
Synthesis, characterisation, water adsorption and proton conductivity of three Cd(II) based luminescent metal–organic frameworks†
Srinivasulu
Parshamoni
,
Himanshu Sekhar
Jena
 ,
Suresh
Sanda
and
Sanjit
Konar
*
,
Suresh
Sanda
and
Sanjit
Konar
*
Department of Chemistry, IISER Bhopal, Indore By-pass Road, Bhauri, Bhopal 462066, Madhya Pradesh, India. E-mail: skonar@iiserb.ac.in; Fax: +91-755-6692392; Tel: +91-755-6692339
First published on 14th July 2014
Abstract
Three hydrogen bonded three-dimensional (3D) metal–organic frameworks (MOFs) namely [Cd(L-tart)(bpy)(H2O)]n·9n(H2O) (1), [Cd(D-tart)(bpy)(H2O)]n·9n(H2O) (2) and [Cd(DL-tart)(bpy)(H2O)]n·6n(H2O) (3) (tart = tartaric acid, bpy = 4,4-bipyridine) have been synthesized by the solvent diffusion technique at room temperature. Compounds 1 and 2 have been characterized by single crystal X-ray analysis, whereas the powder X-ray diffraction patterns show that the structural integrity of compound 3 is similar to 1 and 2. Structural analysis of 1 and 2 shows H-bonded homochiral 3D MOFs, fabricated by the hydrogen bonding interactions between the nearby 2D pillared-layer frameworks through the metal-bound water, metal-bound carboxylate, free carboxylic acid and the hydroxy group of L-/D- tart. The absolute configuration of all the compounds was investigated by solid state circular dichroism (CD) spectroscopy, which signifies that 1 and 2 are enantiomers whereas 3 is racemic. The adsorption studies reveal that compounds 1–3 show a significant amount of uptake for water vapor (∼239 mL g−1 for 1, ∼240 mL g−1 in 2, whereas 184 mL g−1 for 3 at P/P0 ≈ 1 bar) over other solvents (MeOH, EtOH) and an impedance measurement indicates that these compounds show proton conduction (1.3 × 10−6 S cm−1 in 1, 1.3 × 10−6 S cm−1 in 2 and 4.5 × 10−7 S cm−1 in 3) at a higher temperature (358 K) and at 95% relative humidity. The observed conductivity is explained by the so-called vehicle mechanism (activation energy (Ea) = 0.63–0.77 eV). Since all the compounds contain H3O+ cations in the interlayer space, the hydronium ions might act as vehicles to transport the protons in the interlayer space. The photoluminescence properties of all the compounds are also reported.
Introduction
In the past few decades, metal–organic frameworks (MOFs) have undergone explosive growth. Many scientific research groups are devoted to various aspects of MOFs and to exploring their applications in many fields, such as gas storage/separation,1 conductivity,2 luminescence,3 catalysis,4etc. Recently, chiral MOFs have also gained emerging interest not only because of their aesthetically structural architectures and topologies but also because of their potential applications in enantioselective catalysis,5 chiral separation6 and nonlinear optics.7 Generally, two main approaches have been investigated for the construction of chiral MOFs; one is based on the use of chiral sources (chiral organic linkers or metal complexes), while the other is spontaneous resolution of achiral sources. Although some chiral MOFs using achiral ligands have been reported,6c,8 construction of chiral MOFs with enantiopure ligands seems to be straightforward, but it always suffers from complex synthesis and structural limitations of enantiopure ligands.9 In this context, using π-conjugated, rigid organic linkers with enantiopure carboxylates is an effective approach to synthesize homochiral MOFs where chirality is based on the ligand. Although various strategies have been documented for the synthesis of pillared-layer 3D networks, a few H-bonded pillared-layer homochiral 3D networks have been reported so far.10Recently, the development of new materials which show proton conduction has become particularly important because of their potential applications in solid-state electrochemical devices such as batteries and fuel cells.11 Although Nafion is a well-known proton conductor,12 at higher temperatures (>80 °C) its performance decreases due to dehydration.13 In this context, several groups have dedicated their efforts to study various aspects of proton conductivity in MOFs as well as coordination polymers (CPs) and to explore their possible applications in fuel cells as alternatives to Nafion.14 In general, to show proton conduction a material needs proton carriers such as H3O+ or H+ given by acids or OH groups or by incorporation of proton carriers such as imidazole, triazoles, ammonium ions, hydroxonium ions, etc.15 into the frameworks. Amongst the reported MOFs only a very few show conductivity at high temperature (80 °C and 98% RH).16 On the other hand, only a few chiral MOFs exhibiting proton conducting properties have been reported17 and only one of them is claimed to be multifunctional luminescent and proton-conducting as reported by Cabeza et al.18
In the present work, we report a series of three Cd(II) based H-bonded 3D MOFs namely [Cd(L-tart)(bpy)(H2O)]n·9n(H2O) (1), [Cd(D-tart)(bpy)(H2O)]n·9n(H2O) (2) and [Cd(DL-tart)(bpy)(H2O)]n·6n(H2O) (3) using the mixture of L-, D-, DL-tartaric acid (H2tart) and 4,4-bipyridine (bpy) ligands, obtained by slow crystallization under ambient conditions. All the three compounds are characterized by single crystal X-ray diffraction analysis (SXRD), powder X-ray diffraction analysis (PXRD), thermogravimetric analysis (TGA) and the circular dichroism (CD) study. The most remarkable characteristics of these compounds are that they show photoluminescent properties at room temperature and proton conductivity at higher temperature (85 °C) and 95% RH.
Results and discussion
Synthetic aspects
Tartaric acid (Scheme 1) plays an important role in the discovery of chemical chirality and forms discrete chiral complexes, MOFs and CPs with diverse functional properties depending on the metal ions used. The hydroxyl groups present in its backbone assists in shortening the distance between the metal centers and also supports in complexion. Besides, a shorter distance creates a dense or rigid arrangement of metal centers in the system which might cause photoluminescence properties in the d10 system.19 However, the introduction of different pyridyl linkers along with tartrate acid can cause flexibility in the framework and influence their associated properties.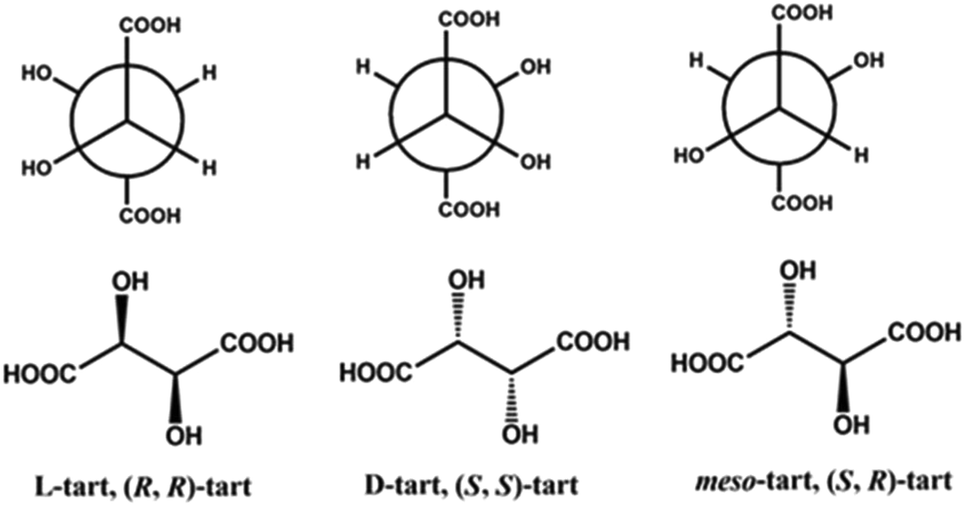 | ||
| Scheme 1 The various tart isomers. Top: Newman projection; bottom: stereo chemical drawing. | ||
In this context, herein three compounds namely [Cd(L-tart)(bpy)(H2O)]n·9n(H2O) (1), [Cd(D-tart)(bpy)(H2O)]n·9n(H2O) (2) and [Cd(DL-tart)(bpy)(H2O)]n·6n(H2O) (3) have been synthesized using a corresponding sodium salt of tartaric acid, bpy and Cd(NO3)2·4H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio through the solvent diffusion technique using an ethanol–water (1:1:1) solvent system at room temperature.
