On the flexibility of carboranylalkylthio substituents in porphyrazines and its relevance to the photophysical properties†
Daniela
Pietrangeli
a,
Alexandra V.
Soldatova‡
b,
Daniele
Casarini
a,
Angela
Rosa
*a and
Giampaolo
Ricciardi
*a
aDipartimento di Scienze, Università della Basilicata, Viale dell'Ateneo Lucano 10, 85100 Potenza, Italy. E-mail: angela.rosa@unibas.it; giampaolo.ricciardi@unibas.it
bCenter for Photochemical Sciences and Department of Chemistry, Bowling Green State University, Bowling Green, OH 43403, USA
First published on 2nd June 2014
Abstract
Ultrafast transient absorption spectrometry and DFT/TDDFT calculations reveal that following photoexcitation, carboranyl- and carboranyl-free alkylthioporphyrazines deactivate by the pathway S1(π,π*)→Sn(Cβ-2pz/Sl.p.,π*)→ground state. The presence of quenching singlet excited states with predominantly Cβ-2pz/Sl.p.,π* character immediately below the primarily photogenerated S1(π,π*) state is a consequence of the electronic structure changes induced by the inherent flexibility of the alkylthio chains.
Alkylthioporphyrazines (ATPzs) are a widely investigated class of tetrapyrroles because of their potential applications in high-tech fields and biomedicine.1–5 Different from alkylthioporphyrins6 and aryl-substituted porphyrazines,7 which show luminescence and 1O2 photosensitizing properties, ATPzs are only weakly fluorescent, if any. According to preliminary photophysical studies, ATPzs undergo fast radiationless decay upon photoexcitation into the intense near-IR Q-band, which makes them promising candidates as sensitizers in photothermal therapy (PTT) of cancer.8
In the present work it is shown that the unusual photophysical behavior of ATPzs is related to the inherent flexibility of the alkylthio groups. The photophysical properties of ATPzs are investigated using 2,3,7,8,12,13,17,18-octakis-(1,2-dicarba closo-dodeca-boran-2-yl)hexylthio-5,10,15,20-(21H,23H)porphyrazine, H2HEHCSPz, as a case study.
The emission spectrum of H2HEHCSPz in CH2Cl2, normalized to the Qx(0,0) band maximum, is characterized by a structureless band centered at 738 nm with a Stokes shift of ∼530 cm−1,† as previously observed in other ATPzs.9 A very low value (0.0017) was obtained for the absolute emission quantum efficiency (qM) from the Strickler–Berg equation. A comparably small qM value was obtained for H2OESPz. Aggregation-induced fluorescence quenching was ruled out on the basis of Lambert–Beer experiments.†
Time-resolved transient absorption changes and kinetic data were acquired in CH2Cl2 at room temperature using 640 nm (Q band envelope) and 340 nm (B band maximum) excitation wavelengths. No dependence on the excitation wavelength was observed. The spectrum formed within the instrument response time (ca. 200 fs) displayed negative signals due to the bleaching of the ground-state absorption bands at 725 nm, 650 nm, and 500 nm, and positive absorption signals between 760–810 nm and 540–600 nm (Fig. 1).
