Visible-light radical reaction designed by Ru- and Ir-based photoredox catalysis
Takashi
Koike
* and
Munetaka
Akita
*
Chemical Resources Laboratory, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: koike.t.ad@m.titech.ac.jp; makita@res.titech.ac.jp; Fax: +81-45-924-5230; Tel: +81-45-924-5230
First published on 2nd July 2014
Abstract
Photoredox catalysis by well-known ruthenium(II) polypyridine complexes and the relevant Ir cyclometalated derivatives has become a powerful tool for redox reactions in synthetic organic chemistry, because they can effectively catalyze single-electron-transfer (SET) processes by irradiation with visible light. Remarkably, since 2008, this photocatalytic system has gained importance in radical reactions from the viewpoint of not only a useful and selective protocol but also green chemistry. In this review, we will describe recent developments of radical reactions involving various carbon-centered radicals through photoredox processes mediated by Ru- and Ir-based photocatalysts.
 Takashi Koike | Takashi Koike was born in 1977 in Toyama, Japan. He received his doctoral degree from the Tokyo Institute of Technology in 2005 under the guidance of Prof. Takao Ikariya. After his graduate career, he joined the Professor Robert H. Grubbs’ group in the California Institute of Technology, USA as a postdoctoral research scholar. In 2007 he returned to the Tokyo Institute of Technology as an assistant professor of Chemical Resources Laboratory. His research interests are in synthetic organic chemistry and organometallic chemistry. Recently, his efforts have been directed to the development of visible-light-driven photocatalysis. |
 Munetaka Akita | Munetaka Akita was born in Fukuoka, Japan, in 1957. He studied organometallic chemistry under the supervision of Professors Makoto Kumada and Kohei Tamao (Kyoto University) and Professors Akira Nakamura and Hajime Yasuda (Osaka University). Soon after receiving his Ph.D. degree in 1984, he joined Professor Yoshihiko Moro-oka's group at the Tokyo Institute of Technology as a research associate. He was appointed as a professor in 2002. The recent research efforts of his group have been devoted to application of carbon-rich organometallics to molecular devices (molecular wires and stimuli-responsive systems), visible-light-promoted catalysis, and supramolecular systems based on anthracene. |
1. Introduction
For the past few years, the visible-light-induced photocatalytic strategy, i.e. photoredox catalysis, with well-defined ruthenium(II) polypyridine complexes and the relevant Ir cyclometalated derivatives has emerged as a powerful tool for redox reactions due to its ability to cause single-electron-transfer (SET) processes under mild conditions by irradiation of visible light (sunlight).1,2 It is attracting the attention of many researchers from the viewpoint of the development of new transformation as well as green and sustainable chemistry. Since 2008, it has represented one of the new trends in organic chemistry (Fig. 1) and is still expanding.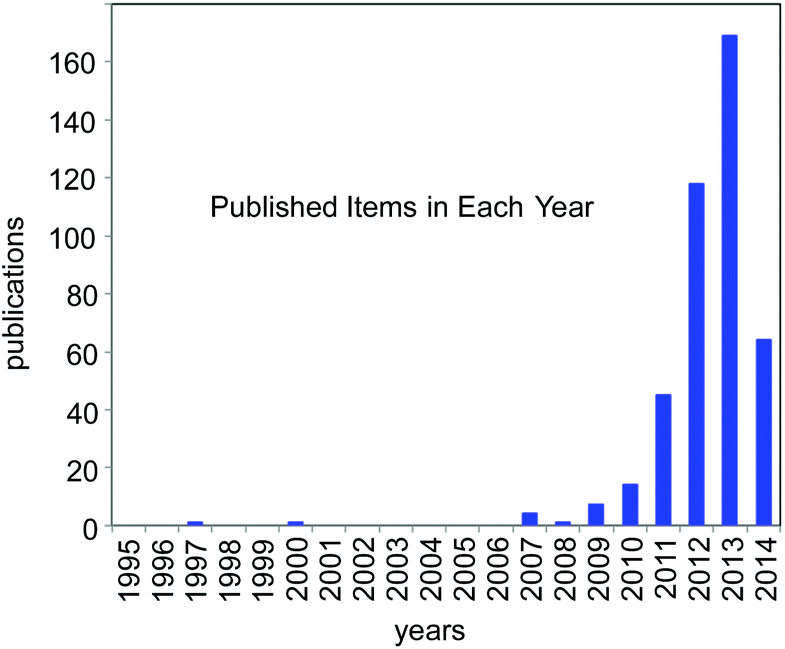 | ||
| Fig. 1 Number of publications in the last 20 years concerned with the study on “photoredox catalysis” (from ISI web of Knowledge; as of 05/09/2014). | ||
Redox reactions are often associated with radical reactions.3 Conventional strategies, such as the use of a stoichiometric amount of oxidant or reductant, electrolysis, and photolysis are accompanied by disadvantages: the formation of considerable amounts of waste derived from the oxidant or reductant or the need for special equipment for electrochemical or photochemical processes. In modern synthetic organic chemistry, protocols need to be easy and safe to use, as well as having efficient and selective outcomes.
Photoredox processes with octahedral Ru and Ir polypyridine complexes are triggered by visible light (sunlight), which is regarded as a mild and abundant stimulus. In addition, normal glassware is allowed to be used. Thus, photoredox catalysis shows merits as a useful tool for generation of organic radicals. In this review, we will deal with the generation of carbon-centered radicals from a variety of precursors and their subsequent radical reactions promoted by metal complex-based photoredox catalysts, especially using octahedral Ru and Ir polypyridine complexes. Before discussing the main subject, we will review the basic photochemical properties of representative photoredox catalysts and strategies for photoredox catalysis.
1.1 Photochemical properties of [Ru(bpy)3]2+
The well-investigated tris(2,2′-bipyridine)ruthenium complex, [Ru(bpy)3]2+, is the most widely used photoredox catalyst because it exhibits outstanding photochemical properties.4 In this section, photochemical properties of [Ru(bpy)3]2+ will be explained as a representative example. Firstly, its absorption maximum (around 450 nm) is in the visible-light region, indicating that it can be easily excited by visible light. Photoexcitation can be performed using a fluorescent light bulb, a LED lamp, a Xe lamp, or even natural sunlight. Secondly, the lifetime of the luminescent triplet excited state (τ = approximately 1 μs), *[Ru(bpy)3]2+, resulting from electron transfer from the dπ orbital of the ruthenium center to the π* orbital of the 2,2′-bipyridine ligand (MLCT) followed by intersystem crossing, is sufficiently long enough for chemical transformations to proceed. Thirdly, the triplet excited state, *[Ru(bpy)3]2+, undergoes single-electron-transfer (SET) to/from external organic molecules, i.e. this triplet state can serve either as a single electron oxidant or reductant. Further successive redox reactions regenerate the initial ground state, [Ru(bpy)3]2+ (Scheme 1). In addition, the photoredox catalyst is air- and moisture-stable and commercially available. Therefore, photoredox catalysis using this Ru photoredox catalyst has become a popular tool for redox reactions of diverse organic compounds.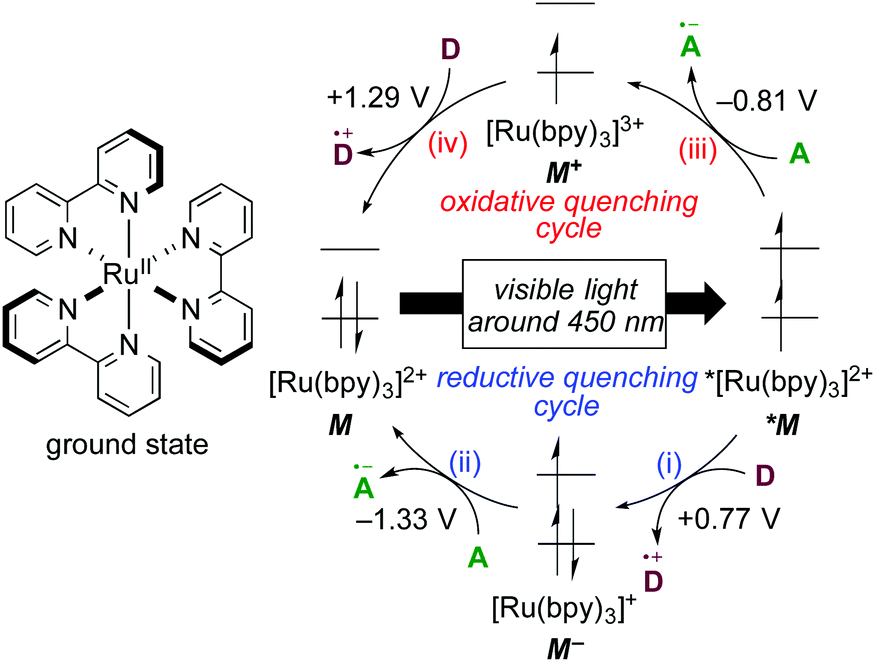 | ||
| Scheme 1 Photoredox cycle of [Ru(bpy)3]2+. | ||
The cycle consisting of a sequence of (i) electron transfer from an electron donor D to *[Ru(bpy)3]2+ (*M) associated with formation of reduced [Ru(bpy)3]+ (M−) and (ii) reduction of an electron acceptor A associated with regeneration of the ground state of the catalyst (M) is called a reductive quenching cycle. On the other hand, the cycle consisting of a sequence of (iii) electron transfer from *[Ru(bpy)3]2+ (*M) to an electron acceptor A associated with formation of the high-oxidation-state [Ru(bpy)3]3+ (M+) and (iv) oxidation of an electron donor D associated with regeneration of the ground state of the catalyst (M) is called an oxidative quenching cycle. Both of these cycles produce D+˙ and A−˙ radicals in a single reactor through SET processes to make, overall, the transformation redox-neutral. The terms reductive and oxidative can be confusing and require explanation. Reductive refers to reduction of the photoexcited species *M, whereas the external electron donor D is oxidized in the same process. Oxidative means oxidation of the photoexcited species *M concomitant with reduction of the external electron acceptor A.
1.2 Basic strategies for photoredox catalysis
Redox potentials of catalytic species and organic reactants are the most important factors to be considered when photoredox reactions are designed. As shown in Scheme 1, the highly reduced species, [Ru(bpy)3]+ (M−), generated upon reductive quenching, serves as a 1e-reductant (M/M− = −1.33 V vs. SCE = Standard Calomel Electrode, in MeCN) and, to be noted, is a stronger reductant than the photoexcited species itself (M+/*M = −0.81 V vs. SCE in MeCN). On the other hand, an oxidative quenching cycle produces the highly oxidized species [Ru(bpy)3]3+ (M+), which turns out to be a strong 1e-oxidant. In the field of photoredox chemistry using [Ru(bpy)3]2+, it is well-known that the combined use of the sacrificial electron donor D (e.g., triethylamine) or acceptor A (e.g., methyl viologen: N,N′-dimethyl-4,4′-bipyridinium) efficiently leads to redox reactions mediated by [Ru(bpy)3]+ (M−) and [Ru(bpy)3]3+ (M+), respectively. Thus, addition of a sacrificial reductant or oxidant has been the classical strategy to promote photoredox reactions.The relevant ruthenium polypyridine derivatives and isoelectronic cyclometalated iridium complexes also exhibit visible-light-induced photoredox catalysis, while the redox potentials of the catalytic species are dependent on the structure of the ligands and the central metal.1h,4a,b,5 In addition, photocatalytic reactions with copper, platinum and gold complexes have been reported.6 Moreover, organic photoredox catalysis, which is mediated by organophotocatalysts, is also an interesting topic;7 however, a long lifetime of excited states of metal-based complexes, derived from the heavy atom effect, can promote chemical transformation more efficiently. Photoredox catalysts featured in this review are summarized in Scheme 2. The tuning of redox potentials by the ligands and the metal is regarded as another strategy for improving the efficiency of catalytic reactions.
 | ||
| Scheme 2 Photoredox catalysts featured in this review. The redox potentials for *[Ir(Fppy)3] were calculated based on literature data.5e,h | ||
Dual catalytic systems combining photoredox catalysis with another catalysis can achieve reactions, which cannot be achieved by just one of the catalytic systems.1n A catalytic system can activate organic molecules (D or A) or intermediates to convert the reactant into reactive species with permissible redox potentials, at which photocatalysts can undergo SET. In particular, this strategy may be applied to selective transformations, such as enantioselective and regioselective reactions.