:1:1 ratio through the solvent diffusion technique using an ethanol–water (1:1:1) solvent system at room temperature.
Compounds 1 and 2 have been characterized by single crystal X-ray diffraction and are found to be isostructural in nature. However, we could not obtain diffraction quality single crystals for compound 3 even after several attempts. In order to confirm the structural integrity of 3, the bulk-phase powder X-ray diffraction patterns (PXRD) of the three compounds were measured and found to be in good agreement with each other, which reflects that compound 3 is isostructural with 1 and 2 (Fig. 1). Furthermore, the PXRD patterns are matched well with the simulated patterns of the single crystal data of both 1 and 2, indicating the phase purity of bulk samples. The differences in intensities of experimental PXRD with the simulated PXRD may be due to the preferred orientation of the crystalline powder sample. The observed difference peak at 2θ = 24° in Fig. 1d in comparison with Fig. 1b and 1c is because two peaks are merged into one broad peak. Additionally we have indexed the powder pattern of compound 3 using the program DICVOL91 that suggests a monoclinic crystal system with unit cell parameters, a = 11.28 Å, b = 11.67 Å, c = 8.24 Å and β = 114.622°.
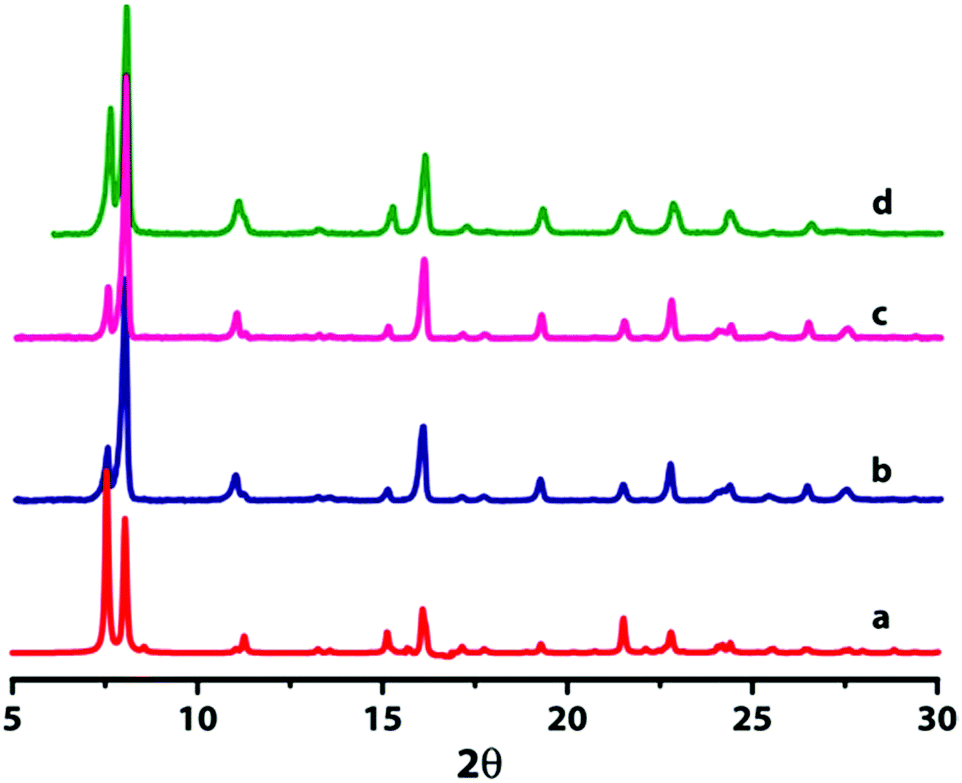 | ||
| Fig. 1 PXRD patterns of compounds 1–3. (a) Simulated PXRD of 1 (red); (b) as-synthesized samples of compounds 1 and (c) compound 2; (d) as-synthesized samples of compound 3, this matches well with compounds 1 and 2. | ||
Compounds 1–3 show similar types of FT-IR spectra (Fig. S1†) and bands are given in Table S1.† A broad band was observed in the region of 3376–3400 cm−1, which specifies the presence of free or coordinated water molecules. The characteristic bands for the asymmetric (v(COO)asym) and symmetric stretching vibrations (v(COO)asym) of the carboxylate groups appear at 1599–1385 cm−1 in 1, 1605–1385 cm−1 in 2, and 1612–1392 cm−1 in 3, respectively. The noted difference (Δν = ν(COO)asym – ν(COO)sym) in the bridging modes of carboxylate groups may confirm the bridging mode of the carboxylate groups found in these compounds.20 The band at 1670 cm−1 may be assigned to the uncoordinated and deprotonated carboxylate group of tart involved in strong hydrogen bonds.
Structural description of compounds 1 and 2
Single crystal X-ray diffraction analysis reveals that compounds 1 and 2 are isostructural and are also enantiomers. Hence only the structure of compound 1 is discussed in detail with a suitable comparison with 2. The compound 1 crystallizes in the chiral orthorhombic system with the space group P21212. Its asymmetric unit comprises of one Cd(II) ion, one L-tart, one bpy and one coordinate water molecule (Fig. S2†). Each Cd(II) centre adapts hepta coordinated with contributions from three oxygen atoms (O1, O1A and O3A) of two different L-tart ligands, one hydroxyl group (O2) of an L-tart ligand, two nitrogen atoms (N1 and N2) from two different bpy linkers and one oxygen atom (O7) from a water molecule and consequently forms a distorted pentagonal bipyramidal geometry (Fig. S3†).It was found that the oxygen atoms of the carboxylate group as well as the hydroxyl group of L-tart and coordinated water molecules are located in the equatorial plane whereas the nitrogen atoms of neutral bpy linkers are occupied in the axial positions. The Cd–O bond lengths are found to be in the range of 2.265(4) Å–2.495(3) Å and Cd–N bond lengths are measured in the range of 2.316(5)–2.325(5) Å. The O–Cd–O and N–Cd–N bond angles lie in the range of 53.1(1)–176.9(2)°. In 1 and 2, a distinctive bridging mode of L-tart was observed which was not documented earlier in the tartrate based 3d metal complexes system. Precisely, in both 1 and 2, out of two hydroxyl and two carboxylate groups of L/D-tart, one hydroxyl and one carboxylate group coordinates to the Cd(II) centre whereas the other two remains uncoordinated. The carboxylate oxygen atoms (O1, O3) of L/D-tart coordinates to the