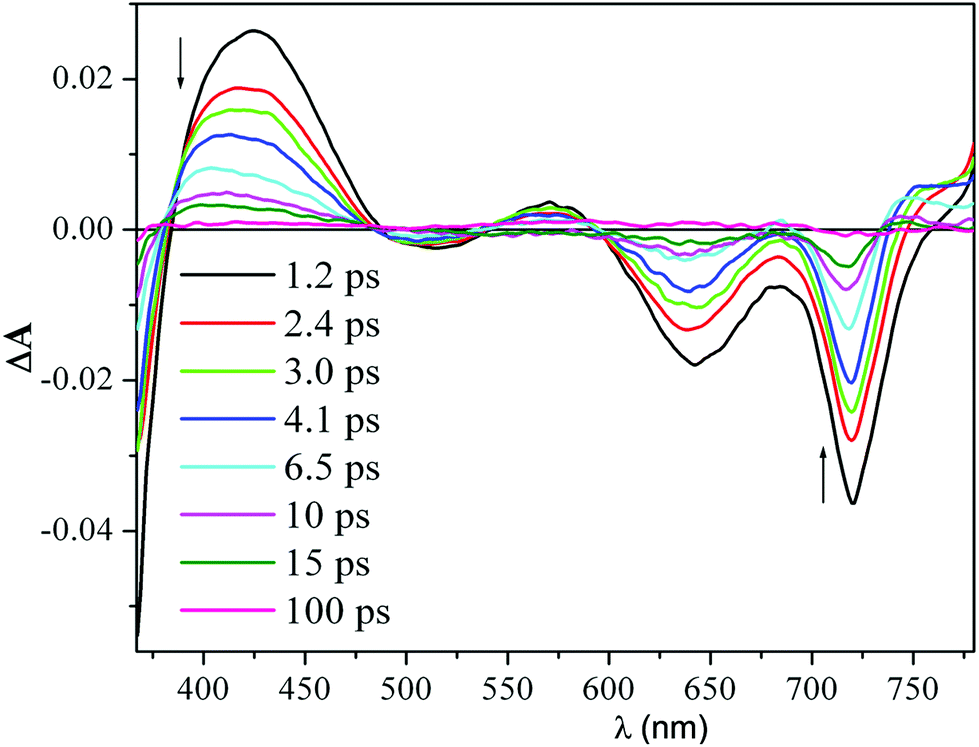 | ||
| Fig. 1 Femtosecond transient absorption difference spectra of a 40 μM solution of H2HEHCSPz in CH2Cl2 at different delay times after 340 nm excitation. | ||
A pronounced photoinduced absorption band at around 420 nm could also be detected superimposed on the strong bleaching signal due to the B band removal (1.2 ps spectrum in Fig. 1). This initially formed transient spectrum could be assigned to the S1(π,π*) state. As the time delay between the pump and the probe increased, the intensity of the transient absorption signal decreased; however, no changes in spectral shape could be detected during decay of the transient, except for the blue shift of the positive signals in the red and blue regions of the probe spectral window. After the blue shift was complete (ca. 8 ps), the transient absorption signal decayed to a small offset with isosbestic behavior. The blue shift of the transient absorption bands is generally associated with vibrational relaxation (cooling) of a vibrationally hot excited state.10 Interestingly, the observed vibrational relaxation process had a similar lifetime, independent of the excitation wavelength. Since the S1(π,π*) state formed within the instrument response time is already in its relaxed energy minimum, there must be other states that lie closely below the S1(π,π*) that might be populated in their high vibrational levels, also within the instrument response time. It is likely that these excited states, which absorb in the same spectral region as the S1(π,π*), decay uniformly to the ground state, resulting in the observed isosbestic behavior of the transient absorption signal.
The decay of the positive absorption as well as the ground-state recovery was best fit by a double exponential function (lifetimes of ca. 3.2 ps and 24 ps) plus a small constant term.† This term can be attributed to the S1(π,π*) state returning to the ground state by a radiative process (fluorescence), although a small contribution to the residual absorption at longer time from the T1(π,π*) state formed in very low yield cannot be ruled out. H2OESPz showed similar spectral and kinetic behavior.†
Variable temperature (VT) 1H NMR experiments provided information on the flexibility of the alkylthio groups. The changes in the H2HEHCSPz 1H NMR spectrum down to −95 °C are shown in Fig. 2. One sharp triplet was detected, at room temperature, at 4.10 ppm for the SCH2 groups, implying the equivalence of the hexyl chains and division of the whole molecule into eight equivalent parts by symmetry. Lowering the temperature down to −65 °C resulted in a broadening of the SCH2 triplet that eventually splits into two broad signals separated by 168 Hz at −95 °C. Line-shape simulation of the experimental traces at various temperatures allowed us to determine an activation free energy, ΔG#, of 9.90 ± 0.15 kcal mol−1 for the twisting of the peripheral chains around the Cβ–S bond.
