Synthesis of semibullvalene derivatives via Co2(CO)8-mediated cyclodimerization of 1,4-dilithio-1,3-butadienes†
Shaoguang
Zhang‡
a,
Ming
Zhan‡
a,
Qifeng
Wang
a,
Chao
Wang
a,
Wen-Xiong
Zhang
a and
Zhenfeng
Xi
*ab
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, CAS, Shanghai 200032, China. E-mail: zfxi@pku.edu.cn
First published on 28th January 2014
Abstract
Co2(CO)8-mediated cyclodimerization of 1,2,3,4-tetrasubstituted 1,4-dilithio-1,3-butadienes readily afforded octa-substituted semibullvalenes. The X-ray crystal structure of 1,2,5,6-tetraethyl-3,4,7,8-tetraphenyl semibullvalene was determined to show an unsymmetrical, localized structure, which is in sharp contrast with the C2 symmetrical structure of its tetramethyl analogue. In addition, this Co2(CO)8-mediated cyclodimerization was found to be dependent on the substitution pattern of 1,4-dilithio-1,3-butadienes. The 1,4-diphenyl dilithio compound gave its corresponding cyclooctatetraene derivative, while the dilithio reagent without substituents at its 1,4-positions afforded a cyclopentadienone [4 + 2] dimer.
Semibullvalene (SBV), a unique strained ring valence isomer of cyclooctatetraene, has long been of fundamental interest both theoretically and experimentally,1–10 because of its skeletal rearrangement,2i,4 its rapid degenerate Cope rearrangement,8 and the predicted existence of a homoaromatic delocalized structure.2c However, although a number of semibullvalene derivatives have been reported by using multi-step classic organic transformations, the diversity of SBVs, especially the substitution pattern on the SBV skeleton, has been much limited due to lack of efficient synthetic approaches to construct SBVs of diversified substitution patterns. Thus, development of alternative synthetic methods as well as construction of new structural types of SBV is needed for further investigation into the physical and chemical properties of SBV.
In 2006, we reported the synthesis of octa-substituted SBVs via CuCl-promoted cyclodimerization of 1,4-dilithio-1,3-butadiene reagents 1 (Scheme 1).4 This is the first synthetic method toward multi-substituted SBVs by metal-mediated C–C bond forming reactions. Though this method could provide a number of SBVs which could not be readily synthesized by other means, the substrate scope was limited and yields of products were not uniformly