In the subsequent sections, methods for generation of organic radicals by photoredox catalysis will be outlined as follows: (i) generation of ionic radicals from redox reaction of alkenes, (ii) oxidatively induced generation of organic radicals and (iii) reductively induced generation of organic radicals (Scheme 3). Judicial choice of appropriate radical precursors, substrates and photocatalysts is the key to construct a sophisticated photocatalytic radical reaction.
 | ||
| Scheme 3 Generation of various C-centered radicals highlighted in this review. | ||
2. Generation of ionic radicals from redox reaction of alkenes
Neutral molecules undergo 1e-redox reaction, i.e. SET, to produce ionic radical species. The group of Yoon has been intensively developing cycloaddition reactions via ionic radicals generated from either 1e-reduction or 1e-oxidation of alkenes by photoredox catalysis. In 2008, they demonstrated that, in the presence of the Ru photocatalyst [Ru(bpy)3]Cl2 (5.0 mol%), a sacrificial electron donor (iPr2NEt), and a Lewis acid (LiBF4), bis(enone) 1 is smoothly reduced to the corresponding radical anion, which is converted to the [2 + 2] intramolecular cycloadduct 2 (Scheme 4).8a In these reactions, addition of an electron donor and a Lewis acid is essential to produce a strongly reducing Ru species M− from the photoexcited species *M and to lower the LUMO level of 1, respectively. Analogous [2 + 2] cyclization by high-energy UV photolysis9 and electrochemical methods10 was reported, but these reaction systems required special reactors. In contrast, the present photocatalytic protocol allows one to conduct the reaction using usual glassware and a readily available light source, including natural sunlight. Furthermore, they extended the present reaction based on the reductive quenching cycle to crossed intermolecular [2 + 2] cycloadditions of enones8b and intramolecular [3 + 2] cycloadditions of aryl cyclopropyl ketones.8c The latter reaction is induced by the action of the Ru photocatalyst and the Lewis acid La(OTf)3. Their report mentioned that the choice of Lewis acid was critical for the SET event from the strongly reducing [Ru(bpy)3]+ (M−) species to carbonyl groups. It should be noted that these systems are typical examples of dual catalysis, i.e. Lewis acid catalysis and photoredox catalysis.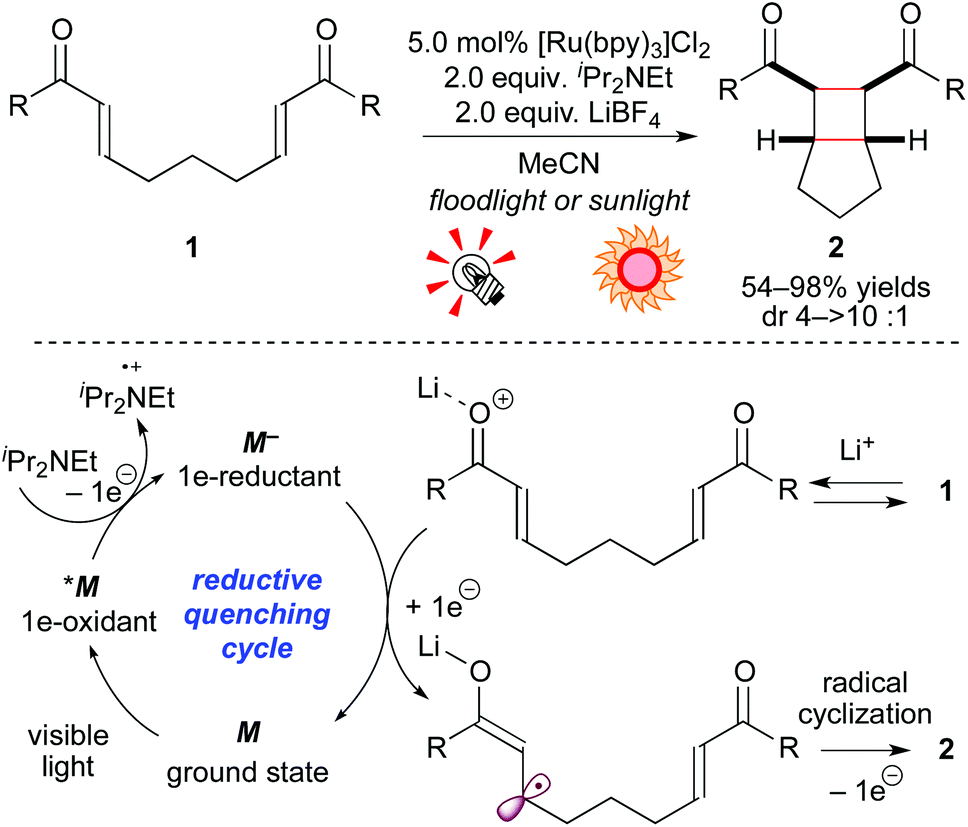 | ||
| Scheme 4 [2 + 2] Cycloadditions of electron-deficient alkenes through the reductive quenching cycle. | ||
As mentioned in the Introduction section, the photoexcited species of metal-based photoredox catalysts can serve both as 1e-oxidants and 1e-reductants. The combination of the Ru photocatalyst with a sacrificial electron acceptor has been proven to affect 1e-oxidation of electron-rich alkenes, leading to radical cations. Yoon and co-workers also described a process complementary to the above-mentioned cycloaddition reactions of electron-deficient enones. The cyclized product 4 was obtained from the reaction of the electron-rich bis(alkene) 3 through the oxidative quenching cycle using MV2+ (methyl viologen) as a sacrificial electron acceptor (Scheme 5).8d
 | ||
| Scheme 5 Photo-oxidative [2 + 2] cycloadditions of electron-rich alkenes. | ||
More recently, it was reported that direct oxidation of the electron-rich styrene 5 through the reductive quenching cycle by [Ru(bpm)3](BArF)2 (bpm: 2,2′-bipyrimidine; BArF: tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), which forms an excited state (*M/M− = +0.99 V vs. SCE in MeCN), is stronger than that using [Ru(bpy)3]2+. The Ru photocatalyst, [Ru(bpm)3](BArF)2, effected crossed intermolecular [2 + 2] cycloaddition using oxygen as the terminal oxidant (Scheme 6a).8e It is notable that, in this reaction, the further stronger oxidizing photocatalyst [Ru(bpz)3]2+ (bpz: 2,2′-bipyrazine, *M/M− = +1.45 V vs. SCE in MeCN) turned out to be sluggish because cycloreversion of the product 6 was induced by the photoactivated [Ru(bpz)3]2+.
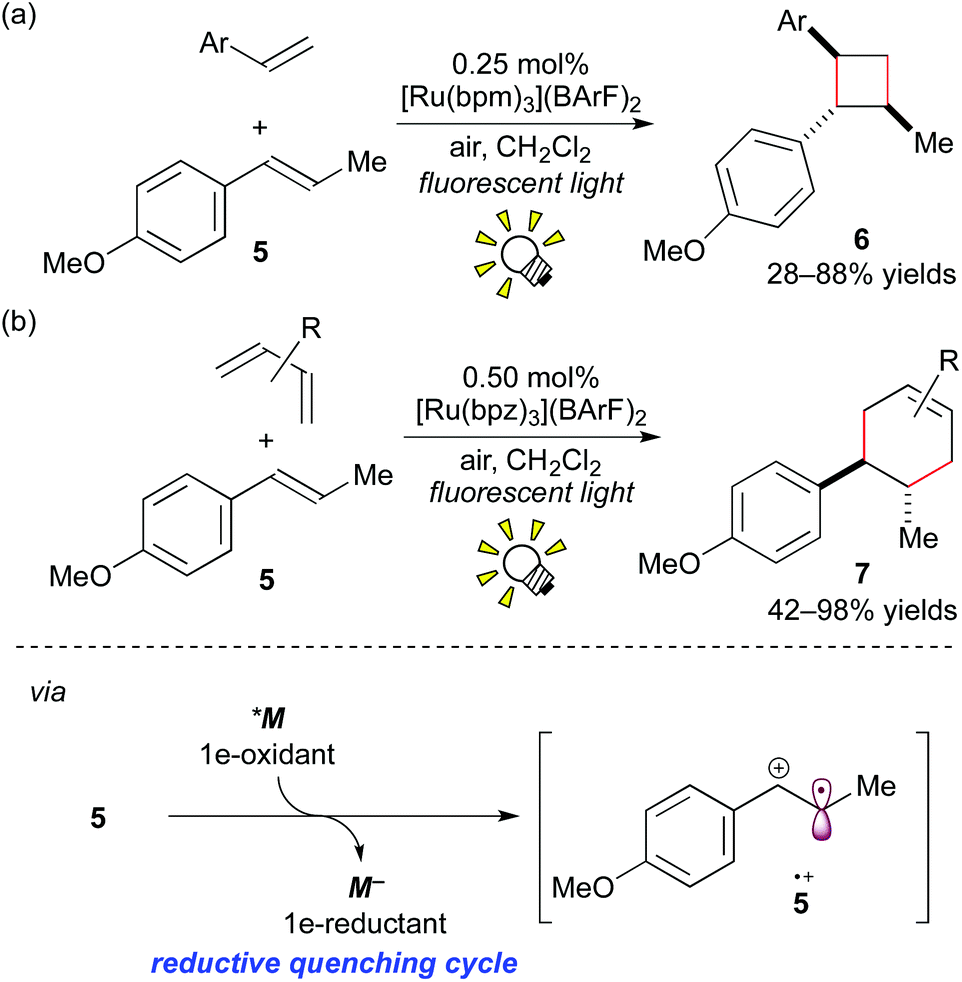 | ||
| Scheme 6 (a) Crossed intermolecular [2 + 2] cycloaddition of styrenes. (b) Radical cation Diels–Alder cycloadditions. | ||
Furthermore, it was revealed that [4 + 2] cycloadditions of electron-rich alkenes can also be promoted through 1e-oxidation of alkenes by photoredox catalysis.8f The Ru photocatalyst, [Ru(bpz)3](BArF)2 (0.50 mol%), effected the Diels–Alder type cycloaddition reactions of styrene 5 with various dienes (Scheme 6b). But it should be noted that this photocatalytic protocol through the cation radical intermediate 5+˙ provides the product 7, which cannot be formed by thermal Diels–Alder cycloaddition.
As is shown, careful tuning of the redox properties of the photocatalysts leads to successful cycloaddition reactions of both electron-rich and -deficient alkenes through ionic radical intermediates.