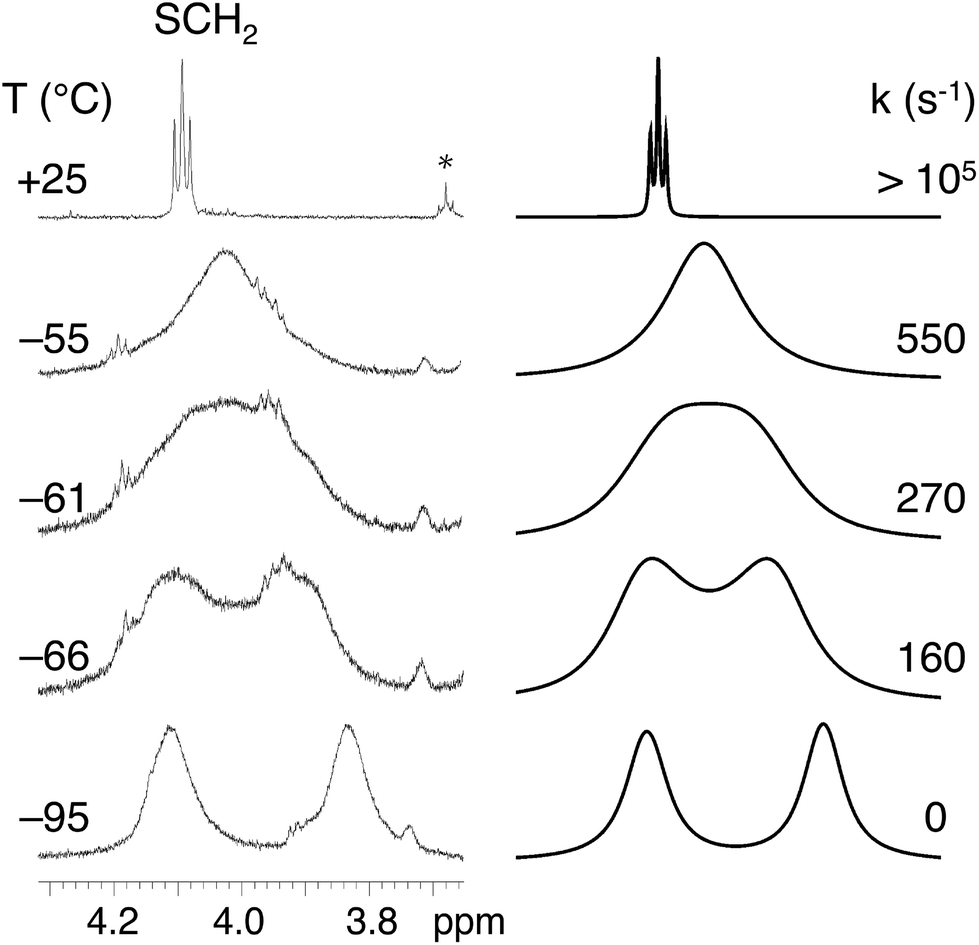 | ||
| Fig. 2 1H NMR experimental traces for H2HEHCSPz in CD2Cl2 showing the temperature dependence of the SCH2 signal at 4.10 ppm (left); the starred signal at 3.7 ppm in the 25 °C trace refers to residual THF in the sample. Simulated traces and computed rate constants (right). | ||
The observed diastereotopic behavior of the SCH2 groups suggests that at low temperatures the elements of symmetry are reduced from eight to four, implying that vicinal SCH2 groups have been locked in an anti arrangement. A comparable free-energy barrier (9.85 ± 0.15 kcal mol−1) was found for H2OESPz,† suggesting that in the investigated ATPzs the flexibility of the peripheral chains is influenced neither by their size nor by the presence of terminal carboranyl groups.
To understand whether the observed flexibility of the alkylthio groups plays a role in determining the unusual photophysical behavior of the investigated ATPzs, DFT/TDDFT calculations of the ground and lowest excited states of the H2OMSPz model system were performed using the Amsterdam Density Functional (ADF) program package.11†
A previous DFT analysis of the conformational behavior of this molecule by some of us revealed that the nearly degenerate C2h and C2v symmetry conformations, characterized by an uu-ud-dd-du (u = up; d = down) and uu-dd-uu-dd orientation of the methyl groups, respectively, are the preferred ones among several choices.12 Therefore, only these two conformers were examined in this study.
To model the electronic effects of the twisting of the methyl groups around the Cβ–S bond, for each conformer two structures were considered, the equilibrium structure, a, and a structure where the methyl groups were constrained to be nearly coplanar with the Pz ring, b.13 The C2h and C2vb structures were, in CH2Cl2 solution, only 11.5 and 13.7 kcal mol−1 less stable than the corresponding equilibrium structures, so they are, in principle, thermally accessible. This fits in with VT 1H NMR data pointing to a facile twisting of the alkyl groups around the Cβ–S bond.