satisfactory. As our continued effort to develop more efficient synthetic methods to provide SBVs of diversified substitution patterns, we attempted to use different metal complexes to mediate the cyclodimerization reaction of dilithio reagents. Herein, we report a new synthetic method toward octa-substituted SBVs via Co2(CO)8-mediated cyclodimerization of 1,4-dilithio-1,3-butadienes (Scheme 1). This study has the following features: (1) Compared with the analogous CuCl-mediated cyclodimerization reaction, this reaction has broader substrate scope with higher yields of products. Thus, for example, the product 2h was obtained in a trace amount by the CuCl-mediated method, but this Co2(CO)8-mediated method gave the product 2h in 41% isolated yield. Furthermore, the X-ray crystal structure of 1,2,5,6-tetraethyl-3,4,7,8-tetraphenyl semibullvalene 2g was determined to have a localized structure at room temperature, which is in sharp contrast with the C2 symmetrical structure of its tetramethyl analogue 2f.4 (2) Cobalt-mediated C–C bond forming reaction has attracted much attention in recent years.11,12 However, formation of C–C bonds from organolithium compounds and cobalt carbonyl complexes is rare.13
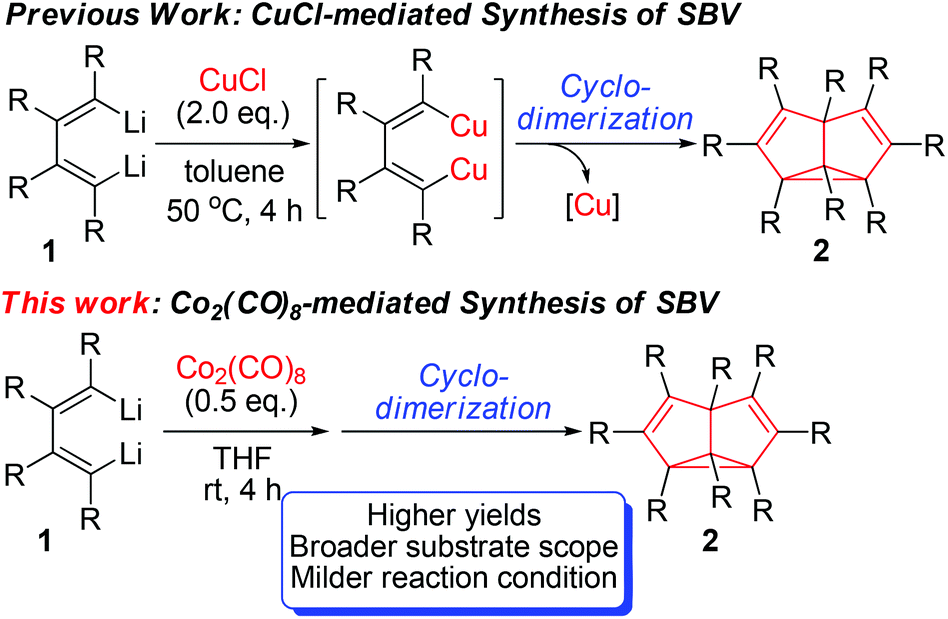 | ||
| Scheme 1 Synthesis of semibullvalenes. | ||
The dilithio reagent 1 was generated in situ from its corresponding 1,4-diiodo compound 3 and t-BuLi in THF (Scheme 2).14a Treatment of 1,2,3,4-tetraethyl-1,4-dilithio-1,3-butadiene 1a with 0.5 equivalents of octacarbonyl dicobalt (Co2(CO)8) at room temperature for 4 h led to a color change from yellow to dark brown along with gas release. The reaction mixture was dried up and subjected to flash chromatography (neutral Al2O3), and thus octaethyl-semibullvalene 2a was isolated in 95% yield.
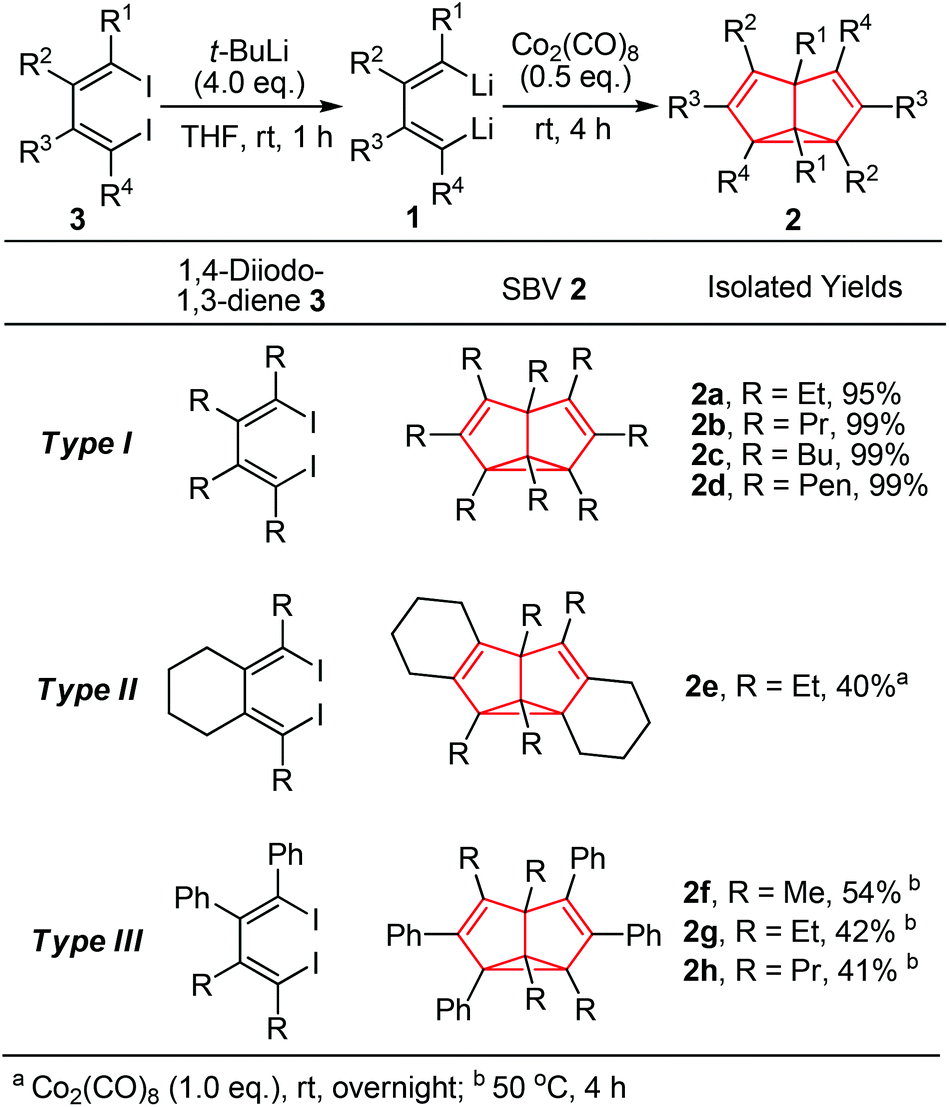 | ||
| Scheme 2 Co2(CO)8-mediated synthesis of semibullvalene 2via cyclodimerization of 1,4-dilithio-1,3-butadienes. | ||
Co2(CO)8 as the promoter is essential. A catalytic amount of Co2(CO)8 (10 mol%) could not lead to a promising yield. Utilization of CoCl2 instead of Co2(CO)8 led to a mixture of products but no SBV derivative was detected.14b Besides, considering the reaction mechanism, the in situ generated LiI might play a key role. For example, when the 1,4-dibromo analogue of diiodo compound 3 was used as the substrate, the