3. Oxidatively induced generation of organic radicals
As mentioned in the above sections, electron-rich tertiary amines are frequently used as sacrificial electron donors to enhance the reducing power of the excited photocatalyst. Recently, photoredox-catalyzed oxidation of electron-rich amines has become a powerful tool for the synthesis of nitrogen atom-containing compounds, including bioactive compounds. One trend involves transformations of iminium ions11 and azomethine ylide intermediates12 through two-electron oxidation of amines by combining a photoredox catalyst with an appropriate oxidant. Reactions of the generated ionic species with a variety of nucleophiles or dipolarophiles have been explored. This review, however, will focus on reactions involving carbon-centered radical species.3.1 Photogeneration of the α-aminoalkyl radical
One-electron oxidation of tertiary amines provides amine radical cations. Subsequent deprotonation produces α-aminoalkyl radicals, which can serve as nucleophiles to react with a variety of electrophiles. Thus, this transformation can achieve elusive C–H functionalization of amines at the α-position.13 In 2012, Pandey and Reiser et al. developed the photoredox reaction between the α-aminoalkyl radical generated from N-aryltetrahydroisoquinoline 8 and an electron-deficient alkene 9 (Scheme 7).14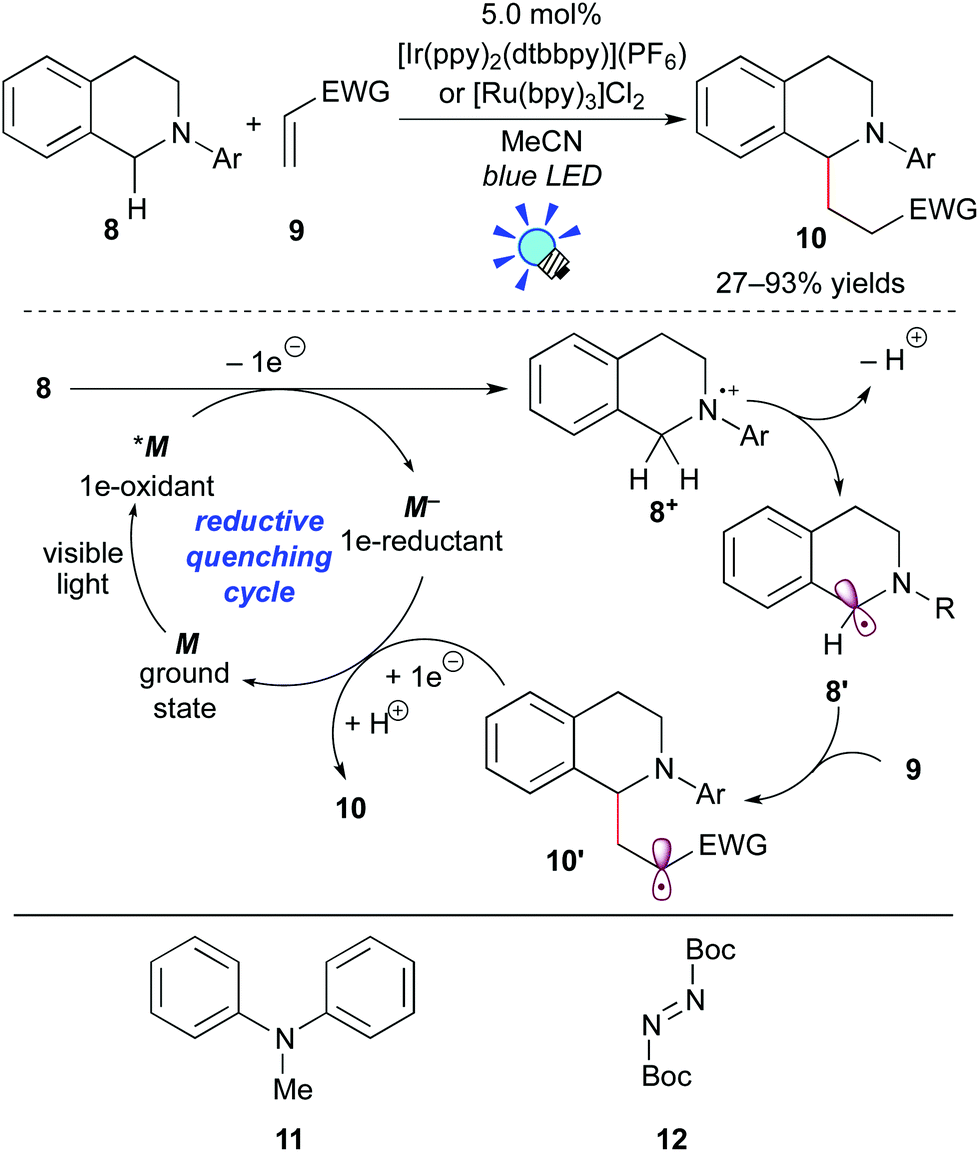 | ||
| Scheme 7 Addition of the α-aminoalkyl radical to electron-deficient double bonds. | ||
The authors showed that both the Ru photocatalyst, [Ru(bpy)3]Cl2, and the Ir photocatalyst, [Ir(ppy)2(dtbbpy)](PF6) (ppy: 2-phenylpyridine; dtbbpy: 4,4′-di-tert-butyl-2,2′-bipyridine), promote the reaction. In some examples, however, the Ir photocatalyst is more effective. A proposed reaction mechanism based on the reductive quenching cycle is illustrated in Scheme 7. The photoexcited species *M undergoes SET from tetrahydroisoquinoline 8 to give the radical cation 8+ and the strong reducing agent M−. Subsequent deprotonation converts 8+ to the α-aminoalkyl radical 8′. Conjugated addition of the nucleophilic radical 8′ to the electron-deficient alkene 9 yields the radical intermediate 10′, which is reduced by the highly reduced species M−, with concomitant regeneration of M. Subsequent protonation of the resulting anionic species produces the adduct 10. It should be noted that this photocatalytic reaction does not require any sacrificial electron donor or acceptor, i.e. a redox neutral process.
Almost at the same time, the Nishibayashi group reported the analogous reaction of aniline 11 with electron-deficient alkenes in the presence of [Ir(ppy)2(dtbbpy)](BF4).15a Moreover, they extended this reaction to C–H amination of amines at the α-position using di-tert-butyl azodicarboxylate 12 as an electrophile.15b
In 2011, MacMillan et al. showed a different pathway to access α-aminoalkyl radicals, i.e. via an oxidative quenching cycle, and achieved C–H arylation of amines at the α-position.16
The photocatalytic reaction of the cyclic or acyclic amine 13 with the cyanoarene derivative 14 in the presence of fac-Ir(ppy)3 (0.5–1.0 mol%) under visible light irradiation afforded the corresponding α-arylated amine 15. The nitrile group serves as a leaving group. A proposed reaction mechanism is depicted in Scheme 8. The photoactivated species *M undergoes SET with the cyanoarene 14, leading to the radical anion 14′ and the strongly oxidizing species M+. The amine 13 is oxidized by M+ to result in the formation of the α-aminoalkyl radical 13′ and the ground state M. The radical coupling of 13′ with 14′ produces the C–C coupled intermediate 15−. Finally, aromatization via elimination of the cyanide anion produces the α-arylated amine product 15. In particular, protected indoline and tetrahydroquinoline architectures, which are prevailing cyclic amine structures in bioactive compounds, are applicable.
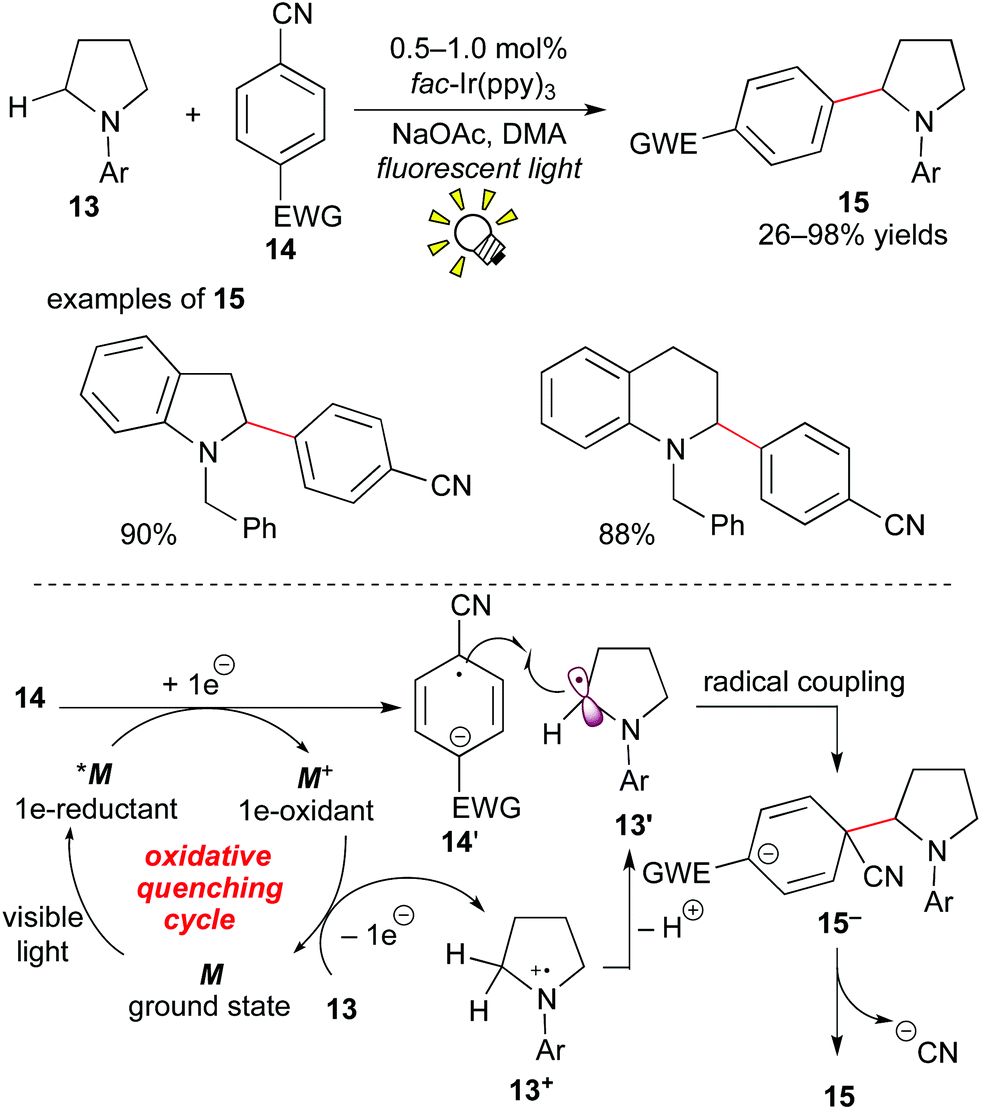 | ||
| Scheme 8 α-C–H arylation of amines. | ||
These reports were followed by related work involving generation of α-aminoalkyl radicals from various electron-rich amines.15c,17
3.2 Photo-oxidation of enamines
Enamines, which are accessible by reaction of carbonyl compounds and secondary amines, are a class of electron-rich tertiary amines, i.e. electron donors. In addition, 1e-oxidation of enamines is connected to “SOMO-activation” studies, leading to functionalization of carbonyl compounds at the α-position.18In 2009, Koike and Akita reported that the Ru photocatalyst, [Ru(bpy)3](PF6)2, effected stoichiometric oxidative coupling of the enamine 16a with TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxy) under visible light irradiation to give the α-oxyaminated aldehyde 17a after hydrolysis (Scheme 9a). Moreover, this system was extended to a photocatalytic system. Catalytic amounts of [Ru(bpy)3](PF6)2 (2.0 mol%) and morpholine (20 mol%) effected direct α-oxyamination of the aldehyde 18. A plausible reaction mechanism, involving a cooperative catalytic cycle consisting of photocatalysis (reductive quenching cycle) and organocatalysis, is shown in Scheme 9b. Morpholine first reacts with the aldehyde 18 to give the enamine 16, which then undergoes 1e-oxidation by the excited species *M. Importantly, the aldehyde 18 cannot be directly oxidized by the photocatalyst because of its very high oxidation potential. Final radical coupling of the cationic radical intermediate 16+ with TEMPO, followed by hydrolysis, gives the product 17 and morpholine.19a
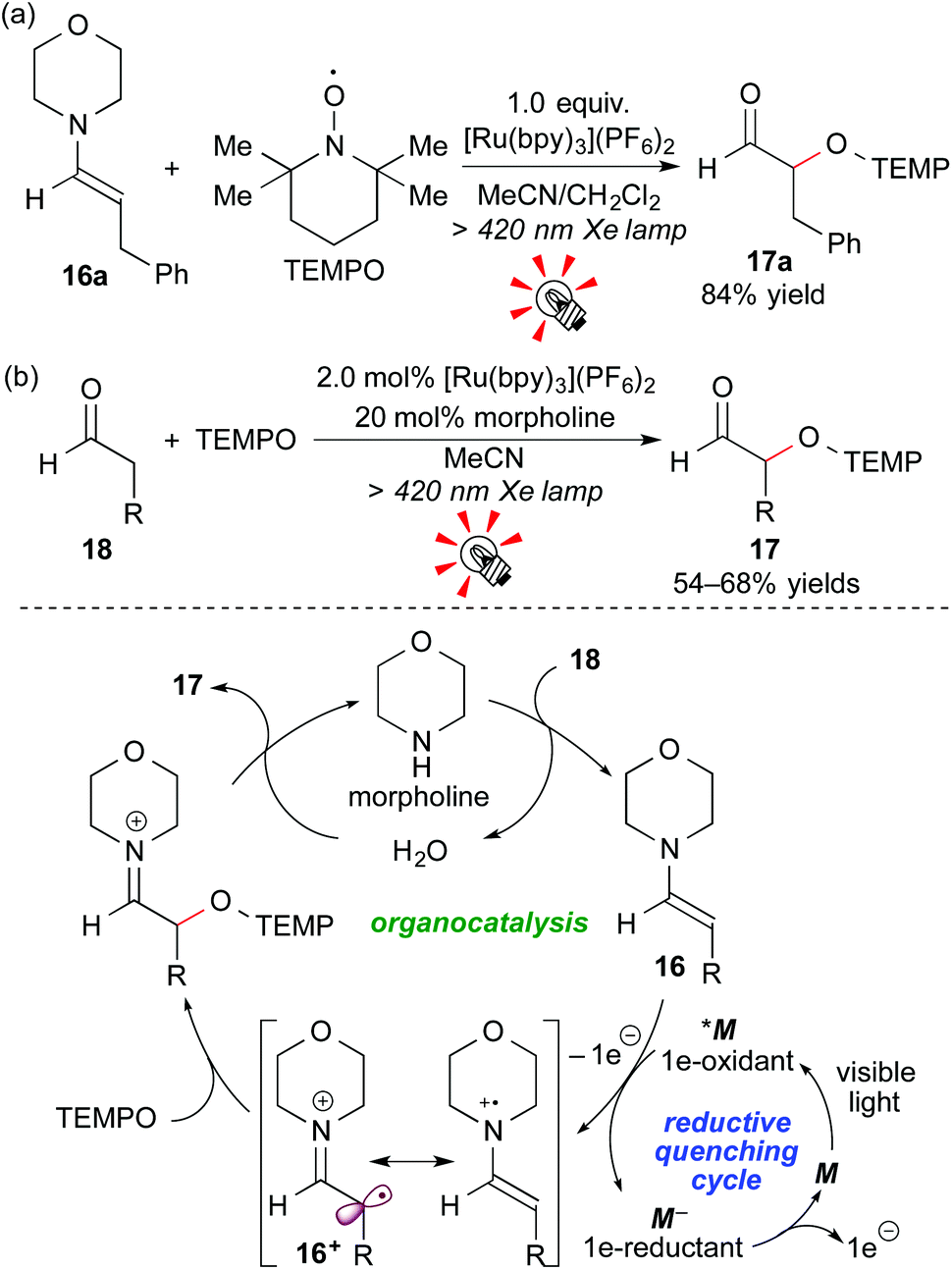 | ||
| Scheme 9 Photocatalytic α-oxyamination of enamine (a) and aldehydes (b). | ||
Furthermore, the group of Koike and Akita extended this reaction to C–C bond formation through the 2e-oxidation process: oxidative coupling of the enamine 16b with silyl enol ether 19. The choice of the electron-acceptor turned out to be crucial in the reaction. The combination of duroquinone 20, an electron acceptor, and the Lewis acid LiBF4 dramatically improved the yield of the γ-diketone product 21. A possible reaction mechanism through the oxidative quenching cycle is illustrated in Scheme 10.