The lowest singlet and triplet excited states of the a and b structures of each conformer were examined in CH2Cl2 solution, at the TDDFT/B3LYP/TZ2P level of theory. The vertical absorption energies, composition, and oscillator strength computed for the whole set of singlet and triplet excited states up to ∼0.2 eV above the S1(Qx) state are gathered in Tables S1–S4.† The S1(Qx) and the lower lying excited states computed for the a and b structures of the C2h conformer, taken as representative, are plotted in Fig. 3.
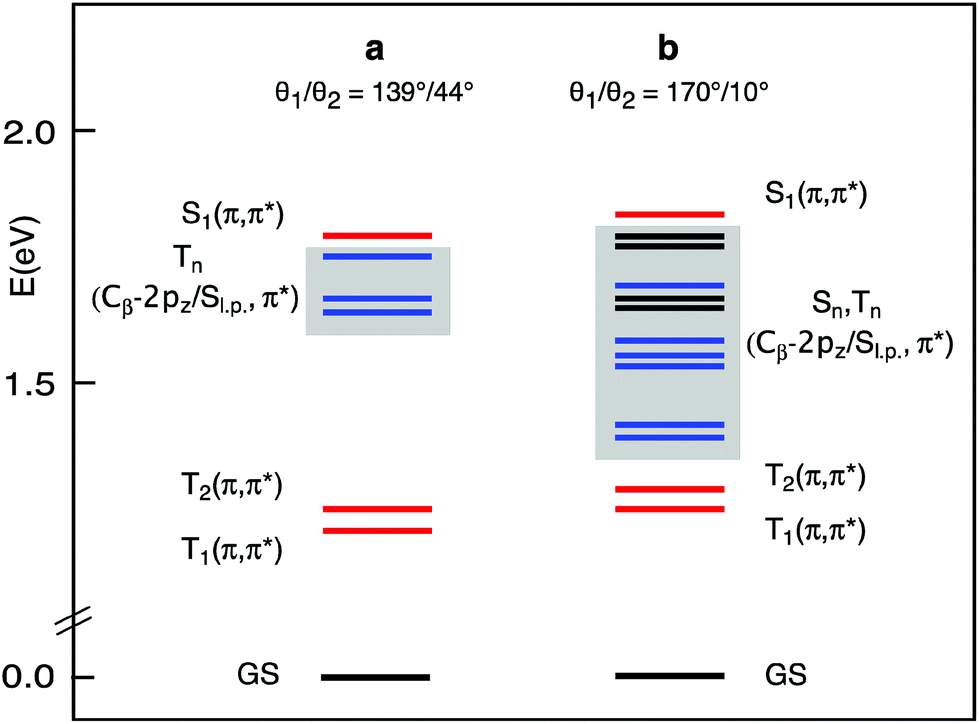 | ||
| Fig. 3 Lowest excited states of the H2OMSPz C2h conformer in the a and b structures obtained at the DFT/B3LYP/TZ2P level in CH2Cl2 solution. The singlet and triplet states with Cβ-2pz/Sl.p.,π* character are printed in black and blue, respectively. | ||
At the equilibrium geometry (Table S1† and Fig. 3), four triplet excited states are found to lie vertically between the S1(π,π*) state, which is located at 676 nm in nice agreement with the experiment, and the normally emissive T1(π,π*) state. Except for the T2(π,π*), which is the triplet corresponding to the S2(Qy) state, these excited states involve transitions out of Cβ-2pz/sulfur lone pair (Sl.p.) π-antibonding MOs into the Gouterman π* MOs.
This scenario changes significantly when the methyl groups are nearly coplanar with the Pz ring, as in the b structure. Now, a manifold of excited states (singlets and triplets) is located between the S1(π,π*) and the T1(π,π*) states (Fig. 3). Except for the T2(π,π*), these excited states are dominated by Cβ-2pz/Sl.p.→π* transitions. In the b structure they end up in this spectral region essentially because of the upshift, visible in the energy level diagram of Fig. 4, of the involved Cβ-2pz/Sl.p MOs, i.e., the 46bu, 41bg, 45bu, and 47ag.