Cd(II) centres in μ-1,1,2-fashion with a Harris notation20,21 of 3.21. Notably one carboxylate oxygen atom (O1) of L/D-tart bridges two Cd(II) centres in chelating bridging mode at a distance of 4.60(4) Å and subtends at an angle of 146.6(1)° (Cd1–O1–Cd1A). Such a bridging mode of carboxylate oxygen extends the hepta coordinated Cd(II) centres along the b direction to form a homochiral 1D chain (P-helical) with a pitch of 8.406 Å (Fig. 2). It was observed that in 1 the L-tart ligands (D-tart for 2) are arranged alternatively around the helical strand. Similarly each neutral pillared bpy linker coordinates to two Cd(II) centres at a distance of 11.72(1) Å and extends along the c direction to form another infinite 1D chain. The aforementioned two 1D chains are perpendicularly interconnected along the bc plane to form a homochiral 2D pillared-layer framework. The resulting 2D framework encompasses continuous rectangular channels of dimensions 4.6 × 11.7 Å2 (Fig. 3). The channels are supported by weak π⋯π stacking interaction (cg⋯cg = 4.552 Å) between pyridine–pyridine moieties of bpy linkers. Interestingly, the coordinated water molecules exhibit bifurcate H-bonding interaction (2.686(5)–2.978(9) Å) with oxygen atoms of coordinated and free carboxylate groups of L/D-tart. The oxygen atoms of the free carboxylate group also H-bonded with the hydroxyl group of L/D-tart. The H-bond parameters are listed in Tables S2 and S3.†
 | ||
| Fig. 2 Illustration of P-helical motifs of 1 (right) and 2 (left) running down the b-axis. Color code: carbon (light gray), hydrogen (gray), nitrogen (blue), oxygen (red) and cadmium (cyan). | ||
 | ||
| Fig. 3 Illustrations of 2D pillared layer frameworks present in 1 along the bc-plane. Color code: same as in Fig. 2. | ||
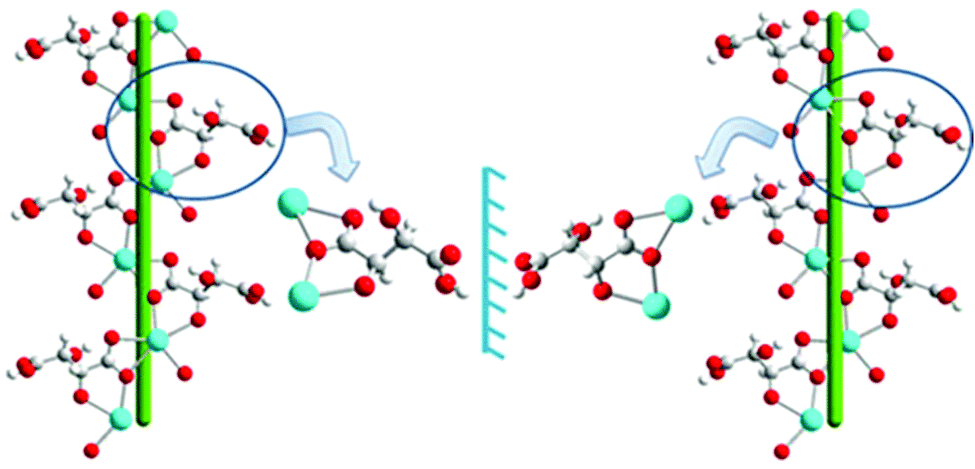
The 3D packing analysis reveals that the above-noted 2D pillared layer frameworks are H-bonded with each other to form a 3D H-bonded supramolecular framework (Fig. 4(a). Precisely in the H-bonded networks (Fig. 4(b)), metal-bound water molecules act as proton donors (O7) and the metal-bound carboxylate oxygen (O3) and the hydroxyl group of L/D-tart as acceptors (O2); however, the oxygen atoms of the free carboxylic acid group of tart ligands act as both a donor (O6) and an acceptor (O5). The noted 3D supramolecular framework enclosed within large square shaped channels (9.9 Å2 × 11.5 Å2) along the ac-plane, which is occupied by guest water molecules. It is interesting to compare the 3D packing in both the compounds (Fig. 5): the bpy pillared linker extend perpendicularly to the [Cd-(L-tart)]n chain (Fig. 5(a)) whereas in 2, they are arranged in a “helical fashion”. Careful analysis of the homochiral 3D supramolecular network of compound 2 revealed that [Cd(D-tart)]n acts as a rod to the helical strand (Fig. 5(b)).
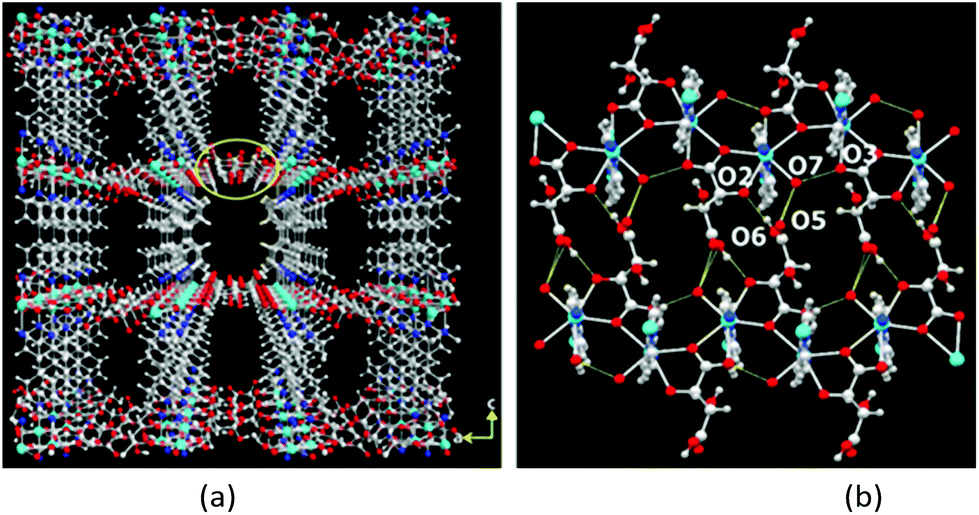 | ||
| Fig. 4 Illustrations of (a) H-bonded homochiral 3D MOF found in 1, (b) H-bonded interactions between metal-bound water and carboxylate groups with free hydroxyl and the carboxylate group of L/D-tart. Color code: same as in Fig. 2. | ||
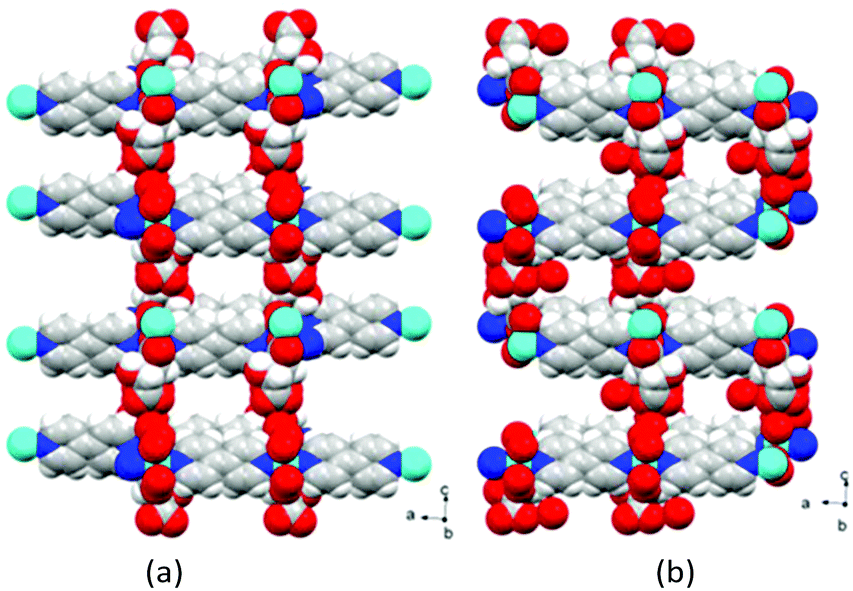 | ||
| Fig. 5 Space-filling diagram of (a) compound 1 and (b) compound 2. Colour code: same as in Fig. 2. | ||
Topological perception of 1 reveals that both the L-tart ligand and the bpy linker are linked to two Cd(II) centers and hence can be regarded as two connected nodes. Each Cd(II) is linked to nearby four Cd(II) centres by two L-tart ligands and two bpy linkers. Therefore, the 2D homochiral porous framework can be simplified as a 4-connected uninodal net with a point symbol of {44·62} exhibiting a sql/Shubnikov tetragonal plane net topology22 (Fig. S4†). PLATON analysis reveals that the compound 1 contains an effective free void volume of about 30.8% of the crystal volume (666.8 Å3 out of the 2165.5 Å3 unit-cell volumes). Nine highly disordered water molecules per formula unit were found within the channels and were modelled with the SQUEEZE routine in PLATON.
Circular dichroism study
In order to confirm the absolute configuration, compounds 1–3 were subjected to the solid state circular dichroism (CD) spectroscopy as shown in Fig. 6.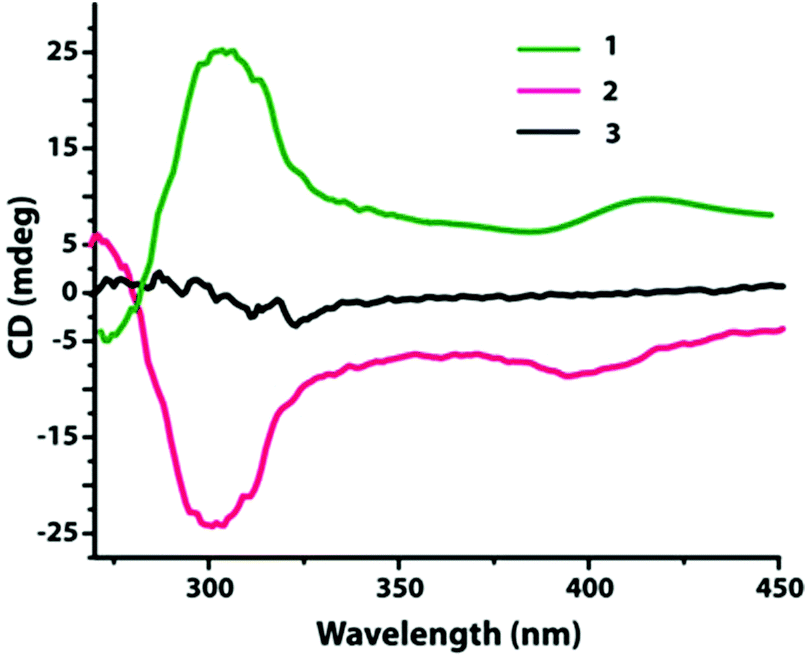 | ||
| Fig. 6 Solid-state CD spectra of compounds 1, 2, and 3. | ||
Compounds 1 and 2 display positive and negative dichroic signals at the wavelength range 270–340 nm, respectively, and are also mirror images to each other. This signifies that they are enantiomers. A similar analysis for compound 3 does not show any dichroic signal in the noted wavelength range which signifies the presence of racemic mixtures of tartrate.
These phenomena imply that the observed homochirality in compounds 1 and 2 was transferred from the corresponding chiral tartrate ligand to the metal center and in turn to the whole framework system.
Thermal stabilities
To examine the thermal stability of compounds 1–3, thermo gravimetric analysis (TGA) was performed in the temperature range of 30–650 °C under a N2 flow with a heating rate of 10 °C min−1. Compounds 1 and 2 show a similar type of profile (Fig. S5 and S6†). A weight loss of 18% (calcd 17%) was observed in the range of 75–180 °C which could be assigned to the loss of one coordinated water and nine lattice water molecules. The dehydrated framework was stable up to 300 °C followed by rapid weight loss due to the decomposition of the framework. Compound 3 shows a gradual weight-loss of 13% (calcd 12%) at around 75–180 °C, which corresponds to the loss of one coordinated water and six lattice water molecules and the dehydrated framework being stable up to 300 °C (Fig. S7†).Adsorption studies
To examine the porous nature of all compounds, we have carried out the adsorption studies with CO2 as well as with different solvent vapors (H2O, EtOH and MeOH). The sorption isotherm of CO2 at 195 K reveals very less uptake (8 cm3 g−1 in 1 and 2, 7 cm3 g−1 in 3) signifying nonporous nature of all compounds (Fig. S8–S10†). This may be due to the presence of smaller channel size compared to the kinetic diameter of CO2 (kinetic diameter, 3.4 Å).23 However, quite interesting results were obtained with the solvent vapour adsorption study.Adsorption isotherms of H2O for both compounds 1 and 2 show less uptake (74 mL g−1) up to P/P0 ∼ 0.50 and it starts rising sharply at P/P0 ∼ 0.63 and reaches a final uptake volume of ∼239 mL g−1 at P/P0 ≈ 1 bar pressure, which is a comparable value with the eminent proton conducting MOFs [Me-FeCr(ox)3] (268 cm3 g−1 at STP). Whereas compound 3 shows almost a linear uptake with the uptake amount of 184 mL g−1 at P/P0 ≈ 1 bar pressure (Fig. 7 and Fig. S11†). Adsorption data of 1 and 2 indicate uptake of 9.3 H2O molecules per formula unit whereas it is 7.3 in the case of 3. The low uptake in all compounds in the low pressure region might be due to the difficulty in occlusion of H2O molecules in small 2D rectangular channels and a sharp uptake indicates reaccumulation of H2O molecules inside the 3D supramolecular pore and filling of unsaturated Cd(II) centers.24 Desorption isotherms of all compounds do not retrace the adsorption profile and show large hysteresis, suggesting strong interaction (H-bonding) with the pendent carboxylate oxygen atoms of the pore surface.23a It is worth mentioning that the adsorption isotherms of MeOH and EtOH for all compounds show a Type-III profile with the uptake amount of 31 mL g−1 and 23 mL g−1 in 1 and 2 (Fig. S12 and S13†), 29 mL g−1 and 28 mL g−1 in 3 at 1 bar pressure respectively (Fig. S14†). The less adsorption can be justified by correlating of their large molecular diameter of MeOH (kinetic diameter 3.8 Å) and EtOH (kinetic diameter 4.3 Å) compared to the effective pore size and unlikely to go inside into the pores, which results only in surface adsorption.25
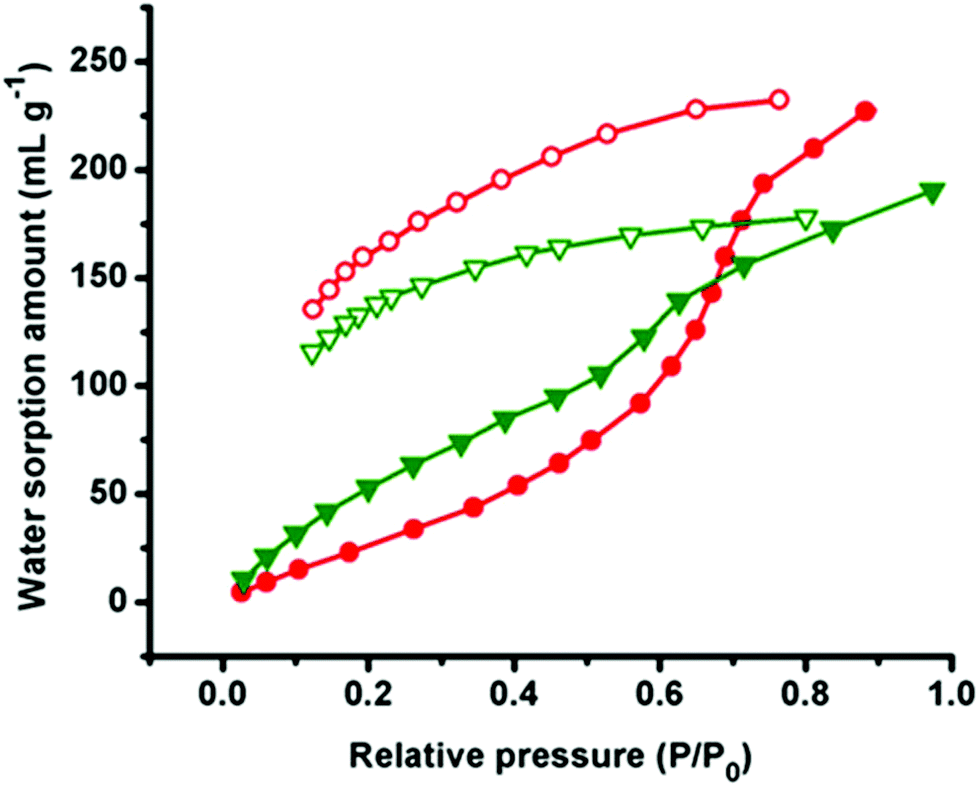 | ||
| Fig. 7 Water vapour adsorption isotherm of compounds 1 (red) and 3 (green). | ||
Proton conductivity study
Although we were unable to identify the disordered water molecules in the framework from X-Ray data, but, in the TG analysis, it has been observed that compounds 1 and 2 contain nine and compound 3 contains six lattice water molecules, respectively. It is worth mentioning that in the frameworks of compounds 1 and 2, the free acidic carboxylates and acidic metal-bound water molecules are orientated towards the interlayer space between the 2D pillared-layer frameworks (Fig. 4(b)). This observation encouraged us to evaluate their proton conductivity properties.We have evaluated the proton conductivity of all compounds 1–3 by ac impedance analysis. All the compounds were exposed and equilibrated at relative humidity for 24 h before measurement. The proton conductivity of all compounds was estimated from the Nyquist plots as shown in Fig. S15–S17.† Compounds 1–3 show proton conductivity values of 5.0 × 10−10 S cm−1 for 1, 5.2 × 10−10 S cm−1 for 2 and 1.3 × 10−10 S cm−1 for 3 at 298 K and 95% relative humidity (RH). In order to assess the effect of humidity on the proton conduction, we performed a humidity sweep impedance measurement and the results show that all the compounds display better conductivity at 95% RH (Table S4†). The observed distorted half-circles in the Nyquist plots might be due to the closeness of relaxation frequencies of different electro active regions resulting only in one distorted or depressed semicircle.26
We have also performed the temperature dependent proton conductivities for all compounds at constant 95% RH which show that the proton conductivities of all the compounds increased with increasing temperature. Compound 1 shows a wide range of proton conductivities from 1.9 × 10−9 S cm−1 (303 K) to 1.3 × 10−6 S cm−1 (358 K), compound 2 shows 1.3 × 10−9 S cm−1 (303 K) to 1.3 × 10−6 S cm−1 (358 K) and compound 3 shows 9.3 × 10−10 S cm−1 (303 K) to 4.5 × 10−7 S cm−1 (358 K) (Fig. 8). Proton conduction in MOFs, particularly at higher temperatures (more than 345 K and 98% RH) is an unusual phenomenon and there are only a few reports available in the literature where MOFs containing lattice H2O molecules, conduct protons at higher temperature (PCMOF-5: 4 × 10−3 S cm−1 at 335 K),16c (MIL-53(Fe)–(NH2)2: 4.1 × 10−8 S cm−1 at 353 K),16d (Ca–BTC–H2O: 3.2 × 10−5 S cm−1 at 345 K).16e
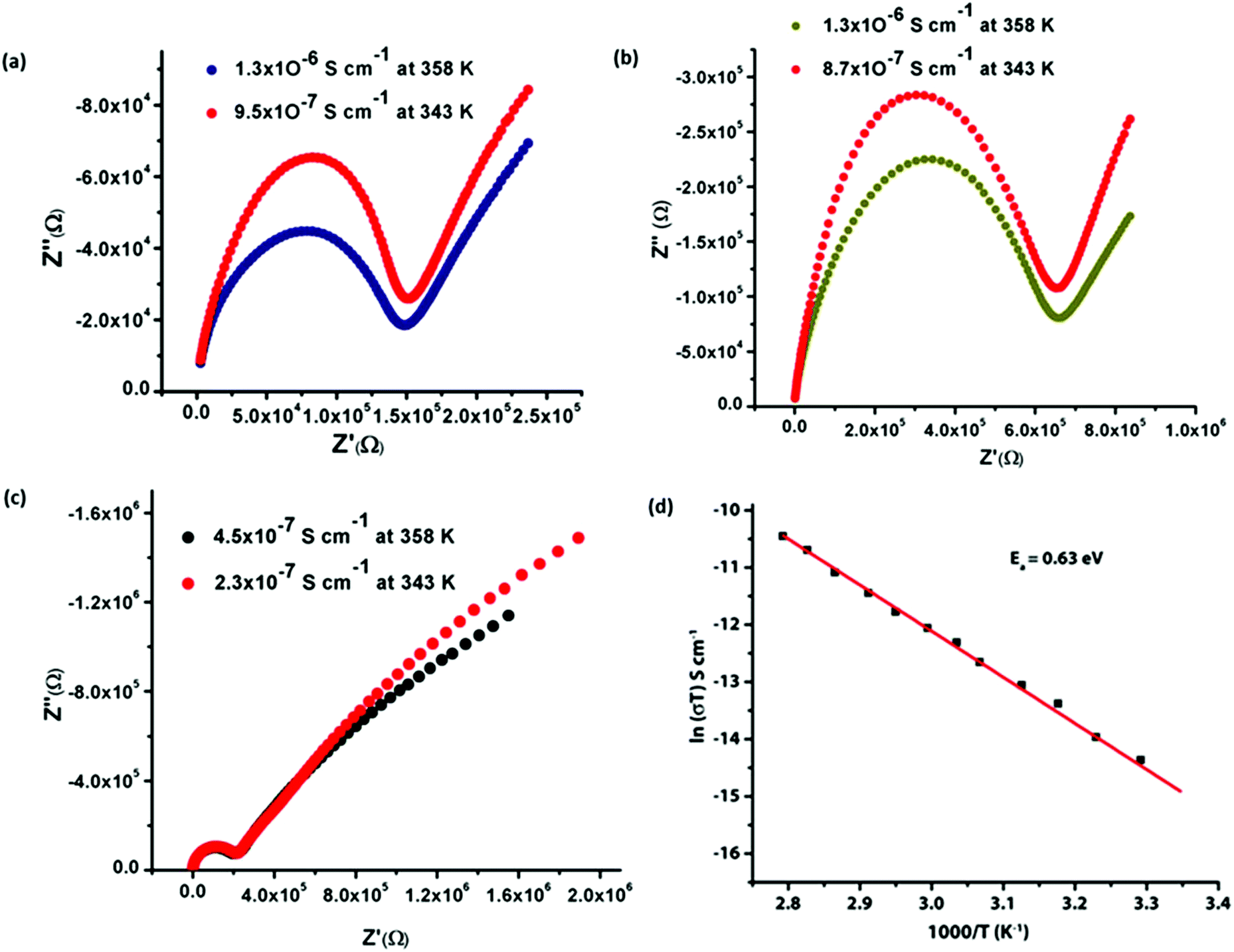 | ||
| Fig. 8 The Nyquist plots for proton conductivity of (a) compound 1, (b) compound 2, (c) compound 3 at different temperature. (d) Arrhenius plot of activation energy for compound 1 showing activation energy value of 0.63 eV. | ||
To ensure the stabilities of the frameworks of all the compounds after exposing at 98% RH for 24 h at room temperature (298 K) and at 358 K, the exposed compounds are subjected to PXRD and TG analysis. The PXRD analysis of the samples 1 and 2 reveals that all major peaks are well matched with as-synthesized samples expecting the peak to be around 2θ = 7–8° (Fig. S18 and S19†). It was found that the two peaks merged into a broad peak, which might be due to the shift of H-bonded layers in the lattice during grinding which produces a different phase than that obtained in the single crystalline form. However in the case of 3 all peaks are well matched with the as-synthesized sample (Fig. S20†). From the TG profile, we observed that sixteen water molecules are present in compounds 1 and 2 at 298 K and 95% RH. Similarly in 3, fourteen water molecules are present under the same conditions. Hence in 1 and 2, six and in 3, seven additional water molecules are accumulating in the framework at 95% RH and room temperature. However at 358 K and 95% RH, in 1 and 2 four and in 3, three additional water molecules are accumulating in the framework (Fig. S21–S23†). The less accumulation of additional water molecules at 358 K might be due to the loss of some weakly H-bonded water molecules from each framework. However, we observed a better proton conductivity of all the compounds at 358 K. The observed relative high proton conductivity at 358 K might be due to the presence of a continuous hydrogen bonding 1D array among the acidic carboxylic acid group, coordinate and lattice H2O molecules.16e Although each framework accumulates more numbers of H2O molecules at 298 K, the less conductivity might be due to the presence of discrete H-bonded clusters in the framework. From the above study, it can be concluded that the free acidic carboxylic acid and acidic metal-bound water molecules act as proton source and lattice water molecules carry the proton in a 1D array in the interlayer space between the 2D networks.
The activation energies for the proton migration of the materials give reasonable insight into the possible mechanism for the movement of protons in solids, i.e. whether it is a Grotthus (<0.6 eV) or a vehicular mechanism (>0.6 eV).27 In the present study, temperature dependencies of the conductivity of all the compounds were measured, and their corresponding Arrhenius plots are shown in Fig. 8d, Fig. S24 and S25†. The activation energies (Ea) for the compounds 1, 2, and 3 were 0.63 eV, 0.67 eV and 0.77 eV, respectively, using eqn (1).
| σT = σ0eEa/kT | (1) |
Although the observed activation energies are found to be low but are well in the range of reported values (0.137 eV–1.16 eV). These values are typically observed for proton conductors that follow the vehicle mechanism. Since all the compounds contain (H3O)+ cations in the interlayer space of the H-bonded 3D networks, the hydronium ions act as a vehicle to transport the protons in the interlayer space.28
Luminescent properties
The solid-state luminescent emission properties of L-, D- and DL-tart, bpy and compounds 1–3 were investigated at room temperature (Fig. 9).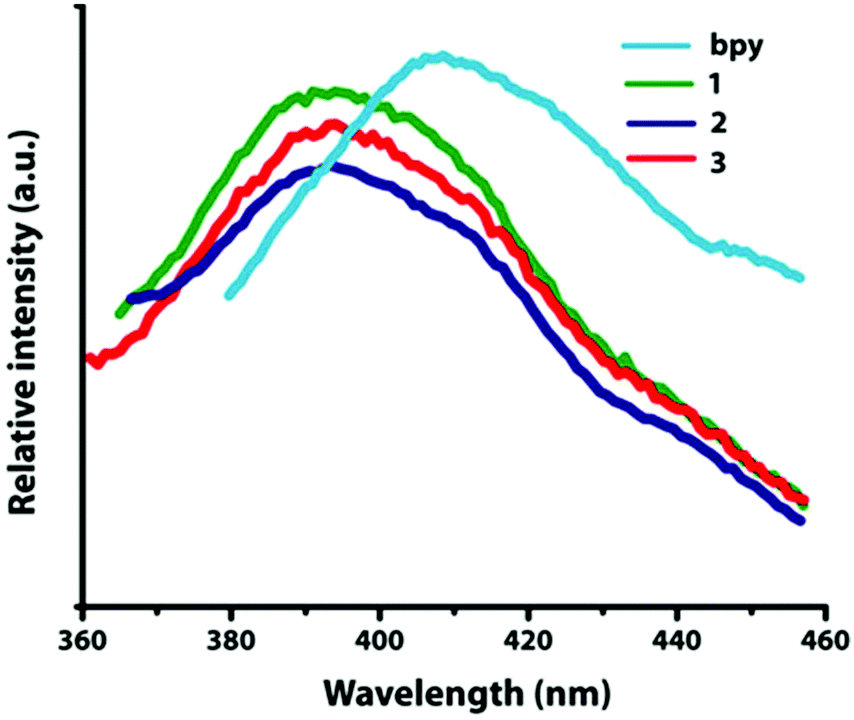 | ||
| Fig. 9 Solid-state emission spectra for the ligand used and compounds 1, 2, and 3. | ||
L-, D- and DL-tart do not show any emission whereas only bpy shows stronger emission at 415 nm (λex = 321 nm), which can be assigned to the π*→π transitions.29 Upon photo excitation, compounds 1–3 display the maximum emission peaks at 392 nm (λex = 281 nm). The emission bands of 1–3 display significant blue shifts, which can be attributed to the charge transfer of pyridine-based ligands to Cd(II) centers (LMCT) in comparison with the emission of the bpy ligand.30
Conclusion
We have successfully synthesised three hydrogen bonded 3D MOFs using the solvent diffusion technique at room temperature. Compounds 1 and 2 have been characterized by single crystal X-ray analysis. All three compounds were characterized by powder X-ray diffraction and thermogravimetric analysis, which shows that they are isostructural in nature. Vapour sorption studies reveal that all compounds show high uptake of water vapour over other solvents (MeOH and EtOH). Impedance measurement indicates that these compounds show proton conduction at higher temperature (358 K) and at 95% relative humidity and the observed conductivity is explained by the so-called vehicle mechanism (activation energy (Ea) = 0.63–0.77 eV). Since all the compounds contain (H3O)+ cations in the interlayer space, the hydronium ions might act as a vehicle to transport the protons in the interlayer space. The photoluminescence properties of all the compounds are also reported.Experimental section
Materials
All the reagents and solvents for synthesis were purchased from commercial sources and used as supplied without further purification. Cd(NO3)2·6H2O, D-, L- and DL-tartaric acid and 4,4-bipyridine were obtained from Sigma-Aldrich Chemical Co. India. The elemental analyses were carried out on a Elementar micro vario cube elemental analyser. FT-IR spectra (4000–500 cm−1) were recorded on a KBr pellet with a Perkin Elmer Spectrum BX spectrometer. Powder X-ray diffraction (PXRD) data were collected on a PANalytical EMPYREAN instrument using Cu-Kα radiation. Solid state UV–Vis spectra, luminescence spectra and CD spectra were recorded using a Shimadzu UV-3101PC spectrometer, a Horiba Jobin-Yvon Fluorolog-3 spectrophotometer and a JASCO J-851-150L CD spectrometer, respectively.Single crystal X-ray diffraction
Single crystal data for compounds 1 and 2 were collected on a Bruker APEX II diffractometer equipped with a graphite monochromator and Mo-Kα (λ = 0.71073 Å, 296 K) radiation. Data collections were performed using φ and ω scans. Non-hydrogen atoms were located from difference Fourier maps and refined anisotropically by full-matrix least-squares on F2, using SHELXS-97.31 All hydrogen atoms were included in the calculated positions and refined isotropically using a riding model. Determinations of the crystal system, the orientation matrix, and cell dimensions were performed according to established procedures. Lorentz polarization and multi-scan absorption correction were applied. All calculations were carried out using SHELXL 97,32 PLATON 99,33 and WinGXsystemVer-1.64.34During the final stages of refinement, some Q peaks having high electron densities were found, which probably correspond to highly disordered solvent water molecules and are removed by the SQUEEZE program.34 From the TG analysis we speculate that compounds 1 and 2 contain nine H2O molecules whereas compound 3 contains six H2O molecules and hence these are included in the molecular formula. Data collection and structure refinement parameters and crystallographic data are given in Table 1 and selected bond lengths and bond angles for compounds 1 and 2 are given in Table S5.†
| Compound | 1 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo. b R w = [∑{w(Fo2 − Fc2)2}/∑{w(Fo2)2}]1/2. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CCDC | 986614 | 986613 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formula | C28H46Cd2N4O23 | C28H46Cd2N4O23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Weight (g mol−1) | 1031.5 | 1031.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal shape | Plate | Plate | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Color | Colorless | Colorless | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Size | 0.42 × 0.37 × 0.24 | 0.42 × 0.37 × 0.24 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal system | Orthorhombic | Orthorhombic | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Space group | P2(1)2(1)2 | P2(1)2(1)2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell length, a (Å) | 21.9953(12) | 21.9737(11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell length, b (Å) | 8.4064(5) | 8.4037(4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell length, c (Å) | 11.7118(7) | 11.7037(6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell angle, α (°) | 90.00 | 90.00 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell angle, β (°) | 90.00 | 90.00 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell angle, γ (°) | 90.00 | 90.00 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell volume, V (Å3) | 2165.53 | 2161.21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell formula units, Z | 4 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Temperature (K) | 296(2) | 296(2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| λ (Mo-Kα) (Å) | 0.71073 | 0.71073 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| μ (mm−1) | 1.037 | 1.039 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| D c (g cm−3) | 1.327 | 1.330 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal_F_000 | 856 | 856 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Measured reflections | 6225 | 5504 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unique reflections | 5664 | 4957 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Goodness-of-fit | 1.038 | 1.118 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Theta range for data collection | 2.54 to 29.82 | 2.54 to 28.51 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R 1[I > 2σ(I)]a | 0.0422 | 0.0474 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R w[I > 2σ(I)]b | 0.1318 | 0.144 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sorption measurements
Gas adsorption measurements were performed using BelSorp-max (BEL Japan) automatic volumetric adsorption instrument. All the gases used were of ultra-pure research grade (99.99%). Before every measurement samples were pretreated for 12 h at 423 K under 10−2 kPa continuous vacuum using BelPrepvac II and purged with N2 on cooling. CO2, CH4 isotherms were measured at 195 K (dry ice–MeOH cold bath). The adsorptions of different solvents like MeOH, H2O and EtOH at 298 K were measured in the vapour state using a BELSORP-aqua3 volumetric or gravimetric analyzer.Conductivity measurements
Proton conductivity measurements of the compounds were measured by the quasi-four-probe method using a Solartron SI 1260 Impedance/Gain-Phase Analyzer and a 1296 Dielectric Interface, in the frequency range of 1 Hz–1 MHz. For sample preparation, ∼130 mg of as-synthesized samples were finely ground and then pressed in a pellet maker to obtain uniform pellets of 0.42–0.46 mm thickness, 13 mm diameter. The temperature and humidity were controlled by a programmable Incubator (JEIOTECH, TH-PE series). Samples were equilibrated for at least 8 h after each step in temperature and 12 h after each step in humidity. All the operations were computer-controlled and automatic. The resistances were calculated from the semicircles of the Nyquist plots. Proton conductivity was measured by the following equation: σ = L/(RA), where σ = proton conductivity, L = thickness of the pellet, R = resistance of the pellet and A = area of the pellet = 4πr2, where r = radius of the pellet. The activation energy (Ea) values were determined from the slopes of Arrhenius plots by least square fitting.:1 H2O and EtOH). Colourless plate shaped single crystals were obtained from the junction of the layers after two weeks. The crystals were separated and washed with EtOH and air-dried (yield 80%). Elemental analysis: Calcd for C28H46Cd2N4O23 (%). C, 32.6; N, 5.4; H, 4.4; Found (%): C, 32.4; N, 5.1; H, 3.9; FT-IR (KBr pellet, cm−1) 3396 (b), 1670 (s), 1599 (s), 1385 (s), 1112 (s), 1073 (s), 529 (b).
:1:1 molar ratio using ethanol as a solvent. Colourless, plate shaped single crystals were obtained from the junction of the layers after two weeks. The crystals were separated and washed with EtOH and air-dried (yield 73%). Elemental analysis: C28H46Cd2N4O23 (%).C, 32.6; N, 5.4; H, 4.4; Found (%): C, 32.4; N, 5.1; H, 3.9; FT-IR (KBr pellet, cm−1) 3376 (b), 1670 (s), 1605 (s), 1385 (s), 1112 (s), 1066 (s), 633 (b).
:1:1 molar ratio using ethanol as a solvent. Colourless crystalline compounds were obtained from the junction of the layers after two weeks. The crystals were separated and washed with EtOH and air-dried (yield 45%). Elemental analysis: Anal. Calcd for C28H40Cd2N4O20 (%). C, 34.4; N, 5.7; H, 4.1; Found (%): C, 34.1; N, 5.2; H, 3.5; FT-IR (KBr pellet, cm−1) 3376 (b), 1667 (s), 1612 (s), 1392 (s), 1112 (s), 1066 (s), 646 (b).
Acknowledgements
SP and SS thank IISER Bhopal for the PhD fellowships and HSJ thanks for the post-doctoral fellowship. SK thanks CSIR, Government of India (project no. 01/(2473)/11/EMRII), and IISER Bhopal for generous financial and infrastructural support. The authors sincerely thank Dr H. N. Gopi, IISER Pune, for CD measurements.References
- (a) M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed., 1997, 36, 1725 CrossRef CAS; (b) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed; (c) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 7, 229 CrossRef CAS PubMed; (d) S. A. Dalrymple and G. K. H. Shimizu, J. Am. Chem. Soc., 2007, 129, 121141 CrossRef PubMed; (e) T. A. Makal, J. R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761 RSC; (f) J. R. Li, R. J. Kuppler and H.–C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (g) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed; (h) H. Hayashi, A. P. Coté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS PubMed; (i) S. Parshamoni, S. Sanda, H. S. Jena and S. Konar, Dalton Trans., 2014, 43, 7191 RSC; (j) S. Biswas, H. S. Jena, S. Goswami, S. Sanda and S. Konar, Cryst. Growth Des., 2014, 14, 1287 CrossRef CAS.
- (a) G. Ferey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J. M. Greneche and J. M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259 CrossRef CAS PubMed; (b) S. Ohkoshi, K. Nakagawa, K. Tomono, K. Imoto, Y. Tsunobuchi and H. Tokoro, J. Am. Chem. Soc., 2010, 132, 6620 CrossRef CAS PubMed; (c) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. M. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef CAS PubMed; (d) C. Y. Duan, M. L. Wei, D. Guo, C. He and Q. J. Meng, J. Am. Chem. Soc., 2010, 132, 3321 CrossRef CAS PubMed; (e) S. C. Sahoo, T. Kundu and R. Banerjee, J. Am. Chem. Soc., 2011, 133, 17950 CrossRef CAS PubMed; (f) S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831 CrossRef CAS PubMed; (g) D. Umeyama, S. Horike, M. Inukai, Y. Hijikata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 1 CrossRef; (h) N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51 CrossRef CAS PubMed.
- (a) Y.-G. Sun, X.-F. Gu, F. Ding, P. F. Smet, E.-J. Gao, D. Poelman and F. Verpoort, Cryst. Growth Des., 2010, 10, 1059 CrossRef CAS; (b) X.-J. Wang, Z.-M. Cen, Q.-L. Ni, X.-F. Jiang, H.-C. Lian, L.-C. Gui, H.-H. Zuo and Z.-Y. Wang, Cryst. Growth Des., 2010, 10, 2960 CrossRef CAS; (c) H.-B. Xu, X.-M. Chen, Q.-S. Zhang, L.-Y. Zhang and Z.-N. Chen, Chem. Commun., 2009, 7318 RSC; (d) S. Sanda, S. Parshamoni, A. Adhikary and S. Konar, Cryst. Growth Des., 2013, 13, 5442 CrossRef CAS.
- (a) A. Corma, H. Garcia and F. X. Llabres i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; (b) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (c) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151 CrossRef CAS; (d) C. D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed; (e) Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315 RSC.
- (a) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS PubMed; (b) K. Nomiya, S. Takahashi, R. Noguchi, S. Nemoto, T. Takayama and M. Oda, Inorg. Chem., 2000, 39, 3301 CrossRef CAS; (c) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (d) R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353 CrossRef CAS PubMed.
- (a) G. Li, W. Yu and Y. Cui, J. Am. Chem. Soc., 2008, 130, 4582 CrossRef CAS PubMed; (b) M. C. Das, Q. S. Guo, Y. B. He, J. Kim, C. G. Zhao, K. L. Hong, S. C. Xiang, Z. T. Zhang, K. M. Thomas, R. Krishna and B. L. Chen, J. Am. Chem. Soc., 2012, 134, 8703 CrossRef CAS PubMed; (c) K. K. Bisht and E. Suresh, J. Am. Chem. Soc., 2013, 135, 15690 CrossRef CAS PubMed.
- (a) X. Duan, Q. Meng, Y. Su, Y. Li, C. Duan, X. Ren and C. Lu, Chem. – Eur. J., 2011, 17, 9936 CrossRef CAS; (b) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS PubMed; (c) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084 CrossRef CAS PubMed.
- (a) K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51, 9577 CrossRef CAS PubMed; (b) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342 RSC; (c) Q.–Y. Liu, Y.-L. Wang, N. Zhang, Y.-L. Jiang, J.-J. Wei and F. Luo, Cryst. Growth Des., 2011, 11, 3717 CrossRef CAS; (d) E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, J. Am. Chem. Soc., 2004, 126, 1419 CrossRef CAS PubMed; (e) X.-L. Tong, T.-L. Hu, L.-P. Zhao, Y.-K. Wang, H. Zhang and X.-H. Bu, Chem. Commun., 2010, 46, 8543 RSC.
- (a) L. Ma, J. M. Falkowski, C. Abney and W. B. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS PubMed; (b) F. J. Song, C. Wang, J. M. Falkowski, L. Q. Ma and W. B. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef CAS PubMed; (c) R. Vaidhyanathan, D. Bradshaw, J.-N. Rebilly, J. P. Barrio, J. A. Gould, N. G. Berry and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2006, 45, 6495 CrossRef CAS PubMed; (d) K. Suh, M. P. Yutkin, D. N. Dybtsev, V. P. Fedinc and K. Kim, Chem. Commun., 2012, 48, 513 RSC.
- (a) S. K. Ghosh, J. Ribas and P. K. Bharadwaj, Cryst. Growth Des., 2005, 5, 623 CrossRef CAS; (b) J.-C. Dai, X.-T. Wu, Z.-Y. Fu, S.-M. Hu, W.-X. Du, C.-P. Cui, L.-M. Wu, H.-H. Zhang and R.-Q. Sun, Chem. Commun., 2002, 12 RSC; (c) D. Sun, R. Cao, Y. Liang, Q. Shi, W. Su and M. J. Hong, J. Chem. Soc., Dalton Trans., 2001, 2335 RSC; (d) C. S. Hong, S. K. Son, Y. S. Lee, M. J. Jun and Y. Do, Inorg. Chem., 1999, 38, 5602 CrossRef CAS PubMed.
- (a) K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637 CrossRef CAS; (b) B. C. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed.
- K. A. Maruitz and R. B. Moore, Chem. Rev., 2000, 104, 4535 CrossRef.
- N. G. Hainovsky, Y. T. Pavlukhin and E. F. Hairetdinov, Solid State Ionics, 1986, 20, 249 CrossRef.
- (a) M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472 CrossRef CAS PubMed; (b) S. Ohkoshi, K. Nakagawa, K. Tomono, K. Imoto, Y. Tsunobuchi and H. Tokoro, J. Am. Chem. Soc., 2010, 132, 6620 CrossRef CAS PubMed; (c) D. Umeyama, S. Horike, M. Inukai, Y. Hijikata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 1 CrossRef; (d) Y. Nagao, R. Ikeda, S. Kanda, Y. Kubozono and H. Kitagawa, Mol. Cryst. Liq. Cryst., 2002, 379, 89 CrossRef CAS; (e) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. M. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef CAS PubMed; (f) J. M. Taylor, R. K. Mah, I. L. Moudrakovski, C. I. Ratcliffe, R. Vaidhyanathan and G. K. H. Shimizu, J. Am. Chem. Soc., 2010, 132, 14055 CrossRef CAS PubMed; (g) S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831 CrossRef CAS PubMed; (h) H. Ōkawa, M. Sadakiyo, T. Yamada, M. Maesato, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 2256 CrossRef PubMed; (i) M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 9906 CrossRef CAS PubMed.
- (a) Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120 CrossRef CAS; (b) M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 9906 CrossRef CAS PubMed; (c) J. A. Hurd, R. Vaidyanathan, V. Thangadurai, C. Ractcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef CAS PubMed; (d) S. Brueekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831 CrossRef PubMed.
- (a) F. Costantino, A. Donnadio and M. Casciola, Inorg. Chem., 2012, 51, 6992 CrossRef CAS PubMed; (b) S. R. Kim, K. W. Dawson, B. S. Gelfand, J. M. Taylor and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 963 CrossRef CAS PubMed; (c) J. M. Taylor, K. W. Dawson and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 1193 CrossRef CAS PubMed; (d) A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034 CrossRef CAS PubMed; (e) A. Mallick, T. Kundu and R. Banerjee, Chem. Commun., 2012, 48, 8829 RSC.
- (a) S. C. Sahoo, T. Kundu and R. Banerjee, J. Am. Chem. Soc., 2011, 133, 17950 CrossRef CAS PubMed; (b) E. Pardo, C. Train, G. Gontard, K. Boubekeur, O. Fabelo, H. Liu, B. Dkhil, F. Lloret, K. Nakagawa, H. Tokoro, S. Ohkoshi and M. Verdaguer, J. Am. Chem. Soc., 2011, 133, 15328 CrossRef CAS PubMed.
- R. M. P. Colodrero, K. E. Papathanasiou, N. Stavgianoudaki, P. Olivera-Pastor, E. R. Losilla, M. A. G. Aranda, L. Leon-Reina, J. Sanz, I. Sobrados, D. Choquesillo-Lazarte, J. M. Garcia-Ruiz, P. Atienzar, F. Rey, K. D. Demadis and A. Cabeza, Chem. Mater., 2012, 24, 3780 CrossRef CAS.
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García Prof. and A. Murcia-Martínez, Chem. – Eur. J., 2006, 12, 3484 CrossRef CAS PubMed.
- S. Parshamoni, S. Sanda, H. S. Jena, K. Tomar and S. Konar, Cryst. Growth Des., 2014, 14, 2022 CAS.
- (a) R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349 RSC; (b) S. Khatua, S. Goswami, S. Parshamoni, H. S. Jena and S. Konar, RSC Adv., 2013, 3, 25237 RSC.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, TOPOS3.2: a new version of the program package for multipurpose crystal–chemical analysis, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- (a) K. L. Gurunatha and T. K. Maji, Inorg. Chim. Acta, 2009, 362, 1541 CrossRef CAS PubMed; (b) A. Hazra, P. Kanoo, S. Mohapatra, G. Mostafa and T. K. Maji, CrystEngComm, 2010, 12, 2775 RSC.
- (a) J. Kumar, P. Kanoo, T. K. Maji and S. Verma, CrystEngComm, 2012, 14, 3012 RSC; (b) S. Sanda, S. Goswami, H. S. Jena, S. Parshamoni and S. Konar, CrystEngComm, 2014, 16, 4742–4752 RSC.
- (a) P. Kanoo, G. Mostafa, R. Matsuda, S. Kitagawa and T. K. Maji, Chem. Commun., 2011, 47, 8106 RSC; (b) S. Sanda, S. Parshamoni and S. Konar, Inorg. Chem., 2013, 52, 12866 CrossRef CAS PubMed.
- (a) V. V. Brus, Semiconductors, 2012, 46, 1012 CrossRef CAS; (b) V. V. Brus, Semicond. Sci. Technol., 2012, 27, 035024 CrossRef; (c) V. V. Brus, M. Zellmeier, X. Zhang, S. M. Greil, M. Gluba, A. J. Töfflinger, J. Rappich and N. H. Nickel, Org. Electron., 2013, 14, 3109 CrossRef CAS PubMed.
- (a) M. Yoon, K. Suh, S. Natarajan and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 2688 CrossRef CAS PubMed; (b) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef CAS PubMed; (c) N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2011, 134, 51 CrossRef PubMed; (d) T. Yamada, M. Sadakiyo and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 3144 CrossRef CAS PubMed.
- (a) X. Y. Dong, R. Wang, J. B. Li, S. Q. Zang, H. W. Hou and T. C. W. Mak, Chem. Commun., 2013, 49, 1059 Search PubMed; (b) K.-D. Kreuer, Chem. Mater., 1996, 8, 610 CrossRef CAS; (c) K. D. Kreuer, A. Rabenau and R. Messer, Appl. Phys. A: Mater. Sci. Process., 1983, 32, 45 CrossRef; (d) A. Telfah, G. Majer, K. D. Kreuer, M. Schuster and Maier, J. Solid State Ionics, 2010, 181, 461 CrossRef CAS PubMed.
- (a) W. Yang, F. Yi, X. Li, L. Wang, S. Dang and Z. Sun, RSC Adv., 2013, 3, 25065 RSC; (b) H. Wang, F. Yi, S. Dang, W. Tian and Z. Sun, Cryst. Growth Des., 2014, 14, 147 CrossRef CAS; (c) R. B. Fu, S. C. Xiang, S. M. Hu, L. S. Wang, Y. M. Li, X. H. Huang and X. T. Wu, Chem. Commun., 2005, 5292 RSC; (d) X. D. Guo, G. S. Zhu, Q. R. Fang, M. Xue, G. Tian, J. Y. Sun, X. T. Li and S. L. Qiu, Inorg. Chem., 2005, 44, 3850 CrossRef CAS PubMed; (e) J. Zhang, W. Lin, Z. F. Chen, R. G. Xiong, B. F. Abrahams and H. K. Fun, J. Chem. Soc., Dalton Trans., 2001, 1806 RSC; (f) C. D. Wu, H. L. Ngo and W. Lin, Chem. Commun., 2004, 1588 RSC.
- (a) L. Y. Zhang, J. P. Zhang, Y. Y. Lin and X. M. Chen, Cryst. Growth Des., 2006, 6, 1684 CrossRef CAS; (b) D. Niu, J. Yang, J. Guo, W. Kan, Q. S. Y. Song, P. Du and J. F. Ma, Cryst. Growth Des., 2012, 12, 2397 CrossRef CAS; (c) X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, N. Hao, C. W. Hu and A. L. Xu, Inorg. Chem., 2004, 43, 1850 CrossRef CAS PubMed; (d) J.-H. Yang, W. Li, S.-L. Zheng, Z.-L. Huang and X.-M. Chen, Aust. J. Chem., 2003, 56, 1175 CrossRef CAS; (e) S.-L. Zheng, J.-M. Yang, X.-L. Yu, X.-M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXTL Program for the Solution ofCrystal of Structures, University of Göttingen, Göttingen, Germany, 1993 Search PubMed.
- G. M. Sheldrick, SHELXL 97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 986613–986614. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00077c |
| This journal is © the Partner Organisations 2014 |