 | ||
| Scheme 10 Photocatalytic oxidative coupling of enamines with silyl enol ethers. | ||
The excited species *M undergoes SET with duroquinone 20, activated by Li+, to be converted into the strongly oxidizing species M+. Oxidation of the enamine 16b by M+ followed by addition to silyl enol ether 19 gives the radical intermediate 21′, and subsequent 1e-oxidation by M+ gives the corresponding coupling product. The resulting compound is converted into the γ-diketone 21 upon hydrolysis. In addition, natural sunlight can also be harnessed as the light source.19b
In 2013, MacMillan and co-workers developed a β-arylation of carbonyl compounds via photoredox-catalyzed generation of an enamine radical cation. The direct β-functionalization of carbonyl compounds has been regarded as a challenging transformation. A proposed mechanism merging photoredox catalysis and organocatalysis is depicted in Scheme 11.
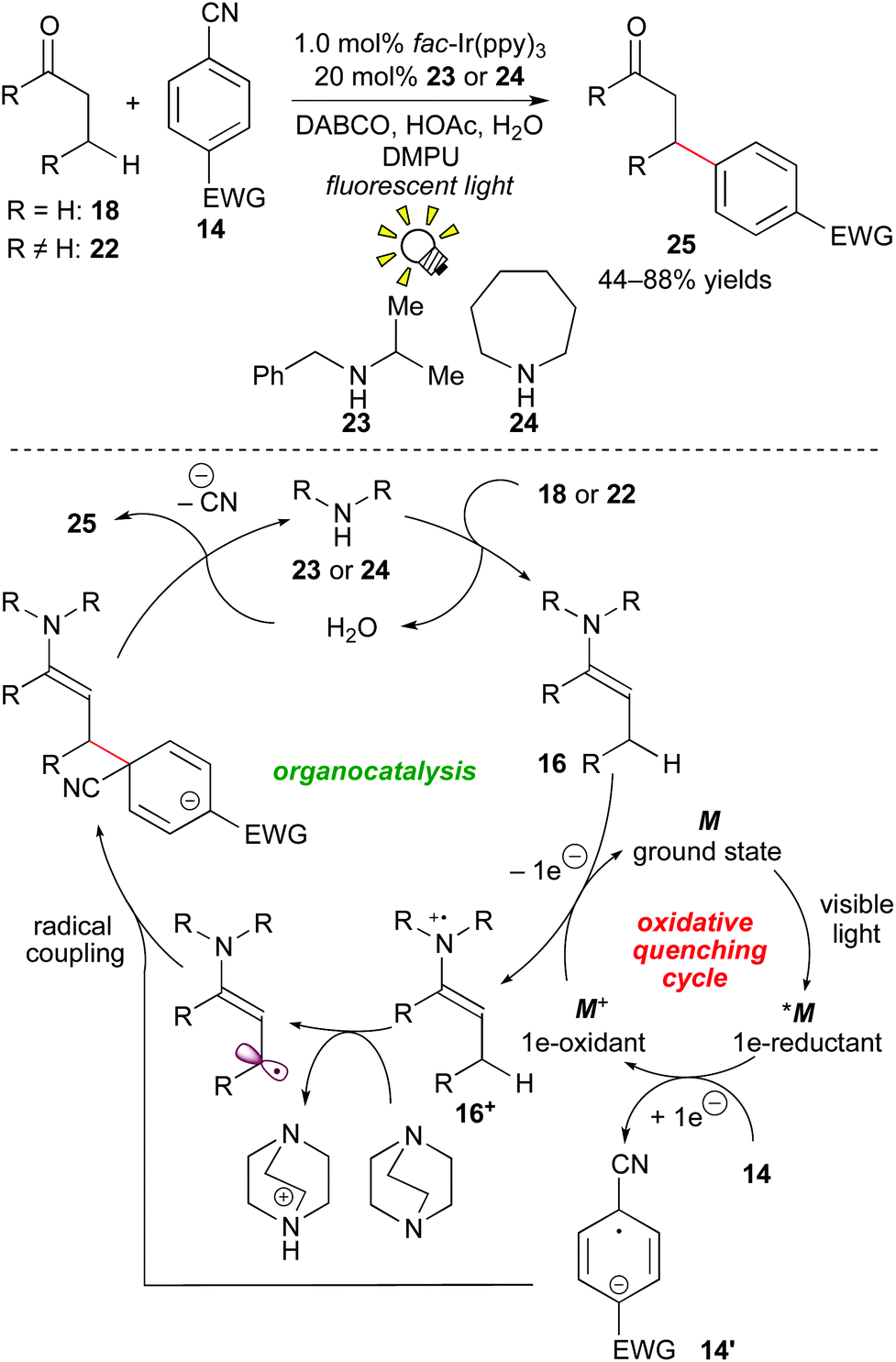 | ||
| Scheme 11 β-Arylation of carbonyl compounds with cyanoarenes. | ||
First, photoactivated species *M undergoes SET to the cyanoarene 14, providing the aryl radical anion 14′ and the highly oxidized species M+. A subsequent SET event from the enamine 16, which is generated from the reaction of the secondary amine catalyst with the aldehyde 18in situ to the oxidant M+ provides the enamine radical cation 16+ and M of the ground state. Importantly, coupling of the enamine cation radical 16+ with the aryl anion radical 14′ does not proceed at the α-position of the carbonyl group. Instead, deprotonation by the base, DABCO (1,4-diazabicyclo[2.2.2]octane), at the β-position gives the β-enaminyl radical intermediate. At this stage, radical coupling produces the β-functionalized product 25 after elimination of the CN group and hydrolysis.20a
More recently, they extended this reaction to the β-hydroxyalkylation of the carbonyl compounds through 1e-reduction of ketones instead of cyanobenzene 14.20b
3.3 Generation of carbon-centered radicals from organoborates
As described above, oxidative generation of nitrogen-containing organic radicals from electron-rich tertiary amines by photoredox catalysis is feasible and enables one to develop unique photocatalytic radical transformations. For the next stage, access to simple organic radicals, such as alkyl and aryl radicals, has remained an interesting topic. Recently, organoborates have attracted great interest as organic radical precursors because they are (i) air- and moisture-stable, (ii) harmless and (iii) tolerable with a variety of functionalities. It has been found that organic radicals can be generated from organoborate derivatives upon oxidation, but previously reported examples required an excess amount of oxidant or co-oxidant.21Akita and co-workers developed photoredox-catalyzed generation of organic radicals from the corresponding organoborates.22a In addition, it was revealed that the reaction of organoborates with electron-deficient alkenes proceeds smoothly in the presence of a catalytic amount of the Ir photoredox catalyst, [Ir(dF(CF3)ppy)2(bpy)](PF6), which exhibits a strong oxidizing ability at the photoexcited state. A plausible reaction mechanism through a reductive quenching cycle is shown in Scheme 12.
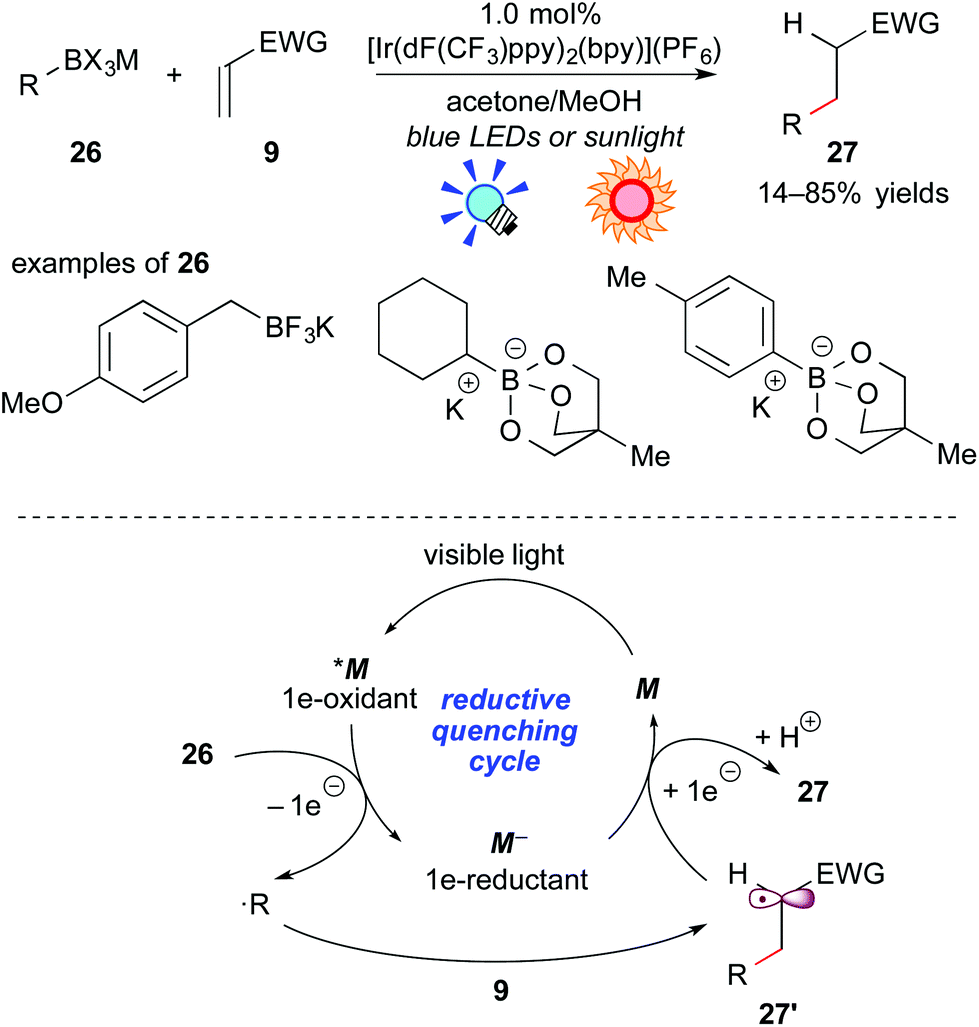 | ||
| Scheme 12 Photocatalytic Giese reaction of organoborates with electron-deficient alkenes. | ||
First, the excited species *M undergoes SET from the organoborate 26 to generate a carbon-centered radical (˙R) and the highly reduced species M−, accompanied by elimination of the boron unit. The carbon-centered radical ˙R reacts with the electron-deficient alkene 9 to afford a C–C coupled radical intermediate 27′. Finally, reduction of 27′ by the reductant M− followed by protonation with methanol gives the product 27 together with M of the ground state. Alkyl- and aryl-borates can be applied to this reaction system. This catalytic system does not require any sacrificial oxidant and reductant, hence a redox neutral process. Furthermore, they showed the generation of an organic radical bearing a heteroatom at the α-position of the radical center, i.e. α-alkoxymethyl radical, from the corresponding organoborate22b and harnessed sunlight as the light source.
More recently, Chen and co-workers reported photocatalytic deboronative alkynylation of primary, secondary, and tertiary alkylborates 26 with alkynylbenzoiodoxoles 28 in the presence of the Ru photocatalyst, [Ru(bpy)3](PF6)2 (Scheme 13).23 They proposed that organic radicals are generated from organoborates through the oxidative quenching mechanism, in which hydroxybenzoiodoxole serves as the electron acceptor. They showed that the reactions of boronic acids also give the corresponding coupling products 29.