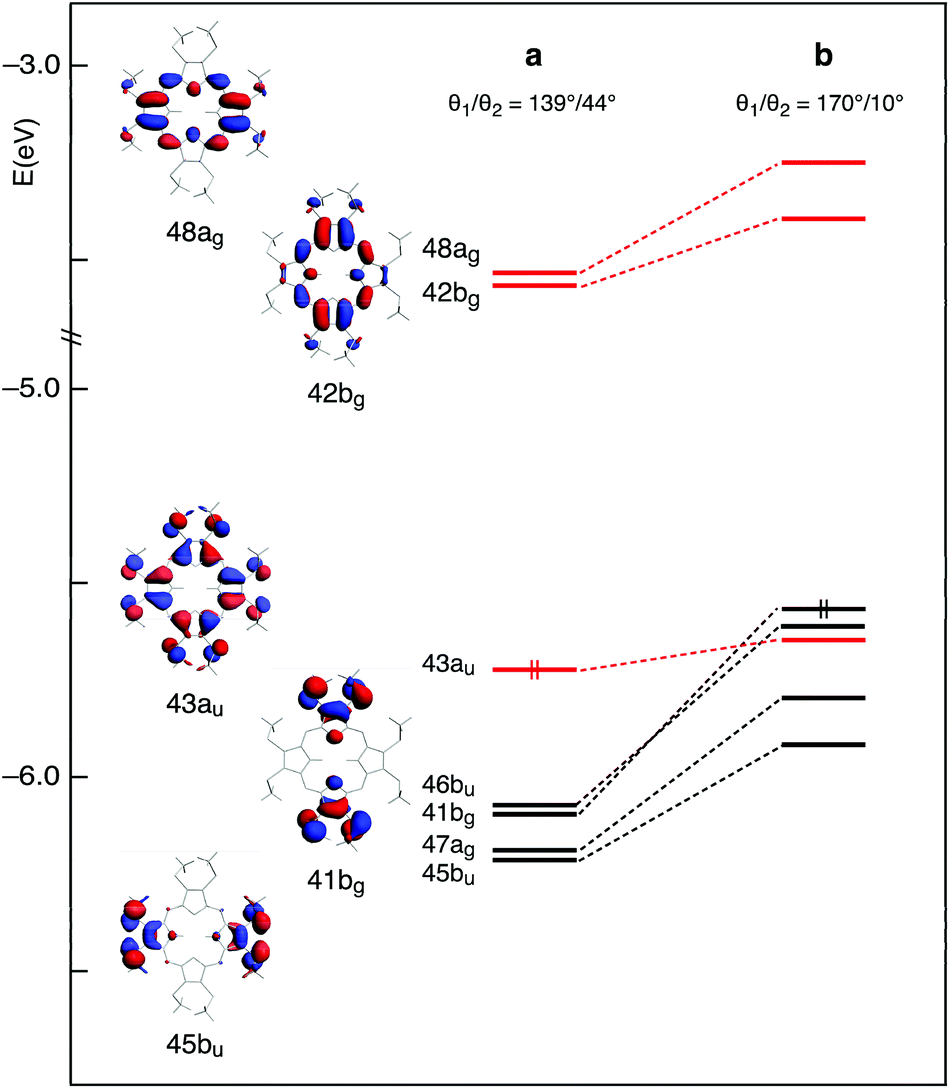 | ||
| Fig. 4 Energy level diagram and molecular orbitals of the H2OMSPz C2h conformer in the a and b structures obtained at the DFT/B3LYP/TZ2P level in CH2Cl2 solution. Double occupancy is indicated for the HOMO only. All lower lying levels are also doubly occupied. The 43au, 42bg, and 48ag Gouterman levels are printed in red. | ||
The destabilization of these MOs can be explained in terms of enhancement of their π-antibonding character (see the plots of the 41bg and 45bu MOs in Fig. 4) in response to the improved overlap between Cβ-2pz and sulfur lone pair orbitals upon twisting of the methyl groups.
The numerous singlet states lying below the S1(π,π*) are likely to facilitate the radiationless decay of this state, as suggested by ultrafast experiments showing that a very small fraction of the photogenerated S1(π,π*) state of the investigated molecules returns directly to the ground state by radiative processes.
