Chiral primary amine catalysed asymmetric conjugate addition of azoles to α-substituted vinyl ketones†
Niankai
Fu
a,
Long
Zhang
ab,
Sanzhong
Luo
*ab and
Jin-Pei
Cheng
ab
aBeijing National Laboratory for Molecular Sciences (BNLMS), CAS Key Laboratory of Molecular Recognition and Function Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P.R. China. E-mail: luosz@iccas.ac.cn; Fax: (+)86-10-6255-4449; Tel: (+)86-10-6255-4446
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin, 300071, China
First published on 19th December 2013
Abstract
We report here the first effective example of asymmetric conjugate addition–protonation reactions of azoles to vinyl ketones by chiral primary amine catalysis. High enantioselectivity can be easily achieved by screening different primary amine catalysts, and the Curtin–Hammett control in the C–N bond formation step was verified by DFT calculations to account for the observed stereoselectivity.
Transformations with nitrogen-containing heterocycles (azoles) have long been a mainstay in organic synthesis due to the ubiquitous nature of azoles in nature and pharmaceuticals as well as in materials.1 Catalytic asymmetric 1,4-conjugate addition of azoles to α,β-unsaturated carbonyls represents a powerful synthetic approach in the area of heterocyclic chemistry.2 Although tremendous advances have been made for this reaction,3 α-branched vinyl carbonyls remain a challenging type of substrate and a catalytic asymmetric aza-addition to α-branched vinyl carbonyls has not been reported so far. The difficulties mainly come from the issues on stereocontrol since no stereocenter is formed during the β-attack of the nucleophiles and instead the carbon stereocenter is generated at the α-position via enolate or enamine protonation, which is a challenging process in asymmetric catalysis in general (Scheme 1).4
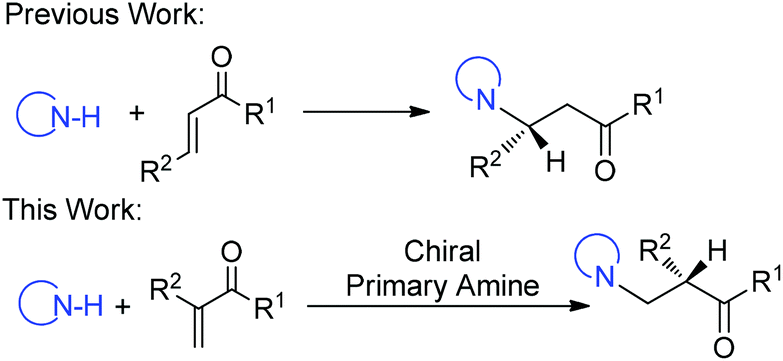 | ||
| Scheme 1 Catalytic asymmetric 1,4-conjugate addition of azoles to α,β-unsaturated carbonyls. | ||
Recently, we have established chiral primary-tertiary diamines as effective catalysts for the iminium activation of α-substituted acroleins and vinyl ketones with a good activity and high enantioselectivity (Scheme 2).5 Our detailed mechanistic studies have also disclosed divergent stereocontrol modes for acroleins and vinyl ketones, wherein the stereoselectivity is determined by the facial selection of the enamine in the cases with acroleins (Step II), and there is Curtin–Hammett control in the C–C bond formation step (Step I) for vinyl ketones (Scheme 2). According to this mechanistic scenario, the notoriously challenging task in manipulating selective proton transfer is transformed into regular selectivity tuning associated with the C–C bond formation. Hence, this infers that successful extensions to other nucleophiles in the reactions with vinyl ketones can be readily achieved.5e With this idea in mind, we have examined the use of azoles as nucleophiles, reacting them with α-branched acroleins and vinyl ketones to give the addition products. Herein, we document the first asymmetric aza-conjugate addition–protonation reactions with α-branched vinyl ketones.
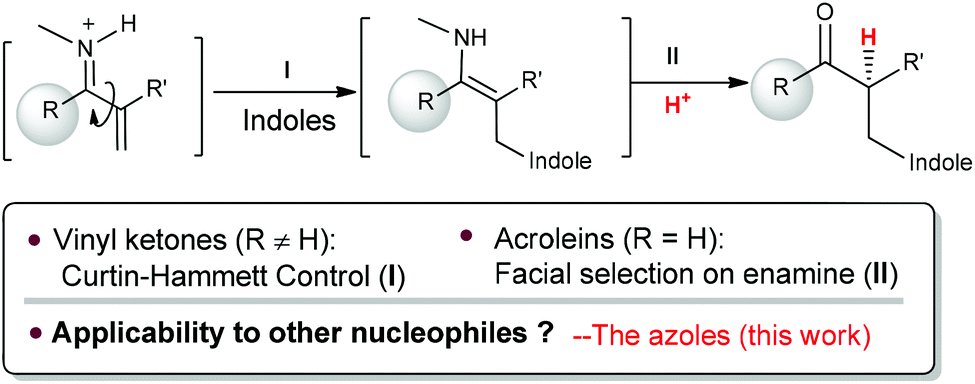 | ||
| Scheme 2 Divergent stereocontrol modes on enamine protonation between α-substituted vinyl ketones and aldehydes. | ||
The reactions of azoles with acrolein 1 were first attempted using chiral primary-tertiary diamines as the catalysts (Chart 1). Though the use of other primary catalyst such as 5b/TfOH gave improved product yields, a noticeable enantioselectivity has not been obtained so far in these reactions (Scheme 3). The lack of stereocontrol in these cases further attests the challenges of this type of transformation, particularly regarding the facially selective delivery of the proton (Step II, Scheme 2).
 | ||
| Chart 1 Chiral primary-tertiary diamine catalysts. | ||
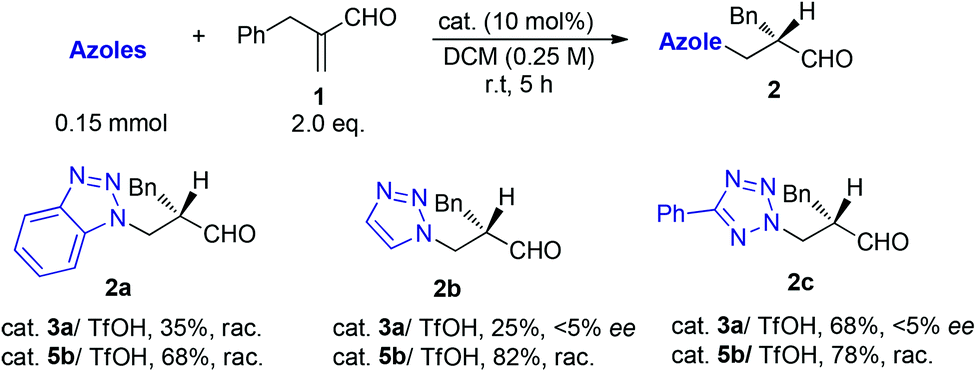 | ||
| Scheme 3 The nonstereoselective reactions of acrolein 1 with azoles. | ||
On the other hand, in the model reaction between vinyl ketone 7a and 1H-benzotriazole 6, a quick screening with our typical primary-tertiary diamine catalysts (Chart 1, Table 1, entries 1–9) led to the identification of 5b/TfOH as the optimal catalyst (Table 1, entry 6). In the presence of 10 mol% 5b/TfOH, the reaction gave the desired product 8a with an 86% isolated yield and in 91% ee (Table 1, entry 6). Further optimization with variations on the acidic additives or solvents did not result in noticed improvements in the enantioselectivity (Table 1, entries 10–12).
|
|
||||
|---|---|---|---|---|
| Entry | Amine | Additive | Yieldb (%) | eec (%) |
| a General conditions: 6 (0.15 mmol), 7a (0.30 mmol), amine/TfOH (10 mol%) in CH2Cl2 (0.60 mL) at 40 °C, 18 h. b Yield of isolated yield. c Determined by HPLC analysis. | ||||
| 1 | 3a | None | 60 | 16 |
| 2 | 3b | None | 25 | <5 |
| 3 | 3c | None | 50 | <5 |
| 4 | 4 | None | 55 | <5 |
| 5 | 5a | None | 85 | 83 |
| 6 | 5b | None | 86 | 91 |
| 7 | 5c | None | 82 | 85 |
| 8 | 5d | None | 83 | 87 |
| 9 | 5e | None | 78 | 84 |
| 10 | 5b | PhCO2H (10 mol%) | 85 | 82 |
| 11 | 5b | H2O (50 eq.) | 40 | 50 |
| 12 | 5b | 2-OMePhOH (10 mol%) | 75 | 88 |
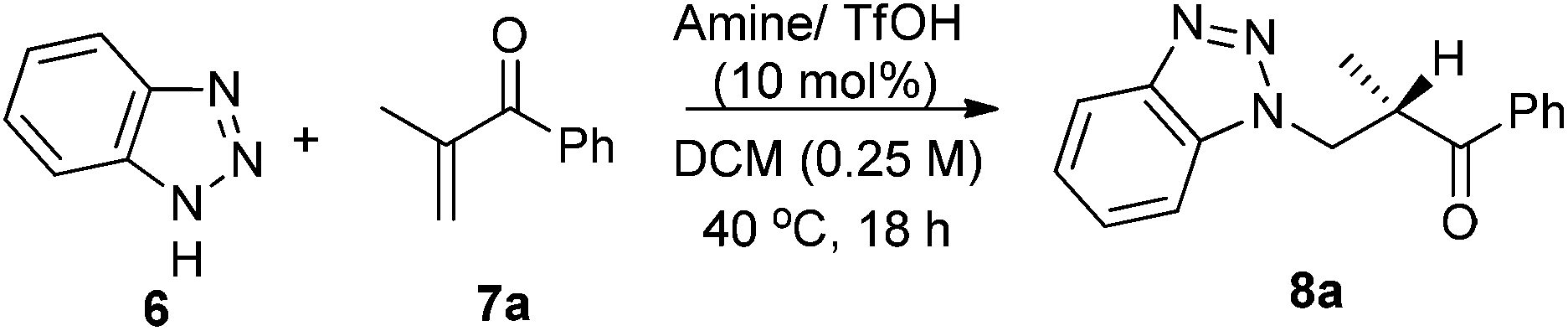
The substrate scope of the reaction of benzotriazole and different α-substituted vinyl ketones was next investigated. As seen from the results (Table 2), aromatic vinyl ketones including a heteroaryl vinyl ketone can be well accommodated in the current catalysis with a good activity and enantioselectivity (Table 2, entries 1–10). Meanwhile, different types of aliphatic enones, such as methyl, ethyl, benzyl and n-butyl vinyl enones can also be applied to this reaction system with a moderate to good enantioselectivity (Table 2, 11–14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yieldb (%) | eec (%) | |
| a General conditions: 6 (0.15 mmol), 7 (0.30 mmol), 5b/TfOH (10 mol%) in CH2Cl2 (0.60 mL) at 40 °C. b Yield of isolated yield. c Determined by HPLC analysis. d At room temperature. | ||||||
| 1 | Ph | Me | 8a | 18 | 86 | 91 |
| 2 | 4-ClC6H4 | Me | 8b | 15 | 82 | 86 |
| 3 | 4-BrC6H4 | Me | 8c | 18 | 84 | 90 |
| 4 | 4-MeC6H4 | Me | 8d | 18 | 85 | 89 |
| 5 | 4-OMeC6H4 | Me | 8e | 24 | 85 | 90 |
| 6 | Ph | Et | 8f | 36 | 73 | 86 |
| 7 | Ph | Bn | 8g | 40 | 75 | 82 |
| 8 | Ph | n-C7H15- | 8h | 30 | 72 | 86 |
| 9 | Ph | Cyclohexyl- | 8i | 72 | 70 | 72 |
| 10 | 2-Thienyl | Bn | 8j | 85 | 70 | 62 |
| 11d | Me | Bn | 8k | 48 | 80 | 74 |
| 12d | Et | Bn | 8l | 72 | 75 | 70 |
| 13d | Bn | Bn | 8m | 75 | 68 | 77 |
| 14d | n-Bu | n-Pr | 8n | 72 | 75 | 40 |
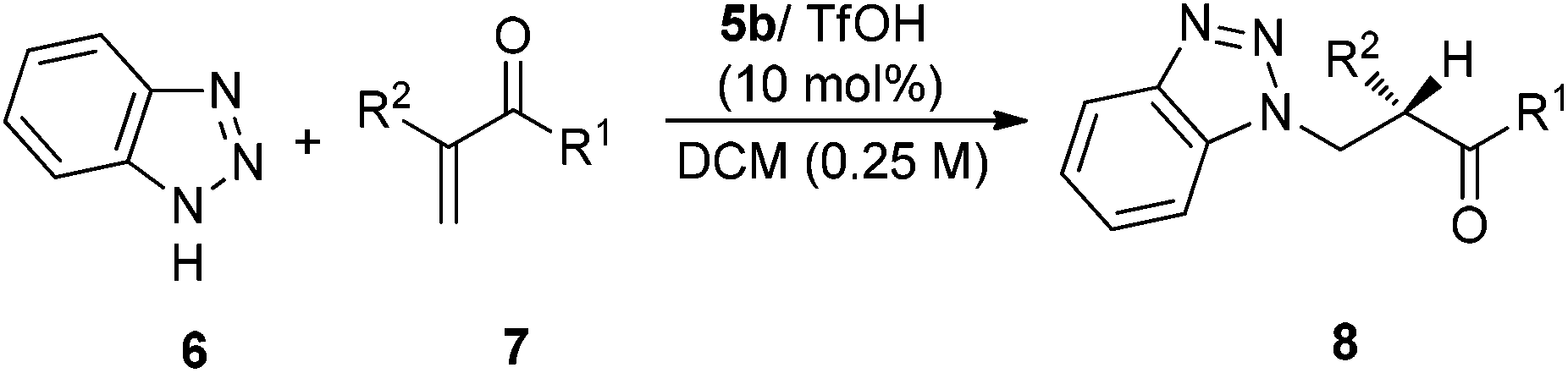
To expand the scope of the reaction, the use of azoles other than 1H-benzotriazole was investigated. In the reaction of 1,2,3-triazole 9, catalyst 5e/TfOH turns out to be the best catalyst, furnishing the best enantioselectivity among differently substituted tertiary amine analogues (Scheme 4). The reaction of 1,2,3-triazole 9 with α-substituted vinyl enones are also quite general and the vinyl ketones 7a–d,g–h,k can be well applied in the reactions to give the addition–protonation products with excellent yields and enantioselectivities (Table 3).
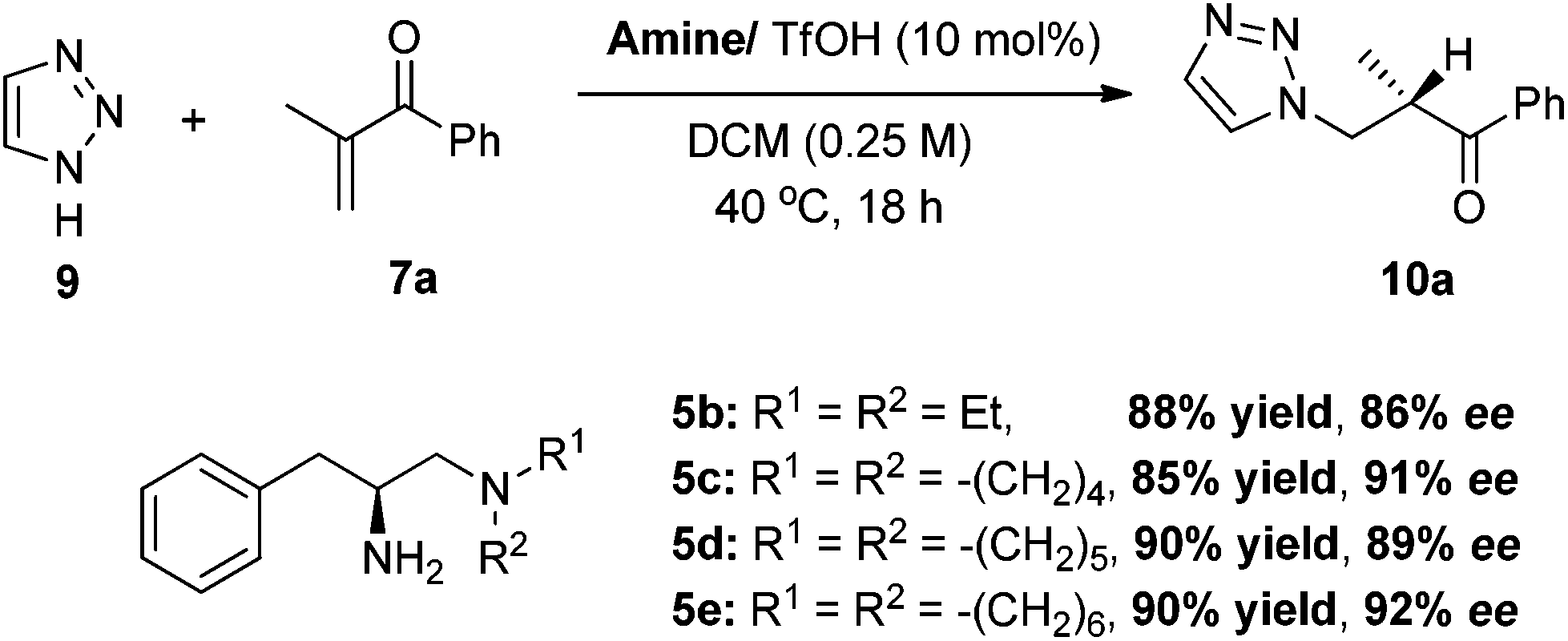 | ||
| Scheme 4 The screening of the primary-tertiary diamine catalysts. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yieldb (%) | eec (%) | |
| a General conditions: 9 (0.15 mmol), 7 (0.30 mmol), 5e/TfOH (10 mol%) in CH2Cl2 (0.60 mL) at 40 °C. b Yield of isolated yield. c Determined by HPLC analysis. d At room temperature. | ||||||
| 1 | Ph | Me | 10a | 18 | 90 | 92 |
| 2 | 4-ClC6H4 | Me | 10b | 18 | 88 | 93 |
| 3 | 4-BrC6H4 | Me | 10c | 18 | 89 | 93 |
| 4 | 4-MeC6H4 | Me | 10d | 18 | 90 | 85 |
| 5 | Ph | Bn | 10e | 40 | 84 | 85 |
| 6 | Ph | n-C7H15- | 10f | 32 | 90 | 87 |
| 7d | Me | Bn | 10g | 48 | 85 | 72 |