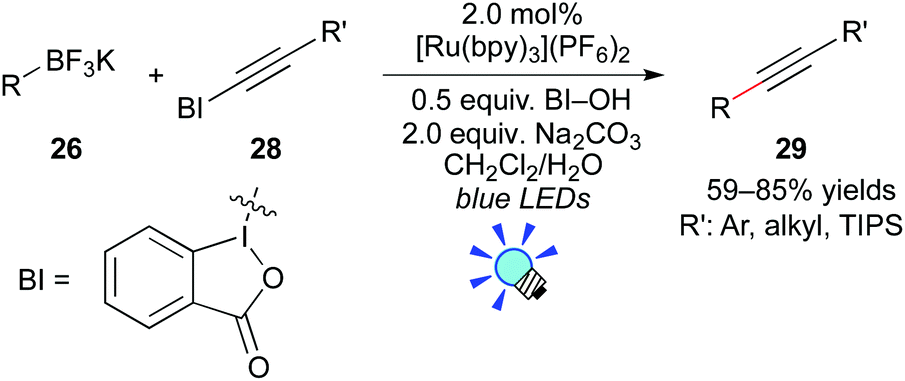 | ||
| Scheme 13 Photocatalytic deboronative alkynylation. | ||
These results suggest that generation of organic radicals with a range of functional groups is accessible by oxidation of organoborates by photoredox catalysis, leading to promising radical initiating systems.
4. Reductively induced generation of organic radicals
One of the most commonly used methods for generation of organic radicals is photolysis or thermolysis of organic halides in the presence of AIBN (azobisisobutyronitrile) and HSnBu3. But the use of explosive AIBN and harmful tin-reagent is a drawback from the viewpoint of safety. It is well-known that 1e-reduction of organic halides and organic onium salts generates organic radicals. As mentioned in “Introduction”, the excited photoredox catalyst can serve as a 1e-reductant under mild reaction conditions, i.e. visible light irradiation at room temperature. Thus, photoredox catalysis can provide a new and green protocol for the generation of organic radicals. In this section, representative photocatalytic radical reactions, which involve organic halides and onium salts as organic radical precursors, will be summarized.4.1 Photo-reduction of organic halides
In 2008, Nicewicz and MacMillan reported an elusive asymmetric intermolecular α-alkylation of the aldehyde 18 with the activated alkyl bromide 30a by elegantly merging Ru photoredox catalysis with chiral amine organocatalysis.24a The reaction mechanism proposed for the synergistic catalysis is illustrated in Scheme 14.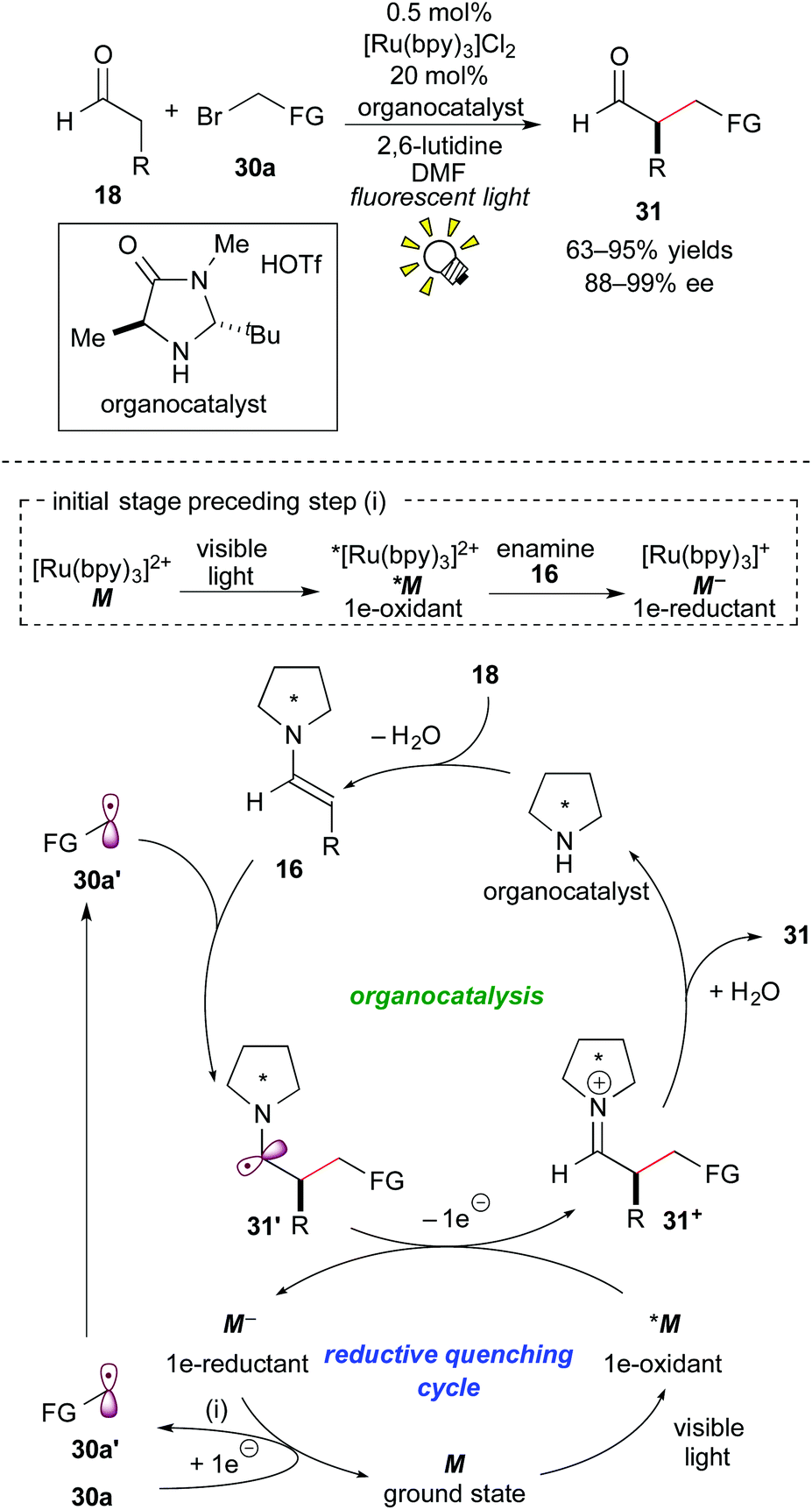 | ||
| Scheme 14 Merger of catalysis for asymmetric α-alkylation of aldehydes. | ||
At the initial stage, the excited species, *[Ru(bpy)3]2+ (*M), is reduced by the enamine 16 derived from the reaction of the secondary chiral amine catalyst with the aldehyde 18. The resulting strong reductant, [Ru(bpy)3]+ (M−), undergoes SET with the alkyl halide 30a, leading to the formation of the alkyl radical 30a′ and regeneration of the ground state [Ru(bpy)3]2+(M). The alkyl radical 30a′ reacts with the preformed enamine 16 to give the C–C coupled radical intermediate 31′. 1e-oxidation of the radical intermediate 31′ by the photoactivated species *M proceeds in the mainstream to afford the iminium intermediate 31+ and the reductant M−, which follows the above-mentioned SET event for generation of the alkyl radical 30a′. Subsequent hydrolysis produces the α-alkylated aldehyde 31 together with the organocatalyst.
The mechanism proposed by the authors is based on the reaction of a SOMOphilic enamine 16 with an electron-deficient radical 30a′, which is in contrast to those mentioned in section 3.2. It is noteworthy that the present catalytic system can provide an easy access to various optically active α-alkylated aldehydes 31 in high yields with excellent enantioselectivity.24
In 2009, Stephenson and co-workers reported an environmentally benign tin-free reductive dehalogenation of alkyl halides in the presence of [Ru(bpy)3]Cl2 and an amine as the hydrogen atom source under visible light irradiation (Scheme 15).25a It was proposed that the alkyl radical is formed via 1e-reduction of the alkyl halide 30b by the action of the photoredox catalyst. Furthermore, they extended their tin-free generation method of organic radicals to radical C–C bond formation with alkenes25b,c and electron-rich hetero-arenes.25d
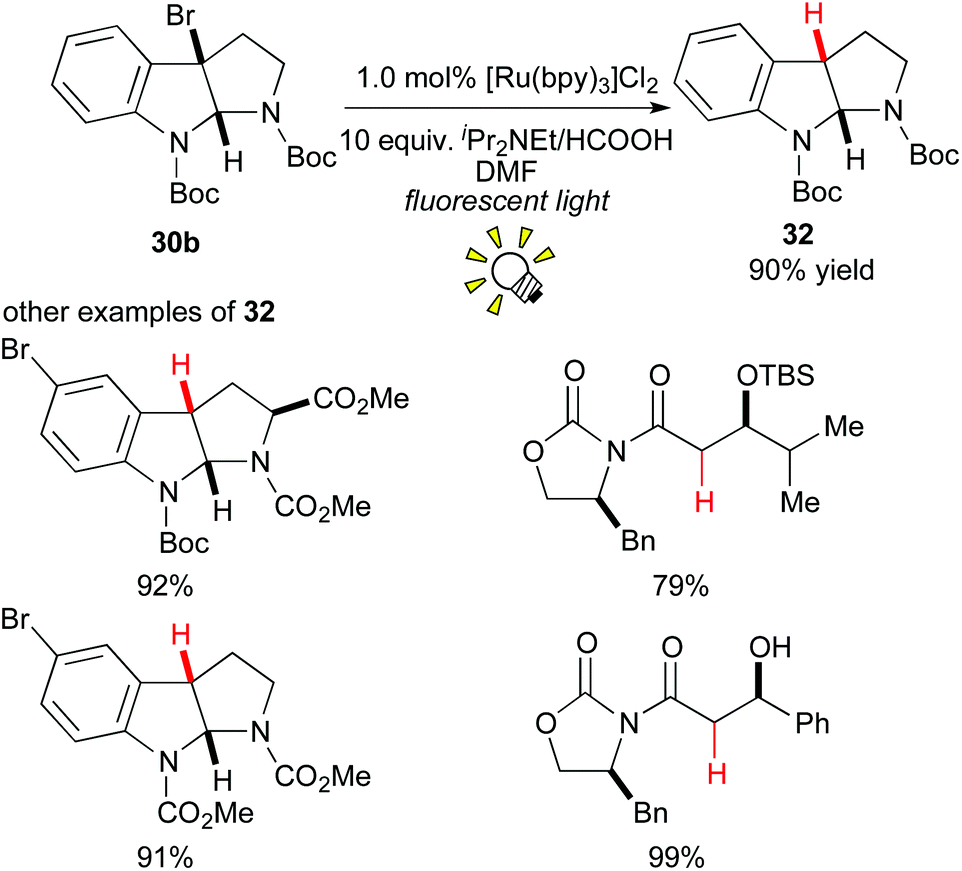 | ||
| Scheme 15 Tin-free reductive dehalogenation. | ||
In 2010, Gagné et al. described an intermolecular radical addition of the glycosyl halide 30c to the electron-deficient alkene 9, leading to the C-glycoside 33 in good yields with high diastereoselectivity (Scheme 16).26
 | ||
| Scheme 16 Tin-free Giese reaction. | ||
These reports were followed by many related studies on photocatalytic radical reaction using activated alkyl halides as the radical precursors.27 Recently, the group of Hawker reported a unique radical polymerization system, whereby generation of organic radicals was controlled by photoredox catalysis.27f
In 2012, Stephenson et al. revealed that the Ir photocatalyst, fac-Ir(ppy)3, affects the generation of organic radicals from unactivated alkyl, alkenyl and aryl iodides (Scheme 17).28a The photoexcited state, *[fac-Ir(ppy)3], has a high reduction potential enough to reduce unactivated organic iodides. Therefore, *M undergoes direct SET to the organic iodide 30d to generate the organic radical 30d′ (˙R). H-atom abstraction and radical cyclization of the resulting radical 30d′ afford the corresponding dehalogenated product 34 and the C–C coupled product 35, respectively. The photocatalyst is regenerated with a reductant, being either nBu3N/HCOOH or Hantzsch ester (diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate).
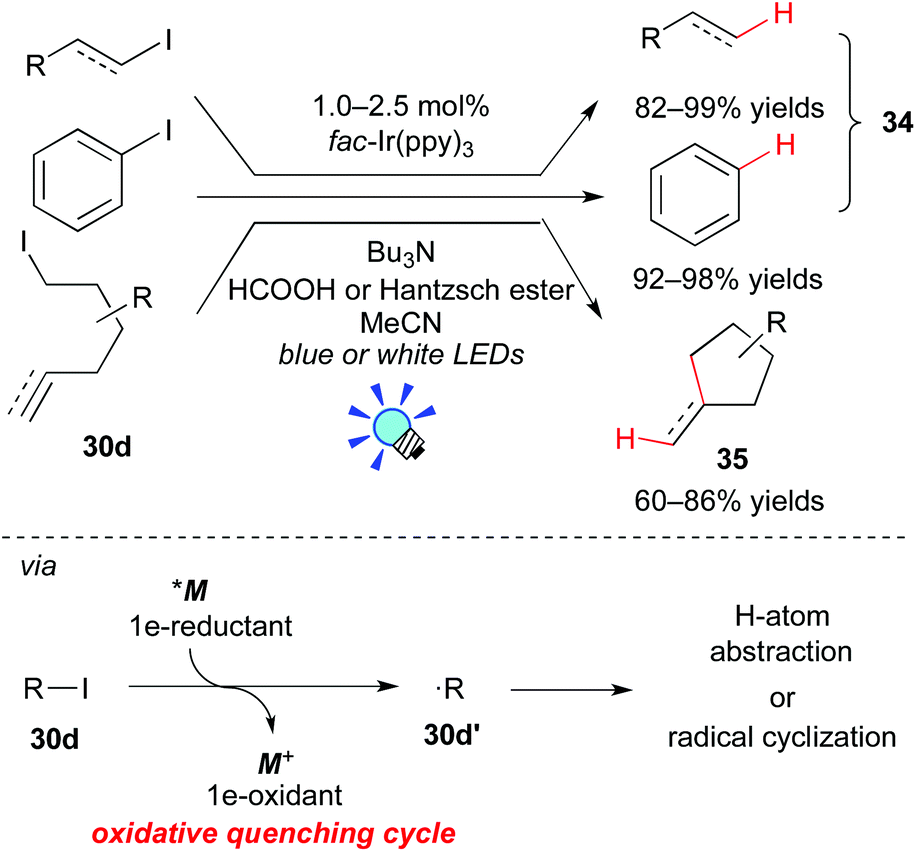 | ||
| Scheme 17 Photogeneration of organic radicals from unactivated organic iodides. | ||
Almost at the same time, Lee and co-workers reported analogous work, i.e. the combination of the Ir photocatalyst, [Ir(ppy)2(dtbbpy)](PF6), and an electron donor, iPr2NEt, affecting the generation of the organic radical from the unactivated organic iodide 30d (Scheme 18a).28b Their reaction proceeds via a reaction pathway different from Stephenson's report, i.e. the reductive quenching cycle. Moreover, the group of Li developed direct arylation of arenes with aryl halides by photoredox catalysis, leading to biaryl 36 (Scheme 18b).28c
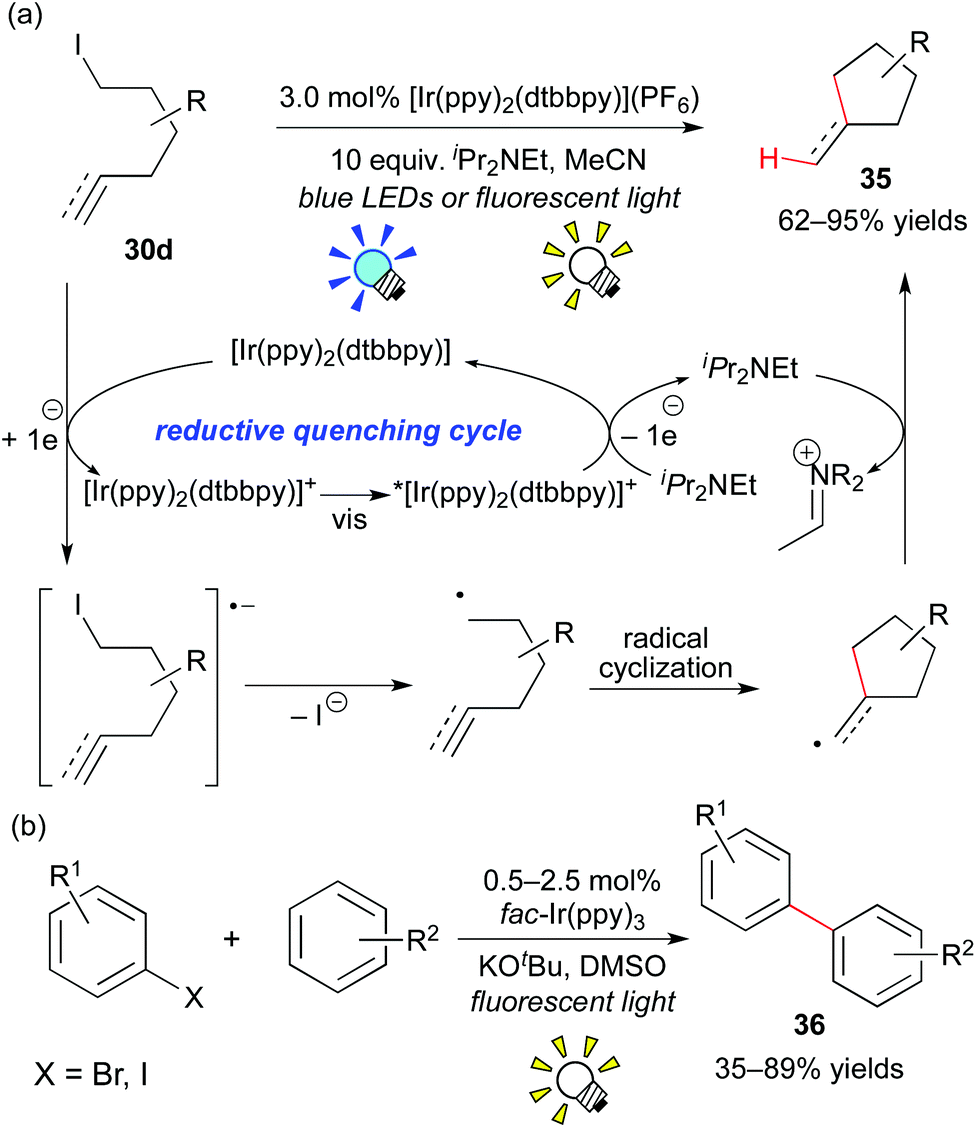 | ||
| Scheme 18 (a) Photocatalytic radical cyclization. (b) Photocatalytic synthesis of biphenyls. | ||
In 2009, the group of MacMillan reported that photoredox catalysis is effective for the generation of a trifluoromethyl radical (˙CF3) from CF3I 30e (Scheme 19).29 The above-mentioned dual catalytic system consisting of photocatalysis and organocatalysis (Scheme 14) also affected asymmetric trifluoromethylation of the aldehyde 18 using CF3I 30e as a CF3 source, leading to the valuable enantioenriched α-trifluoromethylated aldehyde 37, which is known as a useful building block for synthesis of chiral organofluorine compounds.30 In this reaction, the Ru catalyst, [Ru(bpy)3]2+, also afforded the product but in a yield lower than that obtained by the Ir photocatalyst, [Ir(ppy)2(dtbbpy)](PF6). The Ir catalyst exhibits a reduction potential higher than that of the Ru catalyst and can generate the CF3 radical from CF3I more effectively.
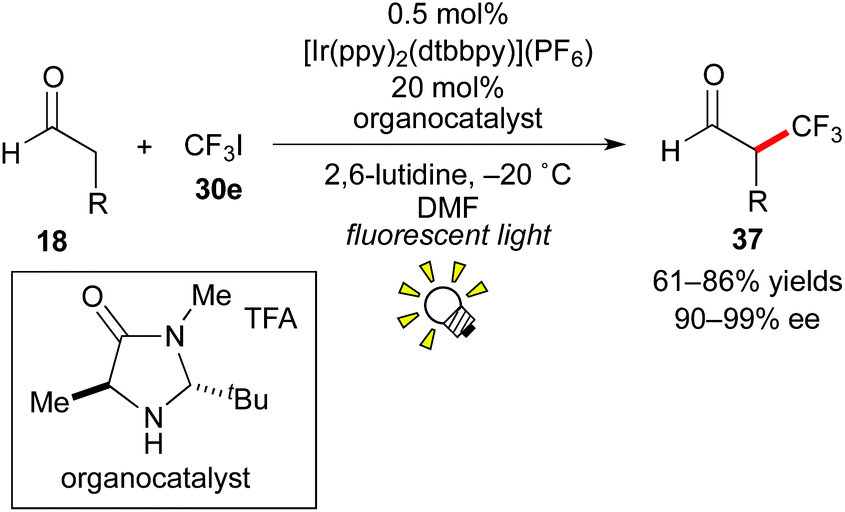 | ||
| Scheme 19 Synthesis of α-CF3-aldehydes by photoredox catalysis. | ||
Furthermore, Nagib and MacMillan reported high-impact work on direct C–H trifluoromethylation of arenes by photoredox catalysis.31a The use of [Ru(phen)3]Cl2 (phen: phenanthroline) or fac-Ir(Fppy)3 (Fppy = 2-(2,4-difluorophenyl)pyridine) as a photocatalyst and CF3SO2Cl 38 as a CF3 source enables the reaction with the broad scope with respect to arene 39, i.e. electron-rich/electron-deficient heteroarenes and unactivated arenes, in the absence of a sacrificial electron donor (Scheme 20).
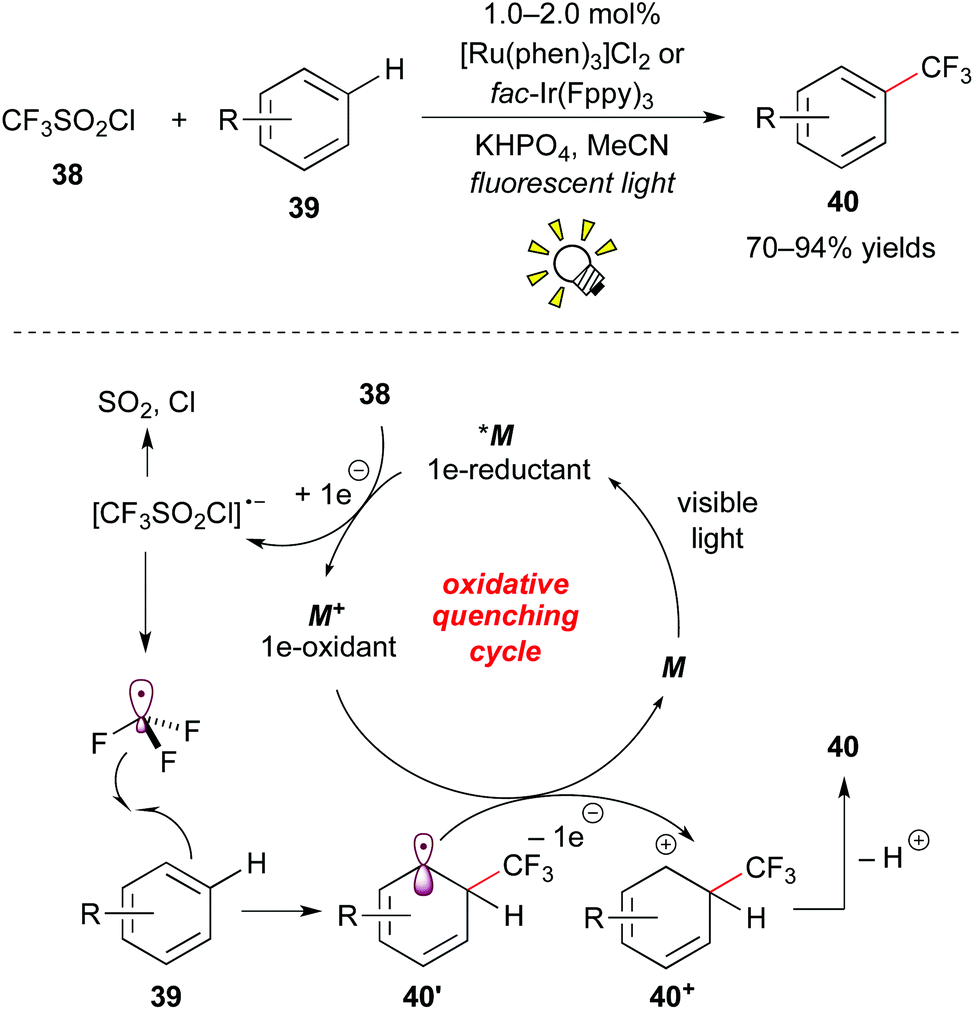 | ||
| Scheme 20 Photocatalytic direct trifluoromethylation of aryl C–H bonds. | ||
The excited species *M reacts with CF3SO2Cl 38 by the first SET process to give ˙CF3 and the strong oxidant M+. The reaction of ˙CF3 with arene 39 affords the corresponding cyclohexadienyl radical 40′, which is oxidized by the oxidant M+ to give the cyclohexadienyl cation 40+ while regenerating M. Deprotonation of the cation intermediate 40+ produces the trifluoromethylated arene 40. It is noteworthy that they showed direct trifluoromethylation of bioactive compounds such as ibuprofen, Vitamin P, and Lipitor and proposed a new approach to the late-stage trifluoromethylation. As remarked in their report, the exploration of CF3I 30e as a CF3 radical source turned out to limit the substrate scope to electron-rich arenes. This approach for trifluoromethylation of π-electron rich arenes such as indole, furan, pyrrole, and thiophene was reported by Cho and co-workers almost at the same time.31b
Recently, a great number of studies on radical trifluoromethylation using CF3I and CF3SO2Cl by photoredox catalysis have appeared.1o,32 Organofluorine compounds have their importance increasing in pharmaceutical and agrochemical fields.33 Thus, photoredox-catalyzed trifluoromethylation can become one of the most promising methods for the synthesis of diverse organofluorine compounds.
4.2 Photo-reduction of organic onium salts
Much attention has been paid to aryl onium salts as radical precursors as an alternative to using aryl halides.34 Additionally, in the photoredox chemistry of [Ru(bpy)3]2+, Kellogg et al. described the generation of aryl radicals by reduction of phenacylsulphonium salts in 1978.35 Furthermore, Pschorr-type cyclization through reduction of aryl diazonium salt derivatives was reported by Cano-Yelo and Deronzier in 1984.36 These reports, along with the recent advancements of organic onium reagents, have stimulated many researchers to develop new photoredox-catalyzed radical transformations using onium reagents.1jIn 2011, Sanford and co-workers reported direct C–H arylation of arene 41 bearing a directing group, such as pyridine, amide, pyrazole, pyrimidine and oxime ether groups, with aryl diazonium 42 by merging photoredox catalysis with Pd catalysis (Scheme 21).37 The ligand-directed C–H arylation by palladium catalysis has been well studied in the past few decades.38 However, the reaction required high temperatures to achieve high yields. The rate-determining step is considered to be formation of the key active Pd–Ar species. They showed that radical arylation of Pd species using aryl diazonium salt 42 by action of the photoredox catalyst, [Ru(bpy)3]Cl2, leads to a C–H arylation under mild reaction conditions. A plausible mechanism based on the dual catalysis process starts with SET from *M to aryl diazonium salt 42, accompanying formation of highly oxidized species M+ and ˙Ar. The reaction of ˙Ar with the palladacycle generated by C–H activation of the substrate affords the PdIII intermediate. The Ru oxidant induces the second SET event from the PdIII intermediate to regenerate the photocatalyst and generate the PdIV intermediate. Finally, C–C bond-forming reductive elimination produces the arylated product 43 together with the original PdII catalyst. This dual catalysis by merging photoredox catalysis and transition-metal catalysis is also fascinating and provides new ways to develop catalytic transformations.39,1n
 | ||
| Scheme 21 C–H arylation based on dual catalysis. | ||
König et al. have intensively developed methods for arylation with aryl diazonium salts using the organic dye eosin Y and a metal-based photoredox catalyst. They reported photoredox-catalyzed arylation of heteroarenes, alkynes and alkenes with aryl diazonium salts.40,41 For example, they recently showed intermolecular amino-arylation of the alkene 44 (Meerwein addition) in the presence of [Ru(bpy)3]Cl2.41c They examined other photocatalysts, including organic dyes such as eosin Y, and revealed that [Ru(bpy)3]Cl2 is the best photocatalyst among them (Scheme 22a).
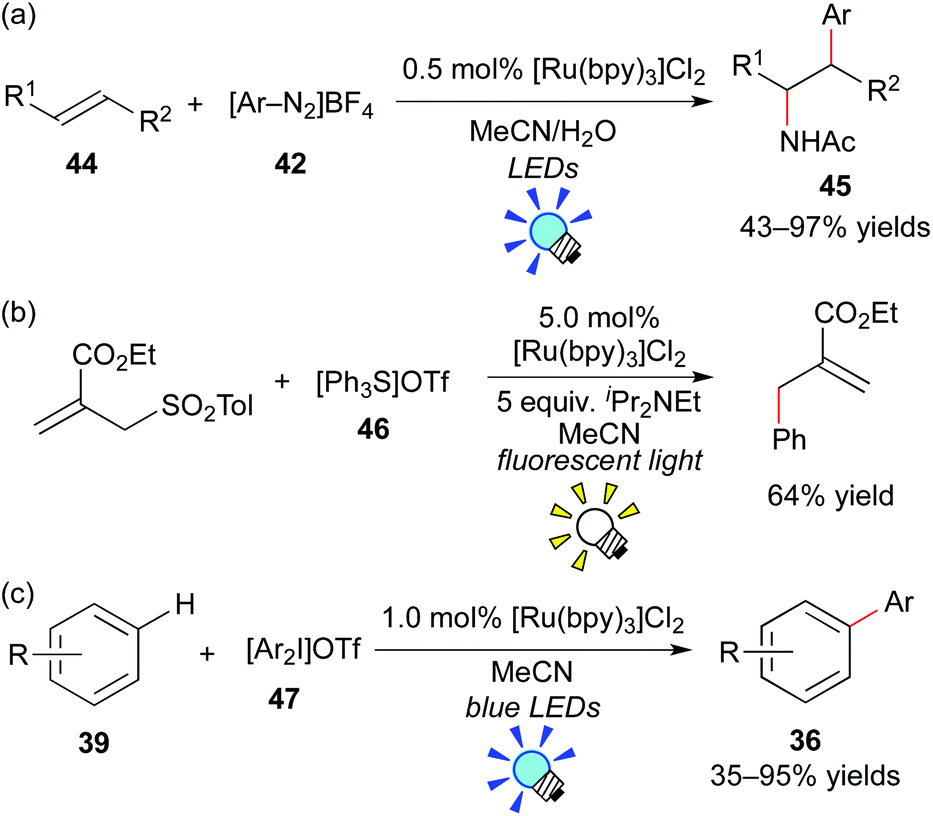 | ||
| Scheme 22 Photogeneration of aryl radicals from aryl onium salts. | ||
In 2013, the group of Fensterbank, Goddard and Ollivier reported that triarylsulfonium salt 46 can serve as a source of aryl radicals by action of the Ru photocatalyst, [Ru(bpy)3]Cl2 (Scheme 22b).42 Moreover, Xiao and co-workers described that diaryliodonium salt 47 can serve as another source of aryl radicals in the presence of [Ru(bpy)3]Cl2 (Scheme 22c).43 Shortly after their reports, a great number of studies related to arylation through generation of aryl radicals from aryl onium salts by photoredox catalysis have been reported.44
On the other hand, the group of Koike and Akita paid attention to electrophilic trifluoromethylating reagents, such as Umemoto's reagent 48 (S-(trifluoromethyl)dibenzothiophenium tetrafluoroborate),45 and Togni's reagent 49 (1-trifluoromethyl-1,2-benziodoxol-3-(1H)-one),46 which are easy to handle in terms of shelf-stable onium reagents at room temperature. They proved that electrophilic trifluoromethylating reagents can serve not only as precursors for the CF3 radical, but also as direct electron acceptors from the photoexcited catalyst. They developed efficient and selective trifluoromethylative difunctionalization of alkenes by photoredox catalysis (Scheme 23). The photoreaction of the alkene 44 with Umemoto's reagent 48 in O-containing solvents, such as H2O, alcohols, or carboxylic acids, in the presence of fac-Ir(ppy)3 under visible light irradiation exclusively afforded the corresponding oxytrifluoromethylated product 50 in a regioselective manner. In addition, the photocatalytic reaction of the alkene 44 with Umemoto's reagent 48 in MeCN containing a small amount of water, which is known as an aminative carbocation-trapping agent, proceeded smoothly via Ritter-type reaction to give the corresponding aminotrifluoromethylated product 51 in good yields. The Ru photocatalyst, [Ru(bpy3)](PF6)2, can promote the transformation in a manner similar to the Ir catalyst.
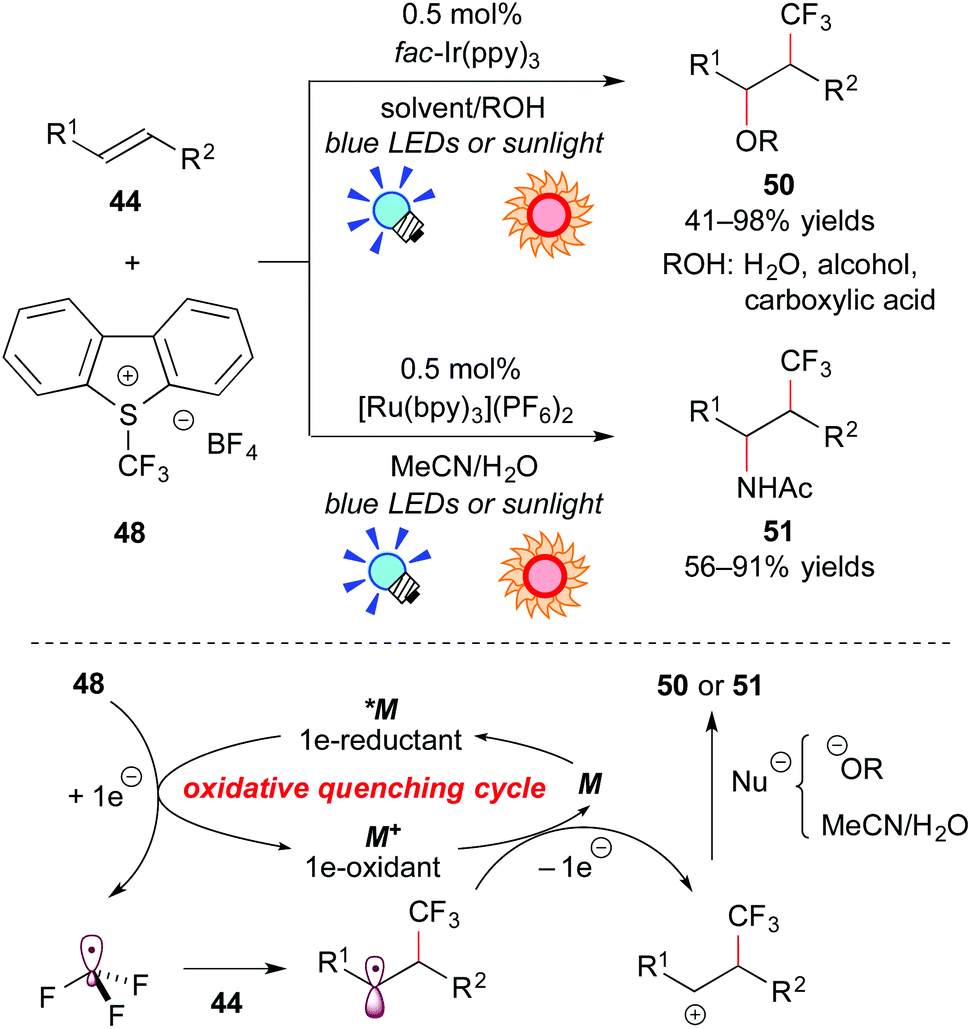 | ||
| Scheme 23 Photoredox-catalyzed difunctionalization of alkenes through construction of a C–CF3 bond. | ||
A plausible reaction mechanism through the oxidative quenching cycle is shown in Scheme 23. First, Umemoto's reagent 48 is reduced by *M to generate ˙CF3. Addition of ˙CF3 to the alkene 44 gives the radical intermediate, which is oxidized by M+ formed through the SET process. Finally, the β-trifluoromethylated carbocation intermediate undergoes solvolytic nucleophilic attack to afford the trifluoromethylated product 50 or 51.47a,c More recently, they extended this photocatalytic reaction to intramolecular trifluoromethylative lactonization of alkenoic acids.47e
The authors also showed that Togni's reagent 49 can serve as a CF3 radical source. Under similar reaction conditions, application of potassium vinyltrifluoroborate 52 to this photocatalytic system provided the trifluoromethylated alkene 53via deboronation (Scheme 24). Their photoredox-catalyzed protocol for construction of Calkenyl–CF3 bonds exhibits a broad scope with excellent efficiency and stereoselectivity.47b
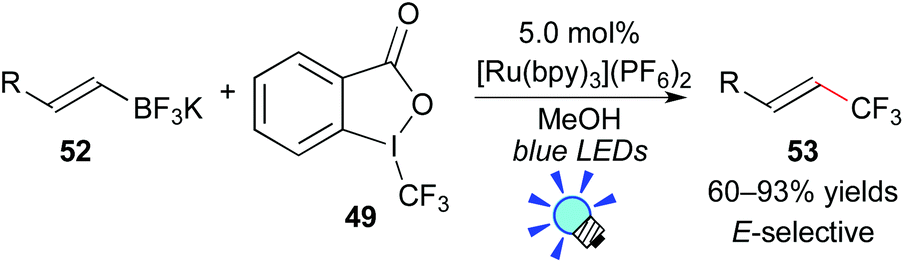 | ||
| Scheme 24 Photocatalytic synthesis of CF3-alkenes via deboronation. | ||
Shortly after their reports, other groups also reported radical trifluoromethylation using electrophilic CF3 reagents in the presence of a photoredox catalyst.1o,48 These onium salt-based CF3 reagents can be handled easily compared to conventional CF3 radical sources, such as gaseous CF3I and hydrolyzable CF3SO2Cl. In addition, different outcomes of reactions have been obtained.