Thus, ultrafast experiments and TDDFT calculations clearly indicate that the twisting-induced π-conjugation between the sulfur lone pairs and the Pz π-system is detrimental to luminescence. That ATPzs appreciably fluoresce from both the Soret and Q bands when the majority of the vicinal alkylthio chains are locked in the most stable anti conformation as it happens in thin “solid” films5 further supports this conclusion. As a final remark, we note that H2HEHCSPz and analogous ATPzs represent relevant examples of tetrapyrrolic compounds where fast radiationless decay of the photogenerated S1(π,π*) state is related to the presence of low-lying ligand-centered excited states, rather than to the presence of low-lying metal-centered excited states, as was shown to occur in several first-row transition-metal tetrapyrroles.10
The results described in this work present necessary background and basic knowledge in support of the prospective evaluation of the photothermal conversion performance14 of H2HEHCSPz and related molecules.
The research was in part financially supported by the Università della Basilicata (RIL-2008 Funds). Spectrometric facilities were provided by the Ohio Laboratory of Kinetic Spectrometry at Bowling Green State University. The authors thank Prof. M. A. J. Rodgers for stimulating discussions. DP and GR thank Dr M. P. Donzello, Università di Roma “La Sapienza,” Italy, and Dr A. Vassallo, Università della Basilicata, Potenza, Italy, for performing microanalyses and mass spectra, respectively.
References
- D. Pietrangeli, A. Rosa, S. Ristori, A. Salvati, S. Altieri and G. Ricciardi, Coord. Chem. Rev., 2013, 257, 2213–2231 CrossRef CAS.
- P. Sun, H. Zong, K. Salaita, J. B. Ketter, A. G. M. Barrett, B. M. Hoffman and C. A. Mirkin, J. Phys. Chem. B, 2006, 110, 18151–18153 CrossRef CAS PubMed.
- H. Zong, P. Sun, C. A. Mirkin, A. G. M. Barrett and B. M. Hoffman, J. Phys. Chem. B, 2009, 113, 14892–14903 CrossRef CAS PubMed.
- Y. Song, H. Zong, E. R. Trivedi, B. J. Vesper, E. A. Waters, A. G. M. Barrett, J. A. Radosevich, B. M. Hoffman and T. J. Meade, Bioconjugate Chem., 2010, 21, 2267–2275 CrossRef CAS PubMed.
- G. Garramone, D. Pietrangeli, G. Ricciardi, S. Conoci, M. R. Guascito, C. Malitesta, D. Cesari, S. Casilli, L. Giotta, G. Giancane and L. Valli, J. Phys. Chem. B, 2008, 112, 11517–11528 CrossRef CAS PubMed.
- K.-I. Sugiura, M. Ravi Kumar, T. K. Chandrashckar and Y. Sakata, Chem. Lett., 1997, 291–292 CrossRef CAS.
- Q. Gan, F. Xiong, S. Li, S. Q. Wang, S. Shen, H. Xu and G. Yang, Inorg. Chem. Commun., 2005, 8, 285–288 CrossRef CAS.
- S. Ristori, A. Salvati, G. Martini, O. Spalla, D. Pietrangeli, A. Rosa and G. Ricciardi, J. Am. Chem. Soc., 2007, 129, 2728–2729 CrossRef CAS PubMed.
- S. Lee, A. J. P. White, D. J. Williams, A. G. M. Barrett and B. M. Hoffman, J. Org. Chem., 2001, 66, 461–465 CrossRef CAS PubMed.
- A. Rosa and G. Ricciardi, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2012, vol. 22, pp. 170–236 Search PubMed.
- ADF2013, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- S. Ristori, G. Ricciardi, D. Pietrangeli, A. Rosa and A. Feis, J. Phys. Chem. C, 2009, 8537–8540 CAS.
- The CMe–S–Cβ–Cβ torsion angles formed by the methylthio groups with the Cβ–Cβ bond of the pyrrole (θ1) and pyrrolenine (θ2) rings were taken as a measure of the degree of twisting of the methyl groups.
- Q. Tian, F. Jiang, R. Zou, Q. Liu, Z. Chen, M. Zhu, S. Yang, J. Wang, J. Wang and J. Hu, ACS Nano, 2011, 5, 9761–9771 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c4qi00055b |
| ‡ Present address: Department of Chemistry, University of Washington, Seattle, WA 98195, USA. |
| This journal is © the Partner Organisations 2014 |