reaction did not give any SBV derivative but only its corresponding diene as a quenched product even at higher temperatures. The in situ generated LiBr might not be able to promote the dimerization process. The purified dilithio compound 1 could react with Co2(CO)8 without additional LiI but the yield of SBV derivative was low. Thus, as a possible reaction pathway (see ESI†), addition of C–Li bond in 1 to Co2(CO)8 would form butadiene bis(lithium carbonylcobaltate), which would be followed by oxidative dimerization to afford SBV 2 with elimination of a lithium carbonylcobaltate species.15
Reaction of Co2(CO)8 with dilithio reagent 1b–d bearing different substituents readily afforded octa-alkyl substituted SBVs 2b–d (Type I) in excellent isolated yields. Similarly, the cyclic dilithio reagent 1e was successfully applied to the synthesis of the fused SBV derivative 2e (Type II) in 40% isolated yield. Furthermore, we tested unsymmetrical 1,2-diphenyl-3,4-dialkyl-1,4-dilithio-1,3-butadienes 1f–h, and the corresponding SBV derivatives 2f–h (Type III) were formed in moderate yields. Besides, when the dilithio reagent 1h was used in the CuCl-mediated synthesis, only a trace amount of SBV product was detected. Thus, the Co2(CO)8-mediated synthesis of SBV derivatives in many cases showed higher efficiency than the CuCl-mediated synthesis via cyclodimerization of dilithio reagents 1.
The solution NMR spectra of isolated SBVs 2a–e (Type I, Type II) all showed degenerated NMR signals, indicating a rapid equilibrium of degenerate Cope rearrangement between two localized structures 2 and 2′. For example, C1/C5, C2/C6, C3/C7 and C4/C8 of herein newly synthesized SBV 2e displayed four singlets at δ = 65.96, 101.43, 128.64, 97.31 ppm, respectively, in the 13C NMR spectrum in C6D6. The chemical shifts in 2e were comparable with those found in octaalkyl-SBV 2a–d.4 However, the NMR spectra of 1,4,5,8-tetraalkyl–2,3,6,7-tetraphenyl semibullvalenes 2g and 2h (Type III) showed different characteristics. In contrast with the symmetrical, degenerated NMR spectra of 2f (R = Me) as we had reported before, the NMR spectra of 2g (R = Et) and 2h (R = Pr) both showed unsymmetrical, non-degenerated NMR signals. The four ethyl groups showed non-equivalent characteristics in the 13C NMR spectrum, which indicated relatively slow degenerate Cope rearrangement in solution.
The substituent-dependent solid-state structure of 2g was also confirmed by X-ray single-crystal structural analysis at room temperature, which was determined as an unsymmetrical, localized structure (Fig. 1). The distances of C2–C8 and C4–C6 were determined as 1.703(4) Å and 2.243 Å, respectively, obviously showing the unsymmetrical, localized structure. The apparent long C2–C8 distance (1.703(4) Å) is comparable with that in other reported SBV derivatives,2d–f but is different from those of unsubstituted SBVs in the gas phase (1.600 Å).2b This much elongated C2–C8 bond together with the normally bonded C1–C2 (1.512(3) Å) and C1–C8 (1.516(3) Å) constituted a strained cyclopropane ring. The other bond lengths of the SBV core are all in the normal range, comparable to other localized structures of SBV derivatives.2d–f The X-ray crystal structure of tetraethyl-substituted SBV 2g was again in sharp contrast with the methyl analogue 2f, which was earlier determined as a C2 symmetrical structure.4 For 2f, its X-ray structural analysis data showed that its corresponding C2–C8 (1.931(3) Å) and C4–C6 (2.084 Å) distances were more similar.
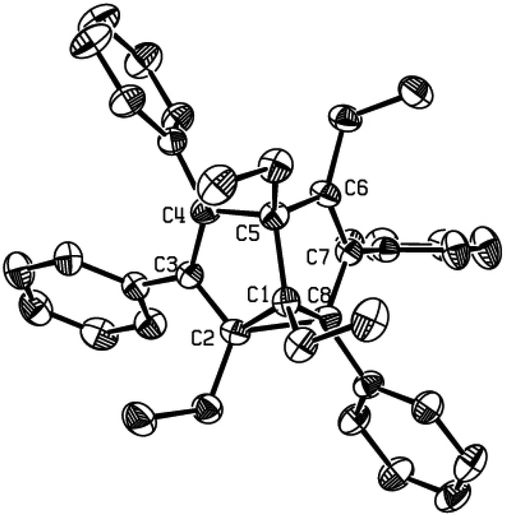 | ||
| Fig. 1 Single-crystal X-ray structure of 2g with 30% thermal ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–C2 1.512(3), C1–C8 1.516(3), C1–C5 1.587(3), C2–C3 1.449(3), C2–C8 1.703(4), C3–C4 1.369(3), C4–C5 1.539(3), C5–C6 1.526(3), C6–C7 1.356(3), C7–C8 1.483(3), C4–C5–C6 94.10(18), C2–C1–C8 68.45(17). | ||
In our previous communication, we reported the X-ray crystal structure of 2b at room temperature as a C2v symmetrical structure.4 We attributed the structural feature to a dynamic or static disorder of two nondegenerate SBV molecules in the solid state. Herein we determined the low-temperature X-ray crystal structure of octapropyl SBV 2b. At −150 °C, 2b crystallized in space group C2/c. Two different structures crystallized in one cell. One showed a C2v symmetrical structure with the distances of C2–C8 and C4–C6 determined as 1.968 Å and 2.043 Å, close to the value of the crystal structure of 2b at room temperature (C2–C8: 1.931(3) Å; C4–C6: 2.084 Å).4 However, another one showed an unsymmetrical localized structure 2b*, which is in sharp contrast with its corresponding crystal structure at room temperature (Fig. 2). The distances of C2–C8 and C4–C6 were determined as 1.690(3) Å and 2.325 Å, respectively, close to the value in 2g as well as other reported SBV derivatives.2d–f The geometry of the cyclopropane ring is also similar to that in 2g. We assumed that at low temperature the proportion of the dominant valence tautomer in Cope rearrangement might increase. This was also observed in the X-ray crystal structure of 1,5-dimethyl-2,4,6,8-semibullvalene tetracarboxylic dianhydride.2f
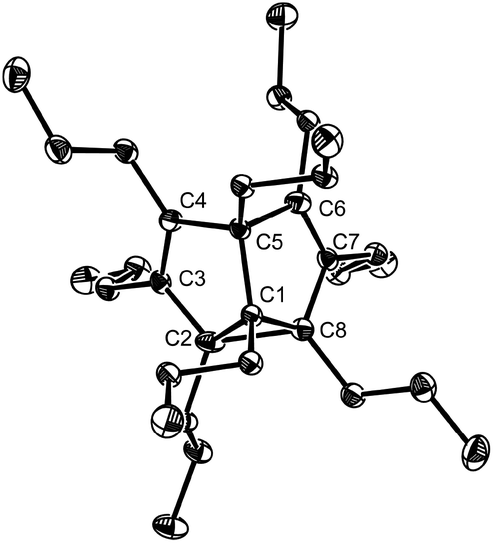 | ||
| Fig. 2 Single-crystal X-ray structure of 2b* at −150 °C with 30% thermal ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–C2 1.512(3), C1–C8 1.514(3), C1–C5 1.557(3), C2–C3 1.460(3), C2–C8 1.690(3), C3–C4 1.347(3), C4–C5 1.545(3), C5–C6 1.525(3), C6–C7 1.356(3), C7–C8 1.474(3), C4–C5–C6 98.47(17), C2–C1–C8 67.91(15). | ||
The substitution pattern on the butadienyl skeleton of dilithio compound 1 has great impact on its reactivity.14 As given in Scheme 3, when 1,4-diphenyl-1,4-dilithio-1,3-butadiene 1i was applied, the cyclooctatetraene 4 was obtained in 53% isolated yield and was structurally characterized (Fig. 3). We assumed that the expected SBV derivative 2i might be formed initially via the Co2(CO)8-mediated cyclodimerization of the dilithio reagent 1i. However, the phenyl groups at 1,5-positions of SBV might destabilize 2i at room temperature and enable 2i to readily rearrange to the thermodynamically more stable 4.6c,7a The thermo-rearrangement of SBVs to their corresponding cyclooctatetraene derivatives has been well studied.2h,4,16
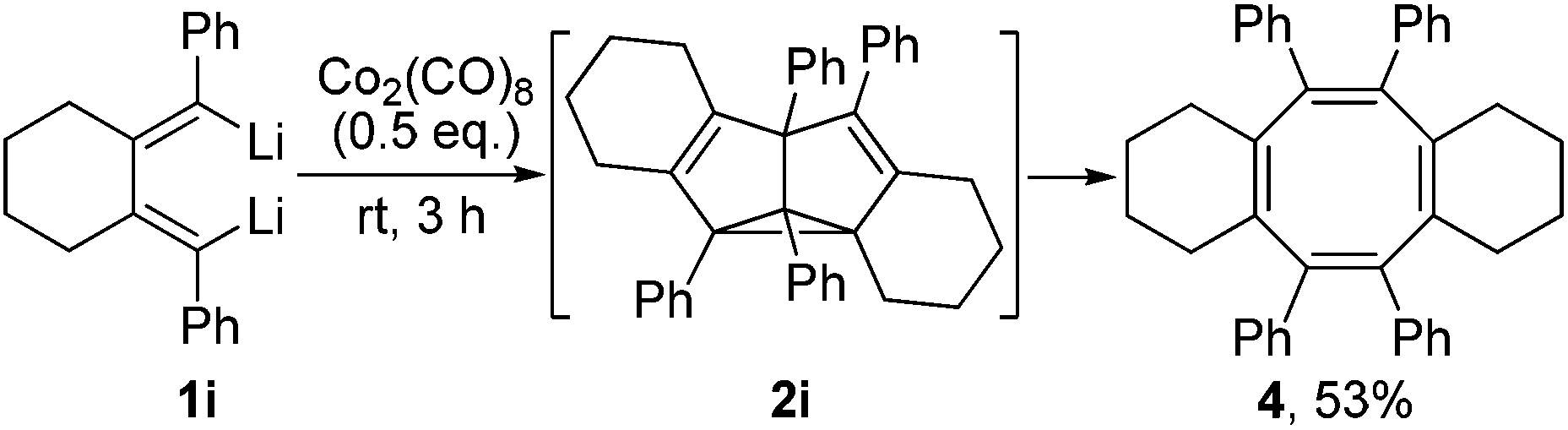 | ||
| Scheme 3 Formation of cyclooctatetraene derivative 4via Co2(CO)8-mediated cyclodimerization of dilithio reagent 1i. | ||
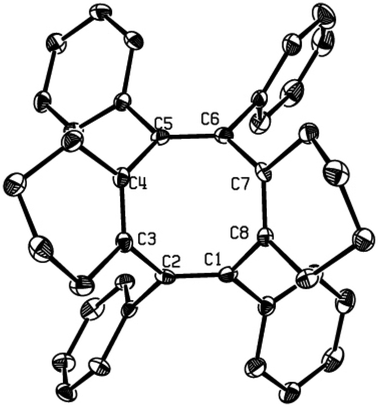 | ||
| Fig. 3 Single-crystal X-ray structure of 4 with 30% thermal ellipsoids. Hydrogen atoms are omitted for clarity. | ||
When 1,4-unsubstituted dilithio compounds 1j–k were subjected to the Co2(CO)8-mediated cyclodimerization reaction, no semibullvalene was formed. Instead, multi-substituted cyclopentadienone dimers 6 were yielded as main products, along with some unknown byproducts (Scheme 4). The structures of 6a and 6b were both characterized by X-ray crystal structural analysis (Fig. 4). We assumed that the 1,4-unsubstituted dilithio compounds 1j–k might be very reactive toward Co2(CO)8. Thus, carbonylation of 1 with Co2(CO)8 took place forming 2,5-unsubstituted cyclopentadienones 5, which afford 6via [4 + 2] cycloaddition.17,18
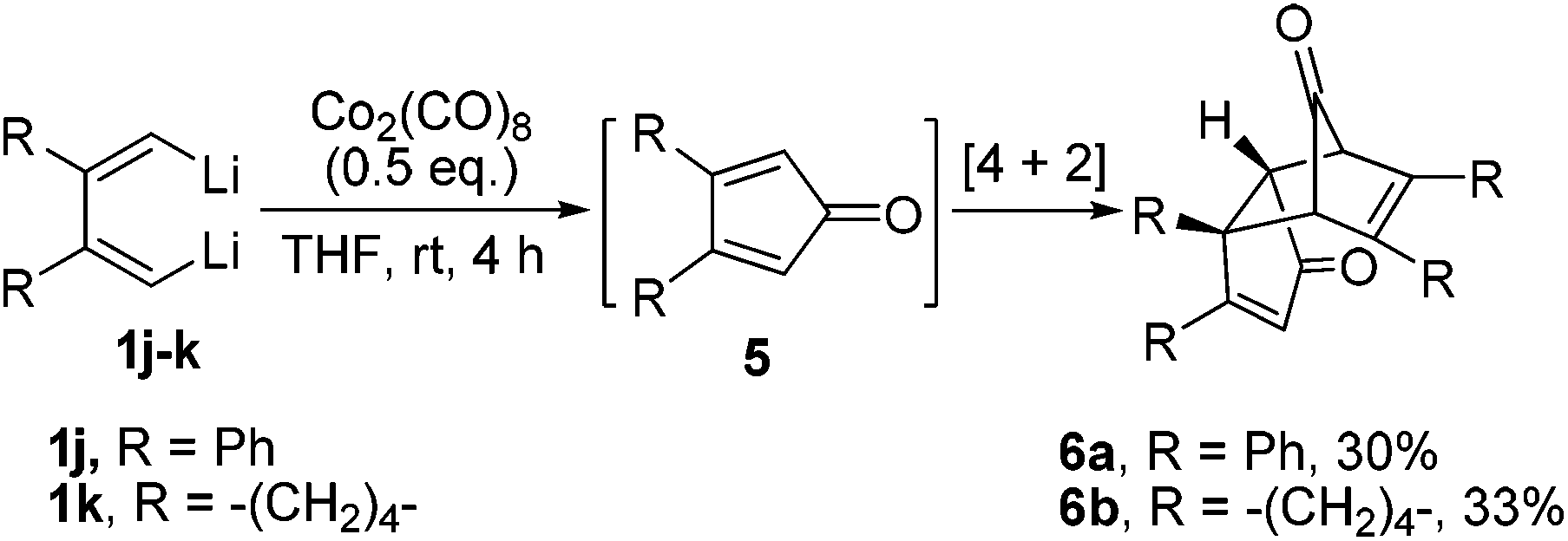 | ||
| Scheme 4 Formation of cyclopentadienone dimer via Co2(CO)8-mediated carbonylation of dilithio reagents and [4 + 2] cycloaddition. | ||
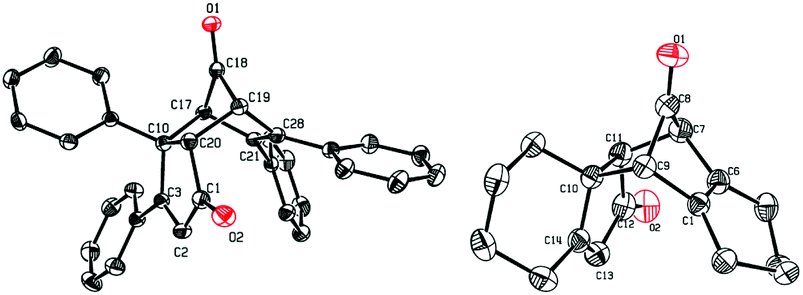 | ||
| Fig. 4 Single-crystal X-ray structures of 6a and 6b with 30% thermal ellipsoids. Hydrogen atoms are omitted for clarity. | ||
In summary, we report a new synthetic method toward octa-substituted SBVs. Several new behaviors of SBVs were disclosed, which are expected to be useful for further study of SBVs with diversified substitution patterns as well as new structural types.
Acknowledgements
This work was supported by the Major State Basic Research Development Program (2011CB808700) and Natural Science Foundation of China. Dedicated to Professor Helmut Quast on the occasion of his 80th birthday.References
- For reviews on semibullvalenes: (a) R. V. Williams, Adv. Theor. Interesting Mol., 1998, 4, 157–202 CrossRef CAS; (b) H. Hopf, Classics in Hydrocarbon Chemistry, Wiley-VCH, Weinheim, Germany, 2000, ch. 10, p. 209 Search PubMed; (c) R. V. Williams, Eur. J. Org. Chem., 2001, 227–235 CrossRef CAS; (d) R. V. Williams, Chem. Rev., 2001, 101, 1185–1204 CrossRef CAS PubMed.
- (a) H. E. Zimmerman and G. L. Grunewald, J. Am. Chem. Soc., 1966, 88, 183–184 CrossRef CAS; (b) Y. C. Wang and S. H. Bauer, J. Am. Chem. Soc., 1972, 94, 5651–5657 CrossRef CAS; (c) A. K. Cheng, F. A. L. Anet, J. Mioduski and J. Meinwald, J. Am. Chem. Soc., 1974, 96, 2887–2891 CrossRef CAS; (d) L. S. Miller, K. Grohmann, J. J. Dannenberg and L. Todaro, J. Am. Chem. Soc., 1981, 103, 6862–6865 Search PubMed; (e) H. Quast, J. Carlsen, R. Janiak, E.-M. Peters, K. Peters and H. G. von Schnering, Chem. Ber., 1992, 125, 955–968 CrossRef CAS; (f) R. V. Williams, V. R. Gadgil, K. Chauhan, D. van der Helm, M. B. Hossain, L. M. Jackman and E. Fernandes, J. Am. Chem. Soc., 1996, 118, 4208–4209 CrossRef CAS; (g) L. M. Jackman, E. Fernandes, M. Heubes and H. Quast, Eur. J. Org. Chem., 1998, 2209–2227 CrossRef CAS; (h) H. Quast, M. Heubes, T. Dietz, A. Witzel, M. Boenke and W. R. Roth, Eur. J. Org. Chem., 1999, 813–822 CrossRef CAS; (i) M. Seefelder, M. Heubes, H. Quast, W. D. Edwards, J. R. Armantrout, R. V. Williams, C. J. Cramer, A. C. Goren, D. A. Hrovat and W. T. Borden, J. Org. Chem., 2005, 70, 3437–3449 CrossRef CAS PubMed; (j) P. R. Griffiths, D. E. Pivonka and R. V. Williams, Chem.–Eur. J., 2011, 17, 9193–9199 CrossRef CAS PubMed.
- (a) H. Jiao and P. v. R. Schleyer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1760–1763 CrossRef; (b) H. Jiao, R. Nagelkerke, H. A. Kurtz, R. V. Williams, W. T. Borden and P. v. R. Schleyer, J. Am. Chem. Soc., 1997, 119, 5921–5929 CrossRef CAS; (c) A. C. Goren, D. A. Hrovat, M. Seefelder, H. Quast and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 3469–3472 CrossRef CAS PubMed; (d) S. C. Wang and D. J. Tantillo, J. Phys. Chem. A, 2007, 111, 7149–7153 CrossRef CAS PubMed.
- C. Wang, J. Yuan, G. Li, Z. Wang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2006, 128, 4564–4565 CrossRef CAS PubMed.
- (a) M. J. S. Dewar, Z. Náhlovská and B. D. Náhlovský, J. Chem. Soc. D, 1971, 1377–1378 RSC; (b) D. R. Greve, J. Phys. Org. Chem., 2011, 24, 222–228 CrossRef CAS.
- (a) C. Schnieders, H. J. Altenbach and K. Müllen, Angew. Chem., Int. Ed. Engl., 1982, 21, 637–638 CrossRef; (b) C. Schnieders, W. Huber, J. Lex and K. Müllen, Angew. Chem., Int. Ed. Engl., 1985, 24, 576–577 CrossRef; (c) B. Düll and K. Müllen, Tetrahedron Lett., 1992, 33, 8047–8050 CrossRef.