5. Conclusions and outlook
In conclusion, photoredox catalysis with Ru and Ir photoredox catalysts has emerged as a powerful tool for the generation of carbon-centered radicals from diverse organic compounds, including conventional radical precursors, such as organic halides and onium salts. More sophisticated photocatalytic systems will be designed by an appropriate choice of photocatalyst, radical precursor and substrate: a redox neutral transformation without addition of an excess amount of oxidant or reductant. We expect that the present photocatalytic protocol expands the research field of radical reactions and stimulates development of novel reactive but storable radical precursors. Furthermore, newly designed photoredox catalysts will be exploited to achieve novel transformations.Notes and references
- For selected reviews on photoredox catalysis, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef; (d) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (e) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (f) S. Maity and N. Zheng, Synlett, 2012, 1851 CAS; (g) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (h) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (i) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (j) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (k) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef; (l) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977 CrossRef PubMed; (m) D. M. Schultz and T. P. Yoon, Science, 2014 DOI:10.1126/science.1239176; (n) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (o) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS PubMed.
- A. Albini and M. Fagnoni, Photochemically-generated Intermediates in Synthesis, John Wiley & Sons, Hoboken, NJ, 2013 Search PubMed.
- Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, 2012, vol. 1–4 Search PubMed.
- (a) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS; (b) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS; (c) Photochemistry and Photophysics of Metal Complexes, ed. D. M. Roundhill, Plenum, New York, 1994 Search PubMed.
- (a) R. C. Young, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1976, 98, 286 CrossRef CAS; (b) R. J. Crutchley and A. B. P. Lever, J. Am. Chem. Soc., 1980, 102, 7128 CrossRef CAS; (c) D. P. Rillema, G. Allen, T. J. Meyer and D. Conrad, Inorg. Chem., 1983, 22, 1617 CrossRef CAS; (d) M.-A. Haga, E. S. Dodsworth, G. Eryavec, P. Seymour and A. B. P. Lever, Inorg. Chem., 1985, 24, 1901 CrossRef CAS; (e) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed; (f) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed; (g) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS; (h) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS; (i) D. Hanss, J. C. Freys, G. Bernardinelli and O. S. Wenger, Eur. J. Inorg. Chem., 2009, 4850 CrossRef CAS.
- For examples of Cu photocatalyst, see: (a) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336 CrossRef CAS PubMed; (b) A. Baralle, L. Fensterbank, J.-P. Goddard and C. Ollivier, Chem. – Eur. J., 2013, 19, 10809 CrossRef CAS PubMed. For examples of Pt photocatalyst, see: (c) J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2013, 19, 6443 CrossRef CAS PubMed. For examples of Au photocatalyst, see: (d) W.-P. To, G. S.-M. Tong, W. Lu, C. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654 CrossRef CAS PubMed; (e) Q. Xue, J. Xie, H. Jin, Y. Cheng and C. Zhu, Org. Biomol. Chem., 2013, 11, 1606 RSC.
- For selected reviews on organic photoredox catalysis, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS; (c) M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710 CrossRef CAS PubMed; (d) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (e) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed; (b) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604 CrossRef CAS PubMed; (c) Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162 CrossRef CAS PubMed; (d) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef CAS PubMed; (e) M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807 RSC; (f) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef CAS PubMed.
- (a) M. T. Crimmins, Chem. Rev., 1988, 88, 1453 CrossRef CAS; (b) T. Bach, Synthesis, 1998, 683 CrossRef CAS PubMed.
- Y. Roh, H.-Y. Jang, V. Lynch, N. L. Bauld and M. J. Krische, Org. Lett., 2002, 4, 611 CrossRef CAS PubMed.
- (a) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed; (b) M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360 RSC; (c) M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 8679 RSC; (d) J. Xuan, Y. Cheng, J. An, L.-Q. Lu, X.-X. Zhang and W.-J. Xiao, Chem. Commun., 2011, 47, 8337 RSC; (e) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed; (f) G. Zhao, C. Yang, L. Guo, H. Sun, C. Chen and W. Xia, Chem. Commun., 2012, 48, 2337 RSC; (g) S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef CAS PubMed; (h) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem. – Eur. J., 2012, 18, 5170 CrossRef CAS PubMed; (i) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed; (j) J. Xuan, Z.-J. Feng, S.-W. Duan and W.-J. Xiao, RSC Adv., 2012, 2, 4065 RSC; (k) Z.-Q. Wang, M. Hu, X.-C. Huang, L.-B. Gong, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2012, 77, 8705 CrossRef CAS PubMed; (l) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425 CrossRef CAS PubMed; (m) S. Zhu and M. Rueping, Chem. Commun., 2012, 48, 11960 RSC; (n) C. L. Mathis, B. M. Gist, C. K. Frederickson, K. M. Midkiff and C. C. Marvin, Tetrahedron Lett., 2013, 54, 2101 CrossRef CAS PubMed; (o) J. Xie, Q. Xue, H. Jin, H. Li, Y. Cheng and C. Zhu, Chem. Sci., 2013, 4, 1281 RSC.
- (a) Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171 CrossRef CAS PubMed; (b) M. Rueping, D. Leonori and T. Poisson, Chem. Commun., 2011, 47, 9615 RSC.
- (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC; (b) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092 CrossRef CAS PubMed.
- P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed.
- (a) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. – Eur. J., 2012, 18, 16473 CrossRef CAS PubMed; (c) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966 RSC.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed.
- (a) S. Cai, X. Zhao, X. Wang, Q. Liu, Z. Li and D. Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 8050 CrossRef CAS PubMed; (b) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed; (c) L. R. Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107 CrossRef PubMed.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef CAS PubMed.
- (a) T. Koike and M. Akita, Chem. Lett., 2009, 38, 166 CrossRef CAS; (b) Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355 RSC.
- (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed; (b) F. R. Petronijevíc, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323 CrossRef PubMed.
- (a) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed; (b) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. D. Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed; (c) J. Wang, S. Wang, G. Wang, J. Zhang and X.-Q. Yu, Chem. Commun., 2012, 48, 11769 RSC; (d) S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302 CrossRef CAS PubMed; (e) T. W. Liwosz and S. R. Chemler, Org. Lett., 2013, 15, 3034 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (b) K. Miyazawa, Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 7249 RSC.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600 CrossRef CAS PubMed.
- (a) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed; (b) J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985 RSC; (c) J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368 CrossRef CAS PubMed; (d) L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104 CrossRef CAS PubMed.
- R. S. Andrew, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef PubMed.
- For selected examples, see: (a) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem. – Eur. J., 2012, 18, 15158 CrossRef CAS PubMed; (b) X. Gu, X. Li, Y. Qu, Q. Yang, P. Li and Y. Yao, Chem. – Eur. J., 2013, 19, 11878 CrossRef CAS PubMed; (c) Q. Liu, H. Yi, J. Liu, Y. Yang, X. Zhang, Z. Zeng and A. Lei, Chem. – Eur. J., 2013, 19, 5120 CrossRef CAS PubMed; (d) H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884 CrossRef CAS PubMed; (e) X.-J. Wei, D.-T. Yang, L. Wang, T. Song, L.-Z. Wu and Q. Liu, Org. Lett., 2013, 15, 6054 CrossRef CAS PubMed; (f) B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850 CrossRef CAS PubMed; (g) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS PubMed.
- (a) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (b) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed; (c) Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed.
- (a) G. Alvernhe, B. Langlois, A. Laurent, I. Le Drean, A. Selmi and M. Weissenfels, Tetrahedron Lett., 1991, 32, 643 CrossRef CAS; (b) H. A. Schenck, P. W. Lenkowski, I. Choudhury-Mukhejee, S.-H. Ko, J. P. Stables, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2004, 12, 979 CrossRef CAS PubMed.
- (a) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (b) N. Iqbal, S. Choi, E. Ko and E. J. Cho, Tetrahedron Lett., 2012, 53, 2005 CrossRef CAS PubMed.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (b) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef CAS PubMed; (c) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem. – Eur. J., 2012, 18, 15158 CrossRef CAS PubMed; (d) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed; (e) N. Iqbal, S. Choi, E. Kim and E. J. Cho, J. Org. Chem., 2012, 77, 11383 CrossRef CAS PubMed; (f) E. Kim, S. Choi, H. Kim and E. J. Cho, Chem. – Eur. J., 2013, 19, 6209 CrossRef CAS PubMed; (g) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed.
- For recent reviews on synthesis of CF3-containing molecules, see: (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (c) J.-A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975 CrossRef CAS PubMed; (d) N. Shibata, S. Mizuta and T. Toru, J. Synth. Org. Chem., Jpn., 2008, 66, 215 CrossRef CAS; (e) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS; (f) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (g) O. A. Tamashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef PubMed; (h) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (i) H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617 CrossRef CAS.
- L. Fensterbank, J.-P. Goddard, M. Malacria and C. Ollivier, Chimia, 2012, 66, 425 CrossRef CAS PubMed.
- (a) D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255 CrossRef; (b) T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellog, J. Org. Chem., 1979, 44, 4953 CrossRef CAS.
- (a) H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Perkin Trans. 2, 1984, 1093 RSC; (b) H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517 CrossRef CAS; (c) H. Cano-Yelo and A. Deronzier, New J. Chem., 1987, 11, 479 CAS.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS PubMed.
- (a) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed; (b) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS PubMed; (c) A. Ariafard, C. J. T. Hyland, A. J. Canty, M. Sharma and B. F. Yates, Inorg. Chem., 2011, 50, 6449 CrossRef CAS PubMed.
- (a) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034 CrossRef CAS PubMed; (b) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed.
- (a) D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed; (b) D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334–5337 CrossRef CAS PubMed.
- (a) P. Schroll, D. P. Hari and B. König, ChemistryOpen, 2012, 1, 130 CrossRef CAS PubMed; (b) T. Hering, D. P. Hari and B. König, J. Org. Chem., 2012, 77, 10347 CrossRef CAS PubMed; (c) D. P. Hari, T. Hering and B. König, Angew. Chem., Int. Ed., 2014, 53, 725 CrossRef PubMed.
- S. Donck, A. Baroudi, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2013, 355, 1477 CrossRef CAS.
- Y.-X. Liu, D. Xue, J.-D. Wang, C.-J. Zhao, Q.-Z. Zou, C. Wang and J. Xiao, Synlett, 2013, 507 Search PubMed.
- (a) W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, J. Org. Chem., 2013, 78, 12202 CrossRef CAS PubMed; (b) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398 CrossRef CAS PubMed; (c) M. Tobisu, T. Furukawa and N. Chatani, Chem. Lett., 2013, 42, 1203 CrossRef CAS; (d) D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J., 2014, 20, 2960 CrossRef CAS PubMed.
- T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS PubMed , and references therein.
- (a) P. Eisenberger, S. Gischig and A. Togni, Chem. – Eur. J., 2006, 12, 2579 CrossRef CAS PubMed; (b) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed; (b) Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 2037 RSC; (c) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed; (d) T. Koike and M. Akita, Synlett, 2013, 2492 CrossRef CAS PubMed; (e) Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Org. Lett., 2014, 16, 780 CrossRef CAS PubMed.
- (a) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gourverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef CAS PubMed; (b) S. Mizuta, K. M. Engle, S. Verhoog, O. Galicia-López, M. O'Duill, M. Médebielle, K. Wheelhouse, G. Rassias, A. L. Thompson and V. Gouverneur, Org. Lett., 2013, 15, 1250 CrossRef CAS PubMed; (c) P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Chem. – Eur. J., 2013, 19, 14039 CrossRef CAS PubMed; (d) P. Xu, A. Abdukader, K. Hu, Y. Cheng and C. Zhu, Chem. Commun., 2014, 50, 2308 RSC; (e) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240 CrossRef CAS PubMed; (f) J. Xie, X. Yuan, A. Abdukader, C. Zhu and J. Ma, Org. Lett., 2014, 16, 1768 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |