characterized by spectroscopic methods, mass spectrometry and elemental analysis. The crystal structures of the gold(III) derivative 3 and the palladium compound 4 have been determined by an X-ray analysis (Fig. 1). Selected bond lengths and angles for both compounds are collected in Table 1.
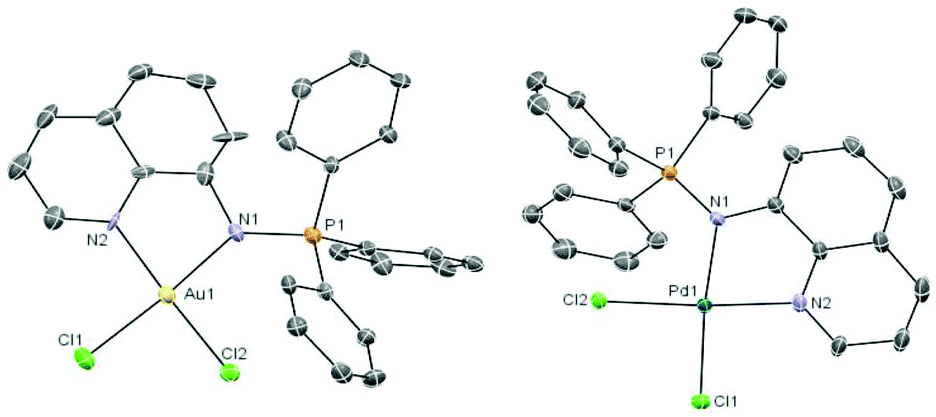 | ||
| Fig. 1 Molecular structure of the cation in the compound [Au((Ph3PN-8-C9H6N)-κ-N, N)Cl2]ClO43 and of the compound [Pd((Ph3PN-8-C9H6N)-κ-N,N)Cl2] 4 with the atomic numbering scheme. | ||
| 3 | 4 | ||
|---|---|---|---|
| Au(1)–N(2) | 1.998(8) | Pd(1)–N(2) | 2.034(5) |
| Au(1)–N(1) | 2.045(13) | Pd(1)–N(1) | 2.060(5) |
| Au(1)–Cl(2) | 2.254(3) | Pd(1)–Cl(2) | 2.2738(15) |
| Au(1)–Cl(1) | 2.278(5) | Pd(1)–Cl(1) | 2.2903(15) |
| P(1)–N(1) | 1.656(11) | P(1)–N(1) | 1.622(5) |
| N(2)–Au(1)–N(1) | 80.5(4) | N(2)–Pd(1)–N(1) | 80.5(2) |
| N(2)–Au(1)–Cl(2) | 172.7(3) | N(2)–Pd(1)–Cl(2) | 170.63(15) |
| N(2)–Au(1)–Cl(1) | 95.1(3) | N(2)–Pd(1)–Cl(1) | 93.62(15) |
| N(1)–Au(1)–Cl(2) | 95.4(3) | N(1)–Pd(1)–Cl(2) | 95.51(13) |
| N(1)–Au(1)–Cl(2) | 175.7(3) | N(1)–Pd(1)–Cl(2) | 173.73(14) |
| Cl(2)–Au(1)–Cl(1) | 88.88(15) | Cl(2)–Pd(1)–Cl(1) | 89.91(6) |
The geometry about the Au(III) and Pd(II) centers is pseudo-square planar with the N(2)–M(1)–N(1) angle of 80.5(4)° (3) and 80.5(2)° (4) suggesting a rigid ‘bite’ angle of the chelating ligand. The Au and Pd centers are on an almost ideal plane with negligible deviations from the least-squares plane.
The main distances M–N(1) or M–N(iminic) and M–N(2) found for the Pd complex 4 are similar to that in related iminophosphorane complexes like [PdCl2(TPAN-C(O)-2-NC5H4)],22 [Pd{k2-C,N-C6H4(PPh2NC6H4Me-4′)-2}{(μ-OAc)}]2,30 [Pd{k2-C,N-C6H4(PPh2NC6H4Me-4′)-2}(tmeda)]ClO4,30 [Pd(C6H4CH2NMe2)(Ph3PNC(O)-2-NC5H4)]ClO4,31 [({Cp-P(Ph2)N-CH2-2-NC5H4}PdCl2)2Fe],24 and [{(Cp-P(Ph2)N-CH2-2-NC5H4)PdCl2}Fe(Cp)].24 The distances Au–N(1), Au–M(2), Au–Cl(1) and Au–Cl(2) in 3 are similar to those obtained by X-ray crystallographic analysis or DFT calculations for [AuCl2(Ph2PyPNC(O)-Ph)]ClO4,32 [(Ph3PN-CH2-2-NC5H4)AuCl2]ClO4,32 [AuCl2(TPAN-C(O)-2-NC5H4)]ClO4,22 [({Cp-P(Ph2)N-CH2-2-NC5H4}AuCl2)2Fe](ClO4)2,24 and [{(Cp-P(Ph2)N-CH2-2-NC5H4)AuCl2}Fe(Cp)]ClO4.24 The distances PN are shorter than in compounds which incorporate a carbonyl group bonded to the iminic nitrogen (due to a smaller delocalized charge density as compared to the CO group).22–24,32
The stability of the compounds in solution can be easily ascertained by 31P{1H} NMR spectroscopy in deuterated solvents (see ESI‡). Most compounds are very stable in CD3CN or CD2Cl2 solution (weeks). The stability in d6-DMSO solution was also evaluated (see ESI‡). Compounds 2 and 3 have half-lives of 4 (2) or 6 (3) hours and they decompose to phosphane oxide and the coordination gold(III) compound based on the deprotonated uninegative bidentate 8-aminoquinoline fragment [Au((N-8-C9H7N)-κ-N,N)Cl2]. The lack of stability for some monometallic gold(III) iminophosphorane coordination complexes23,24 and their decomposition to biologically active gold(III) amino derivatives24 have been described previously. The coordination Pd(II) compound 4 in d6-DMSO solution over time evolves to the endo cyclometalated species [Pd{κ3-C,N,N-C6H4(PPh2N-8-C9H6N)}Cl] (6Scheme 1) by activation of the C–H of one aryl group from the PPh3 fragment of the iminophosphorane ligand. The half-life of 4 is ca. 1.5 hours. The coordination platinum(II) compound 5 evolves to a cyclometalated analogue of 6 to give species [Pt{κ3-C,N,N-C6H4(PPh2N-8-C9H6N)}Cl] (7Scheme 1) by heating DMSO solutions of 5. The new cyclometalated species 6 and 7 have been fully characterized by spectroscopic methods (including 195Pt{1H} NMR), mass spectrometry, conductivity and elemental analysis (see the Experimental section). These species can also be cleanly obtained in high yields (Scheme 1) by reaction of ligand 1 and Pd(AcO)2 in CH2Cl2 followed by treatment with LiCl in MeOH (6) or by refluxing the coordination platinum(II) compound 5 in CH2Cl2 (7). The new cyclometalated species (6, 7) are stable in d6-DMSO solution with half-lives of several weeks. That 6 and 7 are orthometalated species is clear from the downfield shifts undergone by the 31P NMR signals with respect to coordination compounds 4 and 5, respectively. In the 1H and 13C{1H} NMR spectra of 6 and 7 there are also new signals due to the 4 protons (a–d) of the cyclometalated aryl ring and six well resolved peaks (for the carbons of this C6H4-fragment). In addition, the 2JPt–P coupling constant of 426 Hz is visible only for the cyclometalated derivative 7 (its 31P{1H} NMR displays a signal at 44.7 ppm with 195Pt satellites while the signal in the 195Pt{1H}NMR is a doublet at −2899.7 ppm).33 A JPt–P coupling constant is not observable for the coordination iminophosphorane compound 5. We have not seen this coupling constant for other coordination iminophosphorane platinum derivatives either.22 The IM ligand in compounds 6 and 7 acts as a C,N,N-pincer ligand. Urriolabeitia and co-workers have reported on the selective C–H activation by palladium(II) complexes in iminophosphoranes based on the type of iminophosphorane ligand, the solvents employed and the temperature at which the cyclometalation reaction takes place giving endo or exo derivatives.34–37 The semi-stabilized IM ligand described here [Ph3PN-C9H6N] (1) coordinated to a palladium or platinum center(II) in compounds 4 and 5 affords cyclometalated endo derivatives with the IM acting as a C,N,N-pincer ligand in 6 and 7.
The only example of a Pt(II) cyclometalated compound containing an iminophosphorane ligand reported so far is the endo [Pt(C6H4-2-PPh2N-C(O)-2-NC5H4-κ-C,N,N)Cl] derivative obtained by reaction of [PtCl2(NCPh)2] with Ph3PN-C(O)-2-NC5H4 in refluxing 2-methoxyethanol.37
Luminescence studies
We have studied the luminescence of the new metallic complexes 2–7 and of the previously described ligand 127 whose luminescence had not been reported before. All the compounds are luminescent in DMSO solution (5 × 10−4 M) at RT.The excitation and emission data as well as the lifetimes for excited states are summarized in Table 2. The lifetimes are all relatively long (7–12 μs), which indicates that the emission transitions are all forbidden and phosphorescent. Ligand 1, non-cyclometalated complexes 2–4 and cyclometalated palladium complex 6 all show similar optical behavior. The spectra show a broad band with the emission maximum at around 486–521 nm upon excitation between 290 and 410 nm (see ESI‡). Thus, an intraligand transition modified by the coordination to metal is probably responsible for the luminescence in all of the complexes 2–4 and 6. The luminescence profile and features (excitation and emission maxima) of the cyclometalated platinum complex 7 are different. For this compound, an emission band showing some evidence of vibrational structure is observed, whose maximum is red-shifted from 521 nm in the free ligand 1 to 677 nm (see also ESI‡). This spectral shift suggests an 3MLCT character in the excited state of 7.38 On the other hand, unlike what we observed for 1 and complexes 2–6, the absorption spectrum of 7 exhibits a lower-energy band in the 400–500 nm region (see ESI‡) at a substantially longer wavelength than the absorption of the free ligand. This new band leads to the observed emissions and is likely to arise from charge-transfer transitions involving the metal, as has been described for other Pt(II) complexes. Thus, a triplet state of mixed ππ*/MLCT character is probably responsible for the phosphorescence in 7.38–42
| Compound | λ max solution (298 K)a | |
|---|---|---|
| λ exc | λ em [τ (μs)] | |
| a Data of wavelength are given in nm. Data using DMSO as a solvent at room temperature, 5 × 10−4 M. | ||
| 1 | 296, 336, 370, 407 | 521 [10] |
| 2 | 290, 350 sh, 371 sh, 400 | 513 [11] |
| 3 | 294, 337, 370, 404 | 517 [10] |
| 4 | 298 sh, 342, 368 sh | 496 [7] |
| 5 | 306, 343, 377 sh, 390 | 513 [12] |
| 6 | 294, 341 sh, 378, 402 | 486 [10] |
| 7 | 348 sh, 373, 440 | 645, 677 [8], 702 sh |
Non-cyclometalated platinum complex 5 deserves special attention. A DMSO diluted solution of this pure compound is brightly emissive yellow under a common UV lamp (ex 365 nm) at RT. However, the spectrum shows two emissions whose intensities depend on the excitation wavelength and the time of the luminescence measurement (see Fig. S12 and S13 in ESI‡). At 390 nm, it exhibits two emission bands, a broad one with the maximum at 513 nm (similar to that observed for ligand 1 and complexes 2–4 and 6) and another one with a profile and features similar to those observed for the cyclometalated platinum complex 7, with a maximum at 677 nm. Excitation at a longer wavelength, 450 nm, only gives the emission band at 677 nm. When the luminescence of this solution is measured immediately afterwards, the spectrum only shows the emission band at 677 nm. Under a common UV lamp (ex 365 nm) we observed an orange-red emission for the irradiated area while the non-irradiated area remained yellow. This observation seems indicative of the transformation of coordination complex 5 into cyclometalated 7 upon UV irradiation, which has been demonstrated by 31P{1H} and 1H NMR spectroscopy. The transformation is not clean, though, and the NMR spectra of the DMSO solution after irradiation show other signals besides those corresponding to 5 and 7, which have not been identified.
2 and 3 are rare examples of non-cyclometalated gold(III) complexes with chlorido ligands which emit in a fluid solution at room temperature. Two essential control parameters to obtain luminescent (square-planar) gold(III) complexes have been highlighted: the ligand field strength and the rigidity of chelating ligands.43,44 These parameters are also relevant for the preparation of luminescent Pt(II) complexes.38 The iminophosphorane ligands are strong σ-donors, and this is expected to lead to an increase in the ligand field strength, which, in turn, decreases the probability of thermal population of non-emissive d–d states. Importantly, for all complexes, the energies of the excitation and emission bands are potentially of suitable wavelengths for cellular distribution studies.26
We also studied the luminescence properties of the compounds in DMSO solution over time. As expected, the most stable cyclometalated palladium and platinum compounds in DMSO solution (6, 7) and ligand 1 did not change their luminescence significantly over time.
Fig. 2 shows the change in the luminescence properties of compound 6 in DSMO solution at RT over time (24 hour period). The other figures are collected in ESI.‡
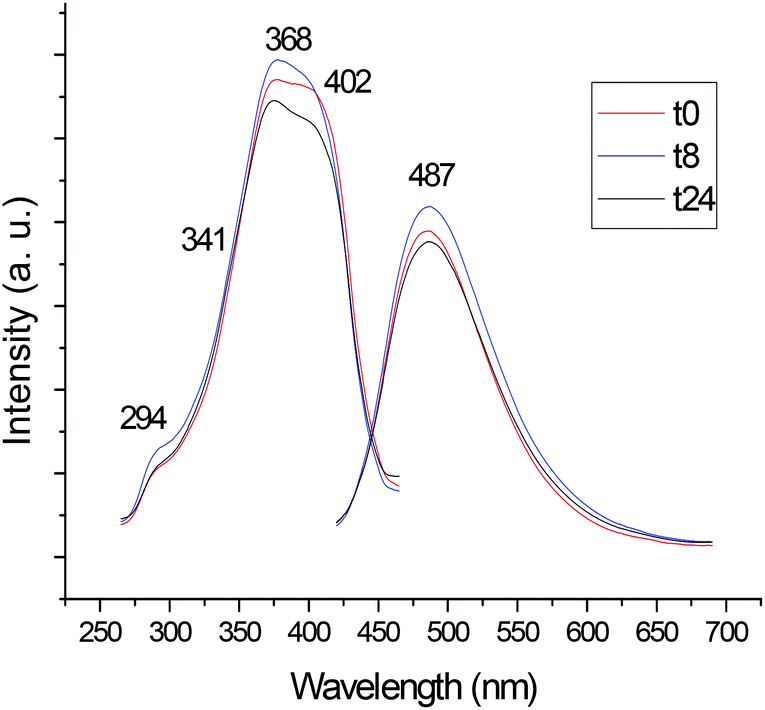 | ||
| Fig. 2 Study of the luminescence of compound 6 in DMSO solution (5 × 10−4 M) at RT over time (24 hours). | ||
In the case of coordination Au(III) complexes, 2 and 3, and coordination Pd(II) complex, 4, the study of the luminescence over time affords results that are in agreement with the stability of these compounds in DMSO solution. As mentioned before, 2 and 3 decompose (with half-lives of ca. 4 (2) and 6 (3) hours in d6-DMSO) to phosphane oxide (PPh3O) and the coordination gold(III) compound [Au((N-8-C9H7N)-κ-N,N)Cl2]. Compound 2 shows a shift of the emission band at 513 nm to lower wavelength (473 nm) concomitant with the appearance of two new bands of lower intensity at 634 and 683 nm after 3.5 hours. The spectrum is the same 24 hours later and therefore it may correspond to that of the decomposition product [Au((N-8-C9H7N)-κ-N,N)Cl2]. Compound 3 is slightly more stable and the change in intensity and position of the bands to give spectra identical to those of 2 over time occurs 8 hours later. Coordination Pd(II) complex 4 shows a shift of the emission band at 496 nm to lower wavelength (486 nm), which corresponds to that of the endo cyclometalated species 6 decomposition product.
The study of the luminescence of compound 7 in a mixture of DMSO–H2O (50:50) was carried out, and a broad emission band at 669 nm was observed, which was similar to that obtained for 7 in DMSO (see Fig. S15 in ESI‡). However, the spectrum was completely different after 6 h, showing two broad emission bands at 430 and 630 nm, which may correspond to decomposition products. NMR studies of solutions of mixtures of 7 in DMSO–H2O close to 1:1 ratio showed decomposition to different products over time. Importantly, the solutions containing these products are still luminescent after 24 hours and their excitation and emission bands are of suitable wavelengths for intramolecular cellular distributions studies.
These preliminary luminescence studies support the decision to select the most intense compounds 3 and 7 as candidates for further intramolecular cellular distribution studies using fluorescence microscopy.
Antiproliferative studies
The antiproliferative properties of ligand 1 and metallic complexes 2–7 were assayed by monitoring their ability to inhibit cell growth using the MTT assay (see the Experimental section). The cytotoxic activity of the compounds was determined as described in the Experimental section in the human ovarian cancer cell line A2780 and in the human lung cancer cell line A549, in comparison with cisplatin. The results are summarized in Table 3. Ligand 1 and its PPh3-based decomposition product (PPh3O) are not cytotoxic in all tested cell lines. The coordination complexes 2 (Au(III)) and 5 (Pt(II)) and cyclometalated monometallic Pt(II) complex 7 are as cytotoxic as or more cytotoxic than cisplatin toward the A2780 cell line (low micromolar range). However, these compounds are poorly selective, being very cytotoxic also towards the non-tumorigenic HEK-293T cell line. The selectivity towards the A2780 cells is more pronounced for the coordination and cyclometalated Pd(II) compounds 4 and 6. Most compounds are poorly cytotoxic towards the human lung cancer cell line A459, with the exception of the cyclometalated monometallic Pt(II) compound 7 which is ca. 3-fold more cytotoxic than cisplatin. However 7 is quite toxic to HEK-293T cell lines as well.
| A2780 | A549 | HEK-293T | |
|---|---|---|---|
| a MTT assay (see the Experimental section). All compounds were dissolved in 1% DMSO and diluted with water before addition to cell culture medium for a 72 h incubation period. Cisplatin was dissolved in H2O. b Since the biological activity of cationic iminophosphorane gold compounds is due to the cation, 2 was used to evaluate the biological activity of compounds 2 and 3 with identical cations. | |||
| PPh3O |
>100 | >100 | >100 |
| 1 | >100 | >100 | >100 |
| 2 | 6.35 ± 0.69 | 25.1 ± 3.8 | 21.6 ± 5.1 |
| 4 | 11.0 ± 1.5 | 62.5 ± 3.7 | 53.5 ± 10.4 |
| 5 | 3.33 ± 0.14 | 19.5 ± 6.5 | 10.8 ± 0.8 |
| 6 | 13.2 ± 2.1 | 86.5 ± 2.5 | 66.0 ± 5.5 |
| 7 | 3.56 ± 0.70 | 4.60 ± 0.50 | 2.64 ± 0.87 |
| Cisplatin | 3.90 ± 1.80 | 8.0 ± 0.5 | 11.0 ± 2.9 |
Studies of intracellular distribution on A2780 cells were attempted with selected compounds (e.g.3 and 7) using fluorescence microscopy, but unfortunately, and in spite of the different conditions tested (different compound concentrations and incubation times with cells), it was impossible to observe any fluorescence.
Reactivity with DNA and HSA (human serum albumin)
Since DNA replication is a key event for cell division, it is among the critically important targets in cancer chemotherapy. It is known that most cytotoxic platinum drugs can form strong covalent bonds with the DNA bases45 and, depending on the ancillary ligands, can also act as DNA intercalators.46 There are also reports on palladium derivatives interacting with DNA in covalent47,48 and non-covalent ways.49,50 Conversely, most gold-based compounds do not display strong interaction with DNA.10,11 Thus, we performed agarose gel electrophoresis studies to unravel the effects of the new compounds 2–7 on plasmid (pBR322) DNA (Fig. 3). Plasmid DNA has two main forms: OC (open circular or relaxed form, Form II) and CCC (covalently closed or supercoiled form, Form I). Changes in electrophoretic mobility of both forms are usually taken as evidence of metal–DNA binding. Generally, the larger the retardation of supercoiled DNA (CCC, Form I), the greater the DNA unwinding produced by the drug.51 Binding of cisplatin to plasmid DNA, for instance, results in a decrease in mobility of the CCC form and an increase in mobility of the OC form (see lanes a–d for cisplatin in Fig. 3).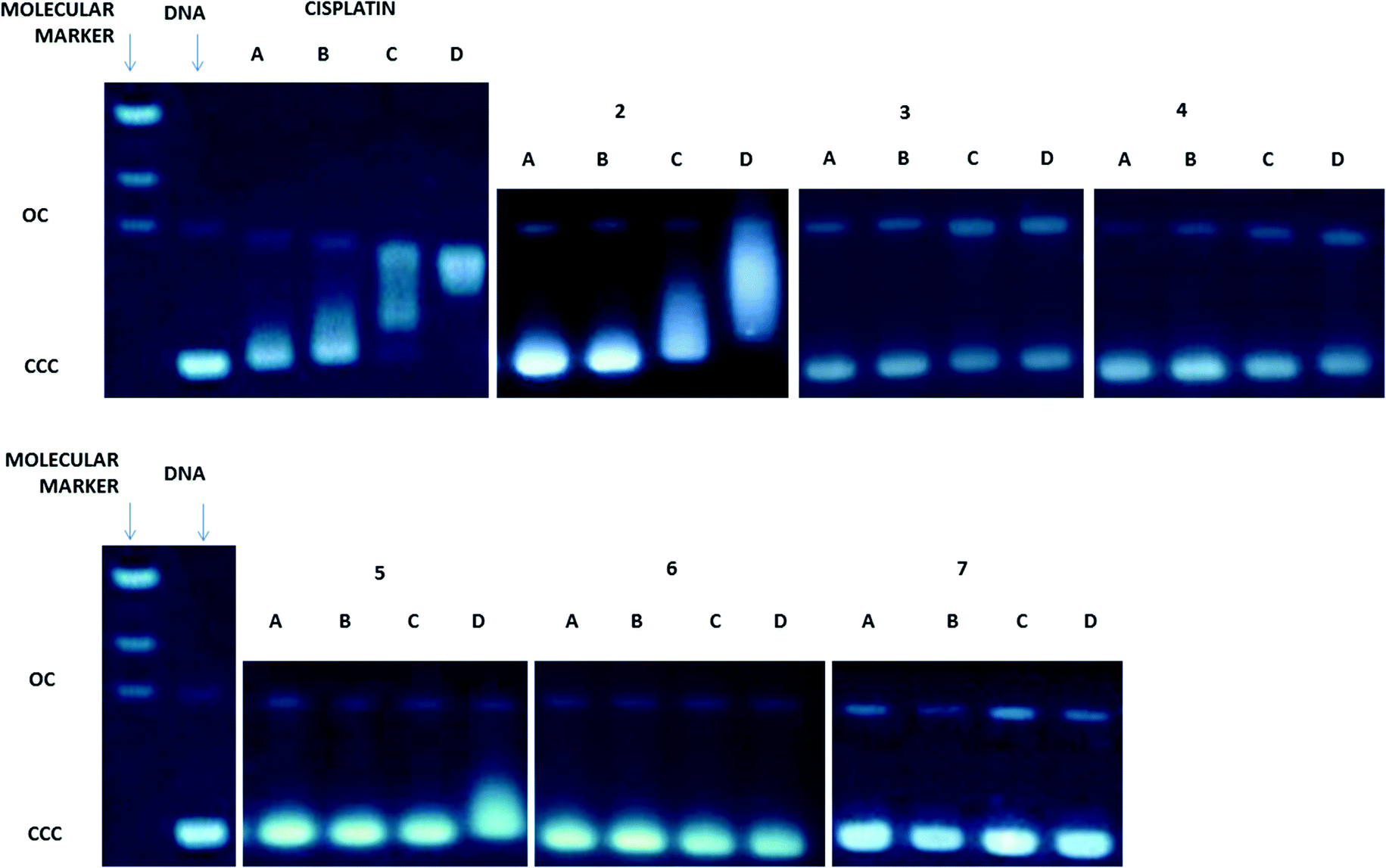 | ||
| Fig. 3 Electrophoresis mobility shift assays for cisplatin and compounds 2–7 (see the Experimental section for details). DNA refers to untreated plasmid pBR322. a, b, c and d correspond to metal/DNAbp ratios of 0.25, 0.5, 1.0 and 2.0 respectively. | ||
Treatment with increasing amounts of compounds 3–7 does not affect the mobility of the faster-running supercoiled form (Form I or CCC) at ratios up to 1.0 metal/DNAbp. In the case of the coordination compounds of Au(III) 3 and Pt(II) 5, at the highest ratio of 2.0 metal/DNAbp, there is an effect of retardation of the faster-running supercoiled form (CCC). We can say that these complexes have no (4, 6, 7) or weak interaction (3, 5) with DNA. In contrast, the coordination gold(III) compound 2 modifies the electrophoretic mobility and the retardation of the faster-running supercoiled form (CCC) in the same way as that described previously for other coordination gold(III) compounds containing IM ligands.23 This is due to the lack of stability of these complexes in DMSO–buffer solutions with the cleavage of the PN from the IM ligand and the subsequent decomposition of the Au(III)–IM complex into PPh3O and cytotoxic square-planar gold(III) compounds which are known to intercalate in DNA.23 Compound 3 is slightly more stable than 2 in solution and thus this effect is less marked. However, oxidative damage of the DNA produced by the metal center cannot be ruled out. The rest of the compounds which are more stable (coordination and cyclometalated compounds of palladium(II) and platinum(II)) have no or weak interaction with plasmid DNA. We have found this to be the case for some coordination and organometallic compounds of gold(III) and organometallic derivatives of palladium(II) containing iminophosphorane ligands.20,22–24 Coordination iminophosphorane complexes of palladium and platinum have displayed interactions with DNA stronger but of a different nature than those exerted by cisplatin.22,24
Human serum albumin (HSA) is the most abundant carrier protein in plasma and is able to bind a variety of substrates including metal cations, hormones and most therapeutic drugs. It has been demonstrated that the distribution, the free concentration and the metabolism of various drugs can be significantly altered as a result of their binding to the protein.52 HSA possesses three fluorophores, these being tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe) residues, with Trp214 being the major contributor to the intrinsic fluorescence of HSA. This Trp fluorescence is sensitive to the environment and binding of substrates, as well as changes in conformation that can result in quenching (either dynamic or static).
Thus, the fluorescence spectra of HSA in the presence of increasing amounts of compounds 2–7 and cisplatin were recorded in the range of 300–450 nm upon excitation of the tryptophan residue at 295 nm (Fig. 4). The compounds caused a concentration dependent quenching of fluorescence without changing the emission maximum or shape of the peaks, as seen in Fig. 4(A) for compound 3.
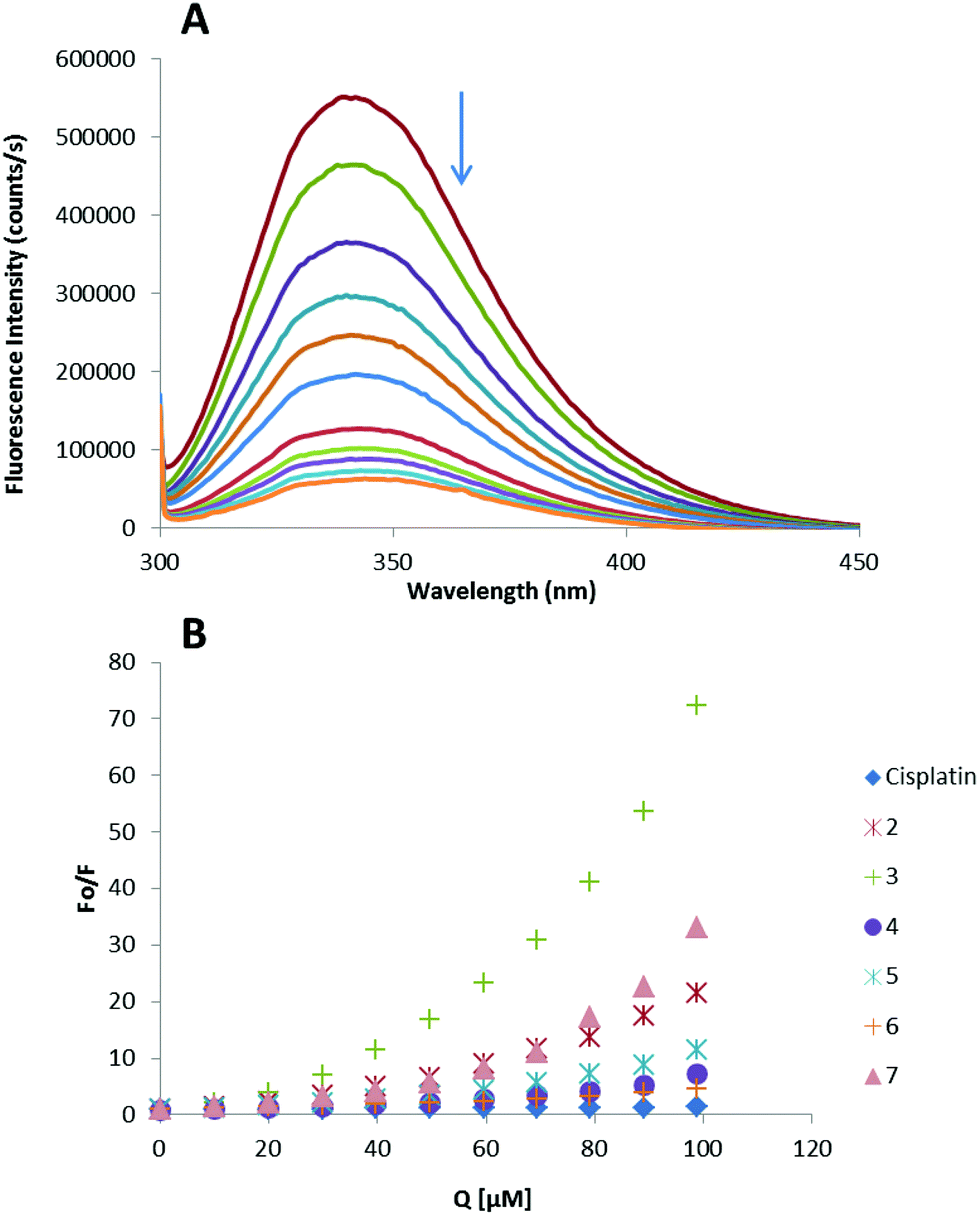 | ||
| Fig. 4 (a) Fluorescence titration curve of HSA with compound 3. Arrow indicates the increase of quencher concentration (10–100 mM). (b) Stern–Volmer plot for HSA fluorescence quenching observed with compounds 2–7 and cisplatin. | ||
The fluorescence data were analyzed by the Stern–Volmer equation. While a linear Stern–Volmer plot is indicative of a single quenching mechanism, either dynamic or static, the positive deviation observed in the plots of F0/F versus [Q] of our compounds (Fig. 4) suggests the presence of different binding sites in the protein.53 Of note, a similar behaviour was observed in the case of some coordination and organometallic iminophosphorane complexes of d8 metals for which we also reported a concentration dependent fluorescence quenching.23,24 In this graph higher quenching by the iminophosphorane complexes was observed compared to that of cisplatin under the chosen conditions, most likely due to the faster reactivity of our compounds with HSA compared to cisplatin.
In the case of [MCl2(TPAN-C(O)-2-NC5H4)] (M = Pd, Pt), isothermal titration calorimetry (ITC)23 showed two different binding interactions which explained the lack of linearity observed in the fluorescence quenching studies, as the Stern–Volmer method assumes all binding sites to be equivalent. We believe that a similar reactivity takes place for the iminophosphorane compounds described here.
Experimental section
Materials and methods
All manipulations involving air-free syntheses were performed at a nitrogen vacuum manifold using standard Schlenk-line techniques under an argon atmosphere or in a glove-box MBraun MOD System. Solvents were purified using a PureSolv purification unit from Innovative Technology, Inc. The phosphine substrate TPA was purchased from Sigma-Aldrich; K[AuCl4], AgClO4, [PdCl2(COD)], Pd(OAc)2 and PtCl2 were purchased from Strem Chemicals and used without further purification. NMR spectra were recorded on a Bruker AV400 (1H NMR at 400 MHz, 13C NMR at 100.6 MHz, 31P NMR at 161.9 MHz, 195Pt NMR at 85.8 MHz). Chemical shifts (δ) are given in ppm using CD3CN, CDCl3, d6-DMSO or D2O as solvents. 1H and 13C resonances were measured relative to solvent peaks considering TMS = 0 ppm. 31P{1H} was externally referenced to H3PO4 (85%) and 195Pt{1H} was referenced to [PtCl6]2−. Infrared spectra (4000–250 cm−1) were recorded on a Nicolet 6700 FT-IR spectrophotometer from nujol mulls between polyethylene sheets. Elemental analyses were performed on a Perkin Elmer 2400 CHNS/O Analyzer, Series II. Mass spectra (ESI) were performed on an Agilent Analyzer and on a Bruker Analyzer. Conductivity was measured using an OAKTON PC 700 pH/conductivity meter in acetone solution. Electrophoresis experiments were carried out in a Bio-Rad Mini sub-cell GT horizontal electrophoresis system connected to a Bio-Rad Power Pac 300 power supply. Photographs of the gels were taken using an Alpha Innotech FluorChem 8900 camera. Fluorescence intensity measurements were carried out on a PTI QM-4/206 SE Spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence using a 1 cm path length quartz cuvette.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N-8-C9H6N (1)27.
An alternative method of synthesis was used: a solution of PPh3 (2.59 mmol) in CH2Cl2 was added dropwise to a solution of 8-azidoquinoline28 (2.59 mmol) in CH2Cl2 at RT and allowed to react for 1 h. The solvent was reduced in vacuo to 2 mL and Et2O (10 mL) was added. A pale-yellow solid precipitated and was filtered off and dried in vacuo. 1 was used without further purification. Yield: 0.857 g, 82%.
N-8-C9H6N)-κ-N,N)Cl2]X [X = PF6− (2); ClO4− (3)].
To K[AuCl4] (0.076 g, 0.2 mmol) in dry MeCN (10 mL), either AgClO4 (0.091 g, 0.44 mmol) for the obtainment of 2 or AgPF6 (0.111 g, 0.44 mmol) for the obtainment of 3 in dry MeCN (5 mL) was added. The reaction mixture was stirred in the dark for 30 min and subsequently filtered through celite (to remove AgCl). 1 (0.081 g, 0.2 mmol) in CH2Cl2 (1 mL) was added and the yellow solutions became purple instantly. After stirring for 30 min the reaction mixture was filtered through celite (to remove KClO4) and then the solvent was reduced in vacuo to ∼2 mL. Upon addition of Et2O (10 mL), a purple solid was obtained, which was washed with MeCN (<1 mL at a time) and Et2O and dried in vacuo. 2: Yield: 0.1198 g, 73%. Anal. calcd for C27H21AuCl2F6N2P2 (817.28): C, 39.68; H, 2.59; N, 3.43. Found: C, 39.21; H, 2.36; N, 3.21. MS(ESI+) [m/z]: 671.0 [M − PF6]+. 31P{1H} NMR (CD3CN): δ 39.6 (s), −142.5 (sept, PF6−). 1H NMR (CD3CN): δ 6.66 (1H, d, J = 7.8 Hz, H7), 7.15 (1H, t, J = 8.0 Hz, H6), 7.62 (1H, d, J = 8. 2 Hz, H5), 7.62–7.82 (9H, m, Hpara + Hmeta), 8.04 (1H, dd, J = 8.3, 5.7 Hz, H3), 8.13 (6H, dd, J = 12.6, 7.6 Hz, Hortho), 8.85 (1H, d, J = 8.3 Hz, H4), 9.44 (1H, d, J = 5.6 Hz, H2). 13C{1H} NMR (CD3CN): δ 121.6 (s, C9H6N), 122.6 (s, C9H6N), 122.9 (d, J = 5.0 Hz, C7), 123.6 (s, C5), 123.9 (s, C3), 128.8 (s, C6), 128.9 (d, J = 13.3 Hz, Cmeta), 131.4 (s, Cipso), 134.2 (d, J = 10.6 Hz, Cortho), 135.1 (d, J = 3.0 Hz, Cpara), 144.4 (s, C4), 146.1 (s, CNP), 148.4 (s, C2). IR (cm−1): ν 370 (Au–Cl), 838 (v br, PF6−), 1266 (PN). Conductivity (acetone): Λ = 110 μS cm−1. 3: Yield: 0.109 g, 69%. Anal. calcd for C27H21AuCl3N2O4P (771.77): C, 42.02; H, 2.74; N, 3.63. Found: C, 42.08; H, 2.96; N, 3.66. MS(ESI+) [m/z]: 671.1 [M − ClO4]+. 31P{1H} NMR (CD3CN): δ 39.6 (s). 1H NMR (CD3CN): δ 6.48 (1H, d, J = 7.7 Hz, H7), 7.06 (1H, t, J = 8.0 Hz, H6), 7.49 (1H, d, J = 8.1 Hz, H5), 7.62–7.82 (9H, m, Hpara + Hmeta), 7.93 (1H, dd, J = 8.0, 5.6 Hz, H3), 8.16 (6H, dd, J = 12.6, 7.6 Hz, Hortho), 8.67 (1H, d, J = 8.3 Hz, H4), 9.41 (1H, d, J = 5.3 Hz, H2). 13C{1H} NMR (CD3CN): δ 121.5 (s, C9H6N), 122.4 (s, C9H6N), 122.6 (s, C7), 124.1 (s, C5), 124.2 (s, C3), 128.4 (s, C6), 130.2 (d, J = 13.3 Hz, Cmeta), 131.5 (s, Cipso), 134.1 (d, J = 10.5 Hz, Cortho), 135.1 (d, J = 2.8 Hz, Cpara), 144.0 (s, C4), 145.8 (s, CNP), 149.0 (s, C2). IR (cm−1): ν 368 (Au–Cl), 1083 (v, br) and 622 (ClO4−), 1267 (PN). Conductivity (acetone): Λ = 128 μS cm−1.
N-8-C9H6N)-κ-N,N)Cl2] (4).
[PdCl2(COD)] (0.041 g, 0.15 mmol) and 1 (0.042 g, 0.15 mmol) were dissolved in dry and degassed CH2Cl2 (4 mL) and allowed to react for 3 hours at room temperature after which an orange precipitate had formed. The solvent was then reduced to a minimum in vacuo and upon addition of Et2O a pale orange solid was obtained which was washed twice with CHCl3 and dried in vacuo. Solution of 1 (0.081 g, 0.2 mmol) in CH2Cl2 was added dropwise to a solution of PdCl2(COD) (0.2 mmol) in CH2Cl2 while stirring at RT and allowed to react for 30 min. The product was then concentrated under vacuum and precipitated with diethyl ether, producing a light orange solid. Yield: 0.098 g, 84%. Anal. calcd for C27H21Cl2N2PPd (581.77): C, 55.74; H, 3.64; N, 4.82. Found: C, 55.38; H, 3.65; N, 4.90. MS(ESI+) 509.0 [M − Cl2]2+, 544.4 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 30.0 (s). 1H NMR (CDCl3): δ 6.27 (1H, d, J = 7.8 Hz, H7), 6.88 (1H, t, J = 7.9 Hz, H6), 7.13 (1H, d, J = 8.0 Hz, H5), 7.47 (1H, dd, J = 8.3, 5.2 Hz, H3), 7.58–7.65 (9H, m, Hpara + Hmeta), 8.11–8.18 (6H, m, Hortho), 8.22 (1H, d, J = 8.3 Hz, H4), 9.26 (1H, dd, J = 5.2, 1.4 Hz, H2). 13C{1H} NMR (CDCl3): δ 118.6 (s, C7), 118.7 (s, C5), 121.8 (s, C3), 125.2 (s, C9H6N), 126.2 (s, C9H6N), 127.2 (s, C6), 129.0 (d, J = 12.9 Hz, Cmeta), 130.0 (s, Cipso), 133.2 (d, J = 3.0 Hz, Cpara), 133.8 (d, J = 9.8 Hz, Cortho), 138.2 (s, C4), 149.7 (s, CNP), 150.2 (s, C2). IR (cm−1): ν 342 (Pd–Cl), 1268 (PN). Conductivity (acetone): Λ = 3.4 μS cm−1.
N-8-C9H6N)-κ-N,N)Cl2] (5).
A solution of 1 (0.1616 g, 0.4 mmol) in 20 mL of CH2Cl2 was added to a suspension of PtCl2 (0.1064 g, 0.4 mmol) in 15 mL and refluxed for 3 h. Upon completion a brown side-product was filtered off and the resulting yellow solution was concentrated under vacuum. Upon addition of diethyl ether, a bright yellow solid was obtained. Yield: 0.1662 g, 62%. Anal. calcd for C27H21Cl2N2PPt (670.43): C, 48.37; H, 3.16; N, 4.18. Found: C, 48.18; H, 3.14; N, 4.15. MS(ESI+) [m/z]: 635.1 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 31.3 (s). 195Pt{1H} NMR (CDCl3): δ −1930.3 (s). 1H NMR (CDCl3): δ 6.23 (1H, d, J = 7.9 Hz, H7), 6.83 (1H, t, J = 7.9 Hz, H6), 7.11 (1H, d, J = 8.0 Hz, H5), 7.46 (1H, t, J = 8.3 Hz, H3), 7.57–7.67 (9H, m, Hpara + Hmeta), 8.14 (6H, dd, J = 8.0, 8.2 Hz, Hortho), 8.30 (1H, d, J = 8.1 Hz, H4), 9.63 (1H, d, J = 5.0 Hz, H2). 13C{1H} NMR (CDCl3): δ 118.8 (d, J = 5.9 Hz, C7), 119.2 (s, C5), 122.1 (s, C3), 125.0 (s, C9H6N), 126.0 (s, C9H6N), 127.0 (s, C6), 129.0 (d, J = 12.6 Hz, Cmeta), 130.2 (s, Cipso), 133.2 (d, J = 2.9 Hz, Cpara), 133.8 (d, J = 9.9 Hz, Cortho), 137.3 (s, C4), 148.6 (s, C2), 151.0 (s, CNP). IR (cm−1): ν 341 (Pt–Cl), 1266 (PN). Conductivity (acetone): Λ = 1.2 μS cm−1.
N-8-C9H6N)}Cl] (6).
A solution of 1 (0.121 g, 0.3 mmol) in 20 mL of CH2Cl2 was added to Pd(OAc)2 (0.067 g, 0.3 mmol) in 10 mL CH2Cl2. The mixture was allowed to react overnight while stirring at RT. Excess LiCl (0.020 g, 0.47 mmol) in MeOH was then added and the mixture was allowed to react for 30 min. Concentration under vacuum and precipitation with diethyl ether afforded an orange solid. Yield: 0.133 g, 81%. Anal. calcd for C27H20ClN2PPd (545.31): C, 59.47; H, 3.70; N, 5.14. Found: C, 58.98; H, 3.72; N, 5.20. MS(ESI+) [m/z]: 544.4 [M], 509.3 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 43.4 (s). 1H NMR (CDCl3): δ 6.73 (1H, dd, J = 6.6, 1.7 Hz, H7), 6.90 (1H, ddd, J = 11.3, 7.7, 1.4 Hz, Hd), 7.05 (1H, ddd, J = 12.8, 7.4, 1.1 Hz, Hc), 7.14–7.20 (2H, m, H5 + H6), 7.26 (1H, t, J = 7.7 Hz, Hb), 7.45 (1H, dd, J = 8.3, 4.8 Hz, H3), 7.53–7.61 (4H, m, Hmeta), 7.65–7.61 (2H, m, Hpara), 7.91–7.99 (4H, m, Hortho), 8.17 (1H, dd, J = 8.3, 1.5 Hz, H4), 8.24 (1H, dd, J = 7.9, 1.6 Hz, Ha), 9.19 (1H, dd, J = 4.7, 1.6 Hz, H2). 13C{1H} NMR (CDCl3): δ 115.7 (d, J = 7.9 Hz, C7), 117.2 (s, C5 + C6), 121.9 (s, C3), 124.6 (d, J = 15.6 Hz, Cd), 125.5 (s, C9H6N), 126.4 (s, C9H6N), 127.4 (s, C5 + C6), 128.8 (d, J = 4.7 Hz, Cc), 129.4 (d, J = 11.9 Hz, Cmeta), 130.7 (s, Cipso), 131.3 (d, J = 3.1 Hz, Cb), 133.1 (d, J = 10.4 Hz, Cortho), 133.6 (d, J = 2.8 Hz, Cpara), 137.7 (s, C4), 138. 6 (d, J = 14.7 Hz, Ca), 143.5 (d, J = 144.7 Hz, Ce, C–P), 147.2 (s, CNP), 148.4 (s, C2), 154.5 (d, J = 19.1 Hz, Cf, C–Pd). IR (cm−1): ν 307 (Pd–Cl), 1285 (PN). Conductivity (acetone): Λ = 30.5 μS cm−1.
N-8-C9H6N)}Cl] (7).
The coordination platinum(II) complex 5 (0.167 g, 0.25 mmol) was dissolved in CH2Cl2 and the resulting solution refluxed for 3 days. After cooling at RT, the solution was concentrated under vacuum and 20 mL of diethyl ether were added. A yellow solid precipitated (7), which was filtered off, dried in vacuo and used without further purification. Yield: 0.1353 g, 87%. Anal. calcd for C27H20ClN2PPt·H2O (651.08): C, 49.74; H, 3.40; N, 4.30. Found: C, 49.77; H, 3.37; N, 4.20. MS (ESI+) [m/z]: 634.08 [M + H]+. 31P{1H} NMR (CDCl3): δ 44.7 (s, 2JPt–P = 426 Hz). 195Pt{1H} NMR (CDCl3): δ −2899.69 (d, 2JP–Pt = 419 Hz). 1H NMR (CDCl3): δ 6.74 (1H, t, J = 4.3 Hz, H7), 6.93 (1H, dd, J = 11.7, 7.6 Hz, Hd), 7.06 (1H, dd, J = 13.4, 6.9 Hz, Hc), 7.14–7.20 (2H, m, H5 + H6), 7.31 (1H, dd, J = 7.6 Hz, Hb), 7.51 (1H, dd, J = 8.3, 4.8 Hz, H3), 7.55–7.60 (4H, m, Hmeta), 7.69 (2H, t, J = 7.2 Hz, Hpara), 7.95 (4H, dd, J = 12.3, 7.6 Hz, Hortho), 8.23–8.29 (2H, m, H4 + Ha), 9.47 (1H, d, J = 4.3 Hz, H2). 13C{1H} NMR (CDCl3): δ 116.1 (s, C7), 117.8 (s, C5 + C6), 122.1 (s, C3), 124.0 (d, J = 15.6 Hz, Cc), 125.2 (s, C9H6N), 126.0 (s, C9H6N), 127.5 (s, C5 + C6), 129.1 (d, J = 4.7 Hz, Cd), 129.5 (d, J = 12.2 Hz, Cmeta), 131.1 (d, J = 3.2 Hz, Cb), 131.3 (s, Cipso), 133.2 (d, J = 10.5 Hz, Cortho), 133.7 (d, J = 2.8 Hz, Cpara), 137.1 (d, J = 13.8 Hz, Ca), 137.6 (s, C4), 142.7 (d, J = 138.4 Hz, Ce, C–P), 147.4 (s, CNP), 147.7 (s, C2), signal corresponding to C–Pt (Cf) not observable. IR (cm−1): ν 319 (Pt–Cl), 1286 (PN). Conductivity (acetone): Λ = 5.7 μS cm−1.
N-8-C9H6N (1)27.
An alternative method of synthesis was used: a solution of PPh3 (2.59 mmol) in CH2Cl2 was added dropwise to a solution of 8-azidoquinoline28 (2.59 mmol) in CH2Cl2 at RT and allowed to react for 1 h. The solvent was reduced in vacuo to 2 mL and Et2O (10 mL) was added. A pale-yellow solid precipitated and was filtered off and dried in vacuo. 1 was used without further purification. Yield: 0.857 g, 82%.
N-8-C9H6N)-κ-N,N)Cl2]X [X = PF6− (2); ClO4− (3)].
To K[AuCl4] (0.076 g, 0.2 mmol) in dry MeCN (10 mL), either AgClO4 (0.091 g, 0.44 mmol) for the obtainment of 2 or AgPF6 (0.111 g, 0.44 mmol) for the obtainment of 3 in dry MeCN (5 mL) was added. The reaction mixture was stirred in the dark for 30 min and subsequently filtered through celite (to remove AgCl). 1 (0.081 g, 0.2 mmol) in CH2Cl2 (1 mL) was added and the yellow solutions became purple instantly. After stirring for 30 min the reaction mixture was filtered through celite (to remove KClO4) and then the solvent was reduced in vacuo to ∼2 mL. Upon addition of Et2O (10 mL), a purple solid was obtained, which was washed with MeCN (<1 mL at a time) and Et2O and dried in vacuo. 2: Yield: 0.1198 g, 73%. Anal. calcd for C27H21AuCl2F6N2P2 (817.28): C, 39.68; H, 2.59; N, 3.43. Found: C, 39.21; H, 2.36; N, 3.21. MS(ESI+) [m/z]: 671.0 [M − PF6]+. 31P{1H} NMR (CD3CN): δ 39.6 (s), −142.5 (sept, PF6−). 1H NMR (CD3CN): δ 6.66 (1H, d, J = 7.8 Hz, H7), 7.15 (1H, t, J = 8.0 Hz, H6), 7.62 (1H, d, J = 8. 2 Hz, H5), 7.62–7.82 (9H, m, Hpara + Hmeta), 8.04 (1H, dd, J = 8.3, 5.7 Hz, H3), 8.13 (6H, dd, J = 12.6, 7.6 Hz, Hortho), 8.85 (1H, d, J = 8.3 Hz, H4), 9.44 (1H, d, J = 5.6 Hz, H2). 13C{1H} NMR (CD3CN): δ 121.6 (s, C9H6N), 122.6 (s, C9H6N), 122.9 (d, J = 5.0 Hz, C7), 123.6 (s, C5), 123.9 (s, C3), 128.8 (s, C6), 128.9 (d, J = 13.3 Hz, Cmeta), 131.4 (s, Cipso), 134.2 (d, J = 10.6 Hz, Cortho), 135.1 (d, J = 3.0 Hz, Cpara), 144.4 (s, C4), 146.1 (s, CNP), 148.4 (s, C2). IR (cm−1): ν 370 (Au–Cl), 838 (v br, PF6−), 1266 (PN). Conductivity (acetone): Λ = 110 μS cm−1. 3: Yield: 0.109 g, 69%. Anal. calcd for C27H21AuCl3N2O4P (771.77): C, 42.02; H, 2.74; N, 3.63. Found: C, 42.08; H, 2.96; N, 3.66. MS(ESI+) [m/z]: 671.1 [M − ClO4]+. 31P{1H} NMR (CD3CN): δ 39.6 (s). 1H NMR (CD3CN): δ 6.48 (1H, d, J = 7.7 Hz, H7), 7.06 (1H, t, J = 8.0 Hz, H6), 7.49 (1H, d, J = 8.1 Hz, H5), 7.62–7.82 (9H, m, Hpara + Hmeta), 7.93 (1H, dd, J = 8.0, 5.6 Hz, H3), 8.16 (6H, dd, J = 12.6, 7.6 Hz, Hortho), 8.67 (1H, d, J = 8.3 Hz, H4), 9.41 (1H, d, J = 5.3 Hz, H2). 13C{1H} NMR (CD3CN): δ 121.5 (s, C9H6N), 122.4 (s, C9H6N), 122.6 (s, C7), 124.1 (s, C5), 124.2 (s, C3), 128.4 (s, C6), 130.2 (d, J = 13.3 Hz, Cmeta), 131.5 (s, Cipso), 134.1 (d, J = 10.5 Hz, Cortho), 135.1 (d, J = 2.8 Hz, Cpara), 144.0 (s, C4), 145.8 (s, CNP), 149.0 (s, C2). IR (cm−1): ν 368 (Au–Cl), 1083 (v, br) and 622 (ClO4−), 1267 (PN). Conductivity (acetone): Λ = 128 μS cm−1.
N-8-C9H6N)-κ-N,N)Cl2] (4).
[PdCl2(COD)] (0.041 g, 0.15 mmol) and 1 (0.042 g, 0.15 mmol) were dissolved in dry and degassed CH2Cl2 (4 mL) and allowed to react for 3 hours at room temperature after which an orange precipitate had formed. The solvent was then reduced to a minimum in vacuo and upon addition of Et2O a pale orange solid was obtained which was washed twice with CHCl3 and dried in vacuo. Solution of 1 (0.081 g, 0.2 mmol) in CH2Cl2 was added dropwise to a solution of PdCl2(COD) (0.2 mmol) in CH2Cl2 while stirring at RT and allowed to react for 30 min. The product was then concentrated under vacuum and precipitated with diethyl ether, producing a light orange solid. Yield: 0.098 g, 84%. Anal. calcd for C27H21Cl2N2PPd (581.77): C, 55.74; H, 3.64; N, 4.82. Found: C, 55.38; H, 3.65; N, 4.90. MS(ESI+) 509.0 [M − Cl2]2+, 544.4 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 30.0 (s). 1H NMR (CDCl3): δ 6.27 (1H, d, J = 7.8 Hz, H7), 6.88 (1H, t, J = 7.9 Hz, H6), 7.13 (1H, d, J = 8.0 Hz, H5), 7.47 (1H, dd, J = 8.3, 5.2 Hz, H3), 7.58–7.65 (9H, m, Hpara + Hmeta), 8.11–8.18 (6H, m, Hortho), 8.22 (1H, d, J = 8.3 Hz, H4), 9.26 (1H, dd, J = 5.2, 1.4 Hz, H2). 13C{1H} NMR (CDCl3): δ 118.6 (s, C7), 118.7 (s, C5), 121.8 (s, C3), 125.2 (s, C9H6N), 126.2 (s, C9H6N), 127.2 (s, C6), 129.0 (d, J = 12.9 Hz, Cmeta), 130.0 (s, Cipso), 133.2 (d, J = 3.0 Hz, Cpara), 133.8 (d, J = 9.8 Hz, Cortho), 138.2 (s, C4), 149.7 (s, CNP), 150.2 (s, C2). IR (cm−1): ν 342 (Pd–Cl), 1268 (PN). Conductivity (acetone): Λ = 3.4 μS cm−1.
N-8-C9H6N)-κ-N,N)Cl2] (5).
A solution of 1 (0.1616 g, 0.4 mmol) in 20 mL of CH2Cl2 was added to a suspension of PtCl2 (0.1064 g, 0.4 mmol) in 15 mL and refluxed for 3 h. Upon completion a brown side-product was filtered off and the resulting yellow solution was concentrated under vacuum. Upon addition of diethyl ether, a bright yellow solid was obtained. Yield: 0.1662 g, 62%. Anal. calcd for C27H21Cl2N2PPt (670.43): C, 48.37; H, 3.16; N, 4.18. Found: C, 48.18; H, 3.14; N, 4.15. MS(ESI+) [m/z]: 635.1 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 31.3 (s). 195Pt{1H} NMR (CDCl3): δ −1930.3 (s). 1H NMR (CDCl3): δ 6.23 (1H, d, J = 7.9 Hz, H7), 6.83 (1H, t, J = 7.9 Hz, H6), 7.11 (1H, d, J = 8.0 Hz, H5), 7.46 (1H, t, J = 8.3 Hz, H3), 7.57–7.67 (9H, m, Hpara + Hmeta), 8.14 (6H, dd, J = 8.0, 8.2 Hz, Hortho), 8.30 (1H, d, J = 8.1 Hz, H4), 9.63 (1H, d, J = 5.0 Hz, H2). 13C{1H} NMR (CDCl3): δ 118.8 (d, J = 5.9 Hz, C7), 119.2 (s, C5), 122.1 (s, C3), 125.0 (s, C9H6N), 126.0 (s, C9H6N), 127.0 (s, C6), 129.0 (d, J = 12.6 Hz, Cmeta), 130.2 (s, Cipso), 133.2 (d, J = 2.9 Hz, Cpara), 133.8 (d, J = 9.9 Hz, Cortho), 137.3 (s, C4), 148.6 (s, C2), 151.0 (s, CNP). IR (cm−1): ν 341 (Pt–Cl), 1266 (PN). Conductivity (acetone): Λ = 1.2 μS cm−1.
N-8-C9H6N)}Cl] (6).
A solution of 1 (0.121 g, 0.3 mmol) in 20 mL of CH2Cl2 was added to Pd(OAc)2 (0.067 g, 0.3 mmol) in 10 mL CH2Cl2. The mixture was allowed to react overnight while stirring at RT. Excess LiCl (0.020 g, 0.47 mmol) in MeOH was then added and the mixture was allowed to react for 30 min. Concentration under vacuum and precipitation with diethyl ether afforded an orange solid. Yield: 0.133 g, 81%. Anal. calcd for C27H20ClN2PPd (545.31): C, 59.47; H, 3.70; N, 5.14. Found: C, 58.98; H, 3.72; N, 5.20. MS(ESI+) [m/z]: 544.4 [M], 509.3 [M − Cl]+. 31P{1H} NMR (CDCl3): δ 43.4 (s). 1H NMR (CDCl3): δ 6.73 (1H, dd, J = 6.6, 1.7 Hz, H7), 6.90 (1H, ddd, J = 11.3, 7.7, 1.4 Hz, Hd), 7.05 (1H, ddd, J = 12.8, 7.4, 1.1 Hz, Hc), 7.14–7.20 (2H, m, H5 + H6), 7.26 (1H, t, J = 7.7 Hz, Hb), 7.45 (1H, dd, J = 8.3, 4.8 Hz, H3), 7.53–7.61 (4H, m, Hmeta), 7.65–7.61 (2H, m, Hpara), 7.91–7.99 (4H, m, Hortho), 8.17 (1H, dd, J = 8.3, 1.5 Hz, H4), 8.24 (1H, dd, J = 7.9, 1.6 Hz, Ha), 9.19 (1H, dd, J = 4.7, 1.6 Hz, H2). 13C{1H} NMR (CDCl3): δ 115.7 (d, J = 7.9 Hz, C7), 117.2 (s, C5 + C6), 121.9 (s, C3), 124.6 (d, J = 15.6 Hz, Cd), 125.5 (s, C9H6N), 126.4 (s, C9H6N), 127.4 (s, C5 + C6), 128.8 (d, J = 4.7 Hz, Cc), 129.4 (d, J = 11.9 Hz, Cmeta), 130.7 (s, Cipso), 131.3 (d, J = 3.1 Hz, Cb), 133.1 (d, J = 10.4 Hz, Cortho), 133.6 (d, J = 2.8 Hz, Cpara), 137.7 (s, C4), 138. 6 (d, J = 14.7 Hz, Ca), 143.5 (d, J = 144.7 Hz, Ce, C–P), 147.2 (s, CNP), 148.4 (s, C2), 154.5 (d, J = 19.1 Hz, Cf, C–Pd). IR (cm−1): ν 307 (Pd–Cl), 1285 (PN). Conductivity (acetone): Λ = 30.5 μS cm−1.
N-8-C9H6N)}Cl] (7).
The coordination platinum(II) complex 5 (0.167 g, 0.25 mmol) was dissolved in CH2Cl2 and the resulting solution refluxed for 3 days. After cooling at RT, the solution was concentrated under vacuum and 20 mL of diethyl ether were added. A yellow solid precipitated (7), which was filtered off, dried in vacuo and used without further purification. Yield: 0.1353 g, 87%. Anal. calcd for C27H20ClN2PPt·H2O (651.08): C, 49.74; H, 3.40; N, 4.30. Found: C, 49.77; H, 3.37; N, 4.20. MS (ESI+) [m/z]: 634.08 [M + H]+. 31P{1H} NMR (CDCl3): δ 44.7 (s, 2JPt–P = 426 Hz). 195Pt{1H} NMR (CDCl3): δ −2899.69 (d, 2JP–Pt = 419 Hz). 1H NMR (CDCl3): δ 6.74 (1H, t, J = 4.3 Hz, H7), 6.93 (1H, dd, J = 11.7, 7.6 Hz, Hd), 7.06 (1H, dd, J = 13.4, 6.9 Hz, Hc), 7.14–7.20 (2H, m, H5 + H6), 7.31 (1H, dd, J = 7.6 Hz, Hb), 7.51 (1H, dd, J = 8.3, 4.8 Hz, H3), 7.55–7.60 (4H, m, Hmeta), 7.69 (2H, t, J = 7.2 Hz, Hpara), 7.95 (4H, dd, J = 12.3, 7.6 Hz, Hortho), 8.23–8.29 (2H, m, H4 + Ha), 9.47 (1H, d, J = 4.3 Hz, H2). 13C{1H} NMR (CDCl3): δ 116.1 (s, C7), 117.8 (s, C5 + C6), 122.1 (s, C3), 124.0 (d, J = 15.6 Hz, Cc), 125.2 (s, C9H6N), 126.0 (s, C9H6N), 127.5 (s, C5 + C6), 129.1 (d, J = 4.7 Hz, Cd), 129.5 (d, J = 12.2 Hz, Cmeta), 131.1 (d, J = 3.2 Hz, Cb), 131.3 (s, Cipso), 133.2 (d, J = 10.5 Hz, Cortho), 133.7 (d, J = 2.8 Hz, Cpara), 137.1 (d, J = 13.8 Hz, Ca), 137.6 (s, C4), 142.7 (d, J = 138.4 Hz, Ce, C–P), 147.4 (s, CNP), 147.7 (s, C2), signal corresponding to C–Pt (Cf) not observable. IR (cm−1): ν 319 (Pt–Cl), 1286 (PN). Conductivity (acetone): Λ = 5.7 μS cm−1.
Luminescence studies
Absorption spectra in solution were recorded using a Unicam UV-4 spectrophotometer. Steady-state photoluminescence spectra were recorded with a Jobin-Yvon Horiba Fluorolog FL-3-11 spectrofluorimeter using band pathways of 3 nm for both excitation and emission. Phosphorescence lifetimes were recorded using a Fluoromax phosphorimeter accessory containing a UV xenon flash tube at a flash rate between 0.05 and 25 Hz. The lifetime data were fitted using the Jobin-Yvon software package and the Origin 7.0 program.Cell culture and inhibition of cell growth
The human lung cancer cell line A549 and the human ovarian cancer cell line A2780 (obtained from the European Centre of Cell Cultures ECACC, Salisbury, UK) were cultured in DMEM (Dulbecco's modified Eagle medium) and RPMI, respectively, containing GlutaMaxI supplemented with 10% FBS and 1% penicillin/streptomycin (all from Invitrogen), at 37 °C under a humidified atmosphere of 95% air and 5% CO2 (Heraeus, Germany). Non-tumoral human embryonic kidney cells HEK-293T were kindly provided by Dr Maria Pia Rigobello (CNRS, Padova, Italy), and were cultivated in DMEM medium, added with GlutaMaxI (containing 10% FBS and 1% penicillin/streptomycin (all from Invitrogen)) and incubated at 37 °C and 5% CO2. For the evaluation of growth inhibition, cells were seeded in 96-well plates (Costar, Integra Biosciences, Cambridge, MA) and grown for 24 h in complete medium. Solutions of the compounds were prepared by diluting a freshly prepared stock solution (in DMSO) of the corresponding compound in aqueous media. Afterwards, the intermediate dilutions of the compounds were added to the wells (100 μL) to obtain a final concentration ranging from 0 to 150 μM, and the cells were incubated for 72 h. DMSO at comparable concentrations did not show any effects on cell cytotoxicity. Following 72 h of drug exposure, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the cells at a final concentration of 0.25 mg mL−1, incubated for 2 h, and then the culture medium was removed and the violet formazan (artificial chromogenic precipitate of the reduction of tetrazolium salts by dehydrogenases and reductases) was dissolved in DMSO. The optical density of each well (96-well plates) was quantified three times in tetraplicates at 540 nm using a multiwell plate reader, and the percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as the mean ± SE of at least three independent experiments.Interaction of metal complexes with plasmid (pBR322) DNA by electrophoresis (mobility shift assay)
10 μL aliquots of pBR322 plasmid DNA (20 μg mL−1) in a buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were incubated with molar ratios between 0.25 and 4.0 of the compounds at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin–DNA adduct were prepared also as controls. After the incubation period, 2 μL of loading dye were added to the samples, and of these mixtures, only 7 μL were finally loaded into the gel. The samples were separated by electrophoresis in 1% (w/v) agarose gel for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterwards, the gel was dyed for 30 min with a solution of GelRed Nucleic Acid stain.Interaction of metal complexes with HSA by fluorescence spectroscopy
The excitation wavelength was adjusted at 295 nm, and the emission spectra were recorded at room temperature in the range of 300–450 nm. The fluorescence intensities of the new compounds, the buffer and the DMSO are negligible under these conditions, and so is the effect of additions of pure DMSO to the fluorescence of HSA. An 8 mM solution of each compound in DMSO was prepared and ten aliquots of 2.5 μL were added successively to a solution of HSA (10 μM) in phosphate buffer (pH = 7.39), the fluorescence being measured after each addition.The data were analyzed using the classical Stern–Volmer equation (F0/F = 1 + KSV[Q]).
Conclusions
In conclusion, we have prepared a series of luminescent iminophosphorane complexes of gold(III), palladium(II) and platinum(II) derived from 8-aminoquinoline. As previously noted, the stability of coordination iminophoshorane complexes is lower than that of their cyclometalated counterparts (especially in the case of gold(III) derivatives). The coordination palladium(II) and platinum(II) compounds can evolve further, under appropriate conditions, to give stable cyclometalated endo species [M{κ3-C,N,N-C6H4(PPh2N-8-C9H6N)}Cl] (M = Pd, Pt) by C–H activation of the phenyl group of the PPh3 fragment. All the cyclometalated compounds are very stable in DMSO solution. The compounds exhibit important cytotoxic effects in the low micromolar range in a human ovarian cancer cell line (A2780S), and in particular the Pd(II) derivatives (monometallic coordination 4 and cyclopaladated 6) show promising selectivity being poorly toxic for the non-tumorigenic human embryonic kidney cell line (HEK-293T).
Studies of the interactions of the compounds with plasmid (pBR322) DNA indicate that (unless they lose the iminophosphorane skeleton in solution) they have no or little interaction with DNA, supporting the idea of different mechanisms of action with respect to cisplatin. The complexes display a concentration dependent fluorescence quenching of HSA which has been correlated to a faster reactivity of our compounds with HSA compared to cisplatin.
Although, unfortunately, the luminescence properties of the compounds did not allow us to detect them in cells via fluorescence microscopy, these results will allow us to design new IM complexes in order to achieve better stability in biological media and to increase the fluorescence detection limits. More specifically, we will focus on the synthesis of cyclometalated IM derivatives containing luminescent phosphines.
Notably, the results described herein and previous reports of our group20–24 support the idea that cyclometalated iminophosphorane compounds of d8 transition metals are an attractive type of compounds which exert anticancer effects in vitro based on different mechanisms of action with respect to cisplatin.
Acknowledgements
Research at Brooklyn College was supported by a grant from the National Institute of General Medical Sciences (NIGMS), SC2GM082307, and a grant from the National Cancer Institute (NCI), 1SC1CA182844 (M.C.). We thank the University of Groningen (Rosalind Franklin Fellowship, A.C.) and the New York Louis Stokes Alliance for Minority Participation (fellowship to undergraduate student F.B.) for funding. The Spanish team thanks the MINECO “Ministerio de Ciencia y Economía” (CTQ2011-22589) and Gobierno de Aragón (Dpto. de Ciencia, Tecnología y Universidad) for financial support (J.J.). EU COST Action CM1105 is gratefully acknowledged for providing opportunities of discussion and for financial support. We thank Dr Esteban P. Urriolabeitia for useful discussions.Notes and references
- A. M. Thayer, Chem. Eng. News, 2010, 88(26), 24–28 Search PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- E. Alessio, Bioinorganic Medicinal Chemistry, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- A. L. Noffke, A. Habtemarian, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219 RSC.
- S. M. Aris and N. P. Farrell, Eur. J. Inorg. Chem., 2009, 1293 CrossRef CAS PubMed.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 41, 279 Search PubMed.
- S. Komeda and A. Casini, Curr. Top. Med. Chem., 2012, 12, 219 CrossRef CAS.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90 CrossRef CAS PubMed.
- A. Casini, J. Inorg. Biochem., 2012, 109, 97 CrossRef CAS PubMed.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550 CAS.
- A. Casini, C. Hartinger, C. Gabbiani, E. Mini, P. J. Dyson, B. K. Keppler and L. Messori, J. Inorg. Biochem., 2008, 102, 564 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817 RSC.
- A. M. Pizarro, A. Habtemarian and P. Sadler, Top. Organomet. Chem., 2010, 32, 21 CrossRef CAS.
- A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135 RSC.
- E. Schuh, C. Pfluger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518 CrossRef CAS PubMed.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863 RSC.
- L. Dalla Via, C. Nardon and D. Fregona, Future Med. Chem., 2012, 4, 525 CrossRef CAS PubMed.
- M. Serratrice, F. Edafe, F. Mendes, R. Scopelliti, S. M. Zakeeruddin, M. Gratzel, I. Santos, M. A. Cinellu and A. Casini, Dalton Trans., 2012, 41, 3287 RSC.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196 CrossRef CAS PubMed.
- N. Shaik, A. Martínez, I. Augustin, H. Giovinazzo, A. Varela, R. Aguilera, M. Sanaú and M. Contel, Inorg. Chem., 2009, 48, 1577 CrossRef CAS PubMed.
- L. Vela, M. Contel, L. Palomera, G. Azaceta and I. Marzo, J. Inorg. Biochem., 2011, 105, 1306 CrossRef CAS PubMed.
- M. Carreira, R. Calvo-Sanjuán, M. Sanaú, X. Zhao, R. S. Magliozzo, I. Marzo and M. Contel, J. Inorg. Biochem., 2012, 116, 204 CrossRef CAS PubMed.
- M. Carreira, R. Calvo-Sanjuán, M. Sanaú, I. Marzo and M. Contel, Organometallics, 2012, 31, 5772 CrossRef CAS PubMed.
- N. Lease, V. Vasileviski, M. Carreira, A. de Almeida, M. Sanaú, P. Hirva, A. Casini and M. Contel, J. Med. Chem., 2013, 56, 5806 CrossRef CAS PubMed.
- D. L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 2 Search PubMed.
- P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966 CrossRef CAS PubMed.
- S. Tasan, O. Zava, B. Bertrand, C. Berhard, C. Goze, M. Picquet, P. Le Gendre, P. Harvey, F. Denat, A. Casini and E. Bodio, Dalton Trans., 2013, 42, 6102 RSC.
- N. Abe, H. Fujii, K. Tahara and M. Shiro, Heterocycles, 2001, 55, 1659 CrossRef CAS PubMed.
- Q. Liu and Y. Tor, Org. Lett., 2003, 5, 2751 CrossRef PubMed.
- J. Vicente, J. A. Abad, R. Clemente, J. Lopez-Serrano, P. G. Jones and D. Bautista, Organometallics, 2003, 22, 4248 CrossRef CAS.
- L. R. Falvello, M. M. Gracia, I. Lazaro, R. Navarro and E. P. Urriolabeitia, New J. Chem., 1999, 35, 227 RSC.
- D. Aguilar, M. Contel, R. Navarro, T. Soler and E. P. Urriolabeitia, J. Organomet. Chem., 2009, 694, 486 CrossRef CAS PubMed.
- M. A. Bennett, M. Contel, D. C. R. Hockless, L. L. Welling and A. C. Willis, Inorg. Chem., 2002, 41, 844 CrossRef CAS PubMed.
- D. Aguilar, M. A. Aragues, R. Bielsa, E. Serrano, R. Navarro and E. P. Urriolabeitia, Organometallics, 2007, 26, 3541 CrossRef CAS.
- R. Bielsa, R. Navarro, E. P. Urriolabeitia and A. Lledos, Inorg. Chem., 2007, 46, 10133 CrossRef CAS PubMed.
- R. Bielsa, A. Larrea, R. Navarro, T. Soler and E. P. Urriolabeitia, Eur. J. Inorg. Chem., 2005, 1724 CrossRef CAS.
- D. Aguilar, R. Bielsa, M. Contel, A. Lledos, R. Navarro, T. Soler and E. P. Urriolabeitia, Organometallics, 2008, 27, 2929 CrossRef CAS.
- J. A. G. Williams, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer-Verlag, Berlin, 2007, vol. 281, p. 205 Search PubMed.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS PubMed and references therein.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Q. Huo, Inorg. Chem., 2010, 49, 5107 CrossRef CAS PubMed.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed.
- V. Sicilia, S. Fuertes, A. Martín and A. Palacios, Organometallics, 2013, 32, 4092 CrossRef CAS.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2001, 219, 489 CrossRef.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409 RSC.
- J. C. Dabrowiak, Metals in Medicine, John Wiley and Sons, Ltd, Chichester, UK, 2009, ch. 4, p. 109 Search PubMed.
- H.-K. Liu and P. Sadler, Acc. Chem. Res., 2011, 44, 349 CrossRef CAS PubMed.
- For example: J. Ruíz, N. Cutillas, C. Vicente, M. D. Villa, G. López, J. Lorenzo, F. X. Avilés, V. Moreno and D. Bautista, Inorg. Chem., 2005, 44, 7365 CrossRef PubMed.
- E. Gao, M. Zhu, L. Liu, Y. Huang, L. Wang, S. Shi, Z. Chuyue and Y. Sun, Inorg. Chem., 2010, 49, 3261 CrossRef CAS PubMed.
- A. Quiroga, J. M. Pérez, I. López-Solera, J. R. Masaguer, A. Luque, P. Román, A. Edwards, C. Alonso and C. Navarro-Ranninguer, J. Med. Chem., 1998, 41, 1399 CrossRef CAS PubMed.
- E.-J. Gao, K.-H. Wang, M.-C. Zhu and L. Liu, Eur. J. Med. Chem., 2010, 45, 2784 CrossRef CAS PubMed.
- K. Fox, Drug-DNA Interact. Protocols. Methods in Mol. Biol, Humana Press Inc., Totowa, NJ, 1997 Search PubMed.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224 CrossRef CAS PubMed.
- J. R. Lacowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999, ch. 8, p. 238 Search PubMed.
Footnotes |
| † In memory of Prof. María Pilar Garcia Clemente, who was an invaluable role model as a female inorganic chemistry professor for many of us at the University of Zaragoza (Spain). |
| ‡ Electronic supplementary information (ESI) available: Table with the crystal data and structure refinement for complexes 3 and 4, luminescence profiles and studies of the luminescence overtime in solution for ligand 1 and compounds 2–7; stability of compounds 2–7 in d6-DMSO solution overtime by 31P{1H} NMR spectroscopy. CCDC 977990 and 977991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00003j |
| This journal is © the Partner Organisations 2014 |