- (a) S. Zhang, J. Wei, M. Zhan, Q. Luo, C. Wang, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2012, 134, 11964–11967 CrossRef CAS PubMed; (b) S. Zhang, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 3485–3489 CrossRef CAS PubMed; (c) S. Zhang, M. Zhan, Q. Luo, W.-X. Zhang and Z. Xi, Chem. Commun., 2013, 49, 6146–6148 RSC.
- (a) K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81–90 CrossRef CAS; (b) N. Graulich, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 172–190 CrossRef CAS; (c) For metal-promoted Cope rearrangements, see: M. R. Siebert and D. J. Tantillo, J. Am. Chem. Soc., 2007, 129, 8686–8687 CrossRef CAS PubMed and references therein.
- S. Winstein, J. Am. Chem. Soc., 1959, 81, 6524–6525 CrossRef CAS.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS; (b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- (a) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed; (b) K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263–3283 CrossRef CAS; (c) H. Yorimitsu and K. Oshima, Pure Appl. Chem., 2006, 78, 441–449 CrossRef CAS; (d) M. E. Welker, Curr. Org. Chem., 2001, 5, 785 CrossRef CAS.
- (a) Z. Mo, Y. Li, H. K. Lee and L. Deng, Organometallics, 2011, 30, 4687–4694 CrossRef CAS; (b) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279–9282 CrossRef CAS PubMed; (c) T. L. Dzwiniel, N. Etkin and J. M. Stryker, J. Am. Chem. Soc., 1999, 121, 10640–10641 CrossRef CAS.
- For coordination or reaction of organolithium compounds with cobalt carbonyls, see: (a) M. G. Richmond and J. K. Kochi, Organometallics, 1987, 6, 777–788 CrossRef CAS; (b) D. Schneider and H. Werner, J. Organomet. Chem., 1990, 384, C33–C37 CrossRef CAS; (c) G. Scholz, R. Gleiter and F. Rominger, Angew. Chem., Int. Ed., 2001, 40, 2477–2479 CrossRef CAS; (d) D. Tews and P. E. Gaede, Organometallics, 2004, 23, 968–975 CrossRef CAS; (e) Q. Wang, W.-X. Zhang and Z. Xi, Organometallics, 2008, 27, 3627–3629 CrossRef CAS; (f) Q. Wang, W.-X. Zhang, C. Lin and Z. Xi, Chin. Sci. Bull., 2010, 55, 2915–2923 CrossRef CAS.
- (a) Z. Xi, Acc. Chem. Res., 2010, 43, 1342–1351 CrossRef CAS PubMed; (b) J. Wei, Z. Wang, W.-X. Zhang and Z. Xi, Org. Lett., 2013, 15, 1222–1225 CrossRef CAS PubMed.
- (a) L. T. Mika, R. Tuba, I. Tóth, S. Pitter and I. T. Horváth, Organometallics, 2011, 30, 4751–4764 CrossRef CAS; (b) K. Uehara, S. Hikichi, A. Inagaki and M. Akita, Chem.–Eur. J., 2005, 11, 2788–2809 CrossRef CAS PubMed; (c) H. Bukowsky, E. Uhlemann and W. Klaui, Anal. Chim. Acta, 1996, 319, 271–274 CrossRef CAS; (d) G. Jenner and E. M. Nahmed, J. Organomet. Chem., 1991, 407, 135–142 CrossRef CAS.
- C. Wang and Z. Xi, Chem. Commun., 2007, 5119–5133 RSC.
- (a) C. F. H. Allen and J. A. VanAllan, J. Am. Chem. Soc., 1950, 72, 5165–5167 CrossRef CAS; (b) M. A. Ogliaruso, M. G. Romanelli and E. I. Becker, Chem. Rev., 1965, 65, 261–367 CrossRef CAS.
- (a) Q. Luo, C. Wang, W.-X. Zhang and Z. Xi, Chem. Commun., 2008, 1593–1595 RSC; (b) Z. Xi and Q. Song, J. Org. Chem., 2000, 65, 9157–9159 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials including experimental procedures, copies of 1H and 13C NMR spectra of all new products and crystallographic data for 2b*, 2g, 4, 6a, 6b. CCDC 966204, 966203, 971720, 966206, 966207. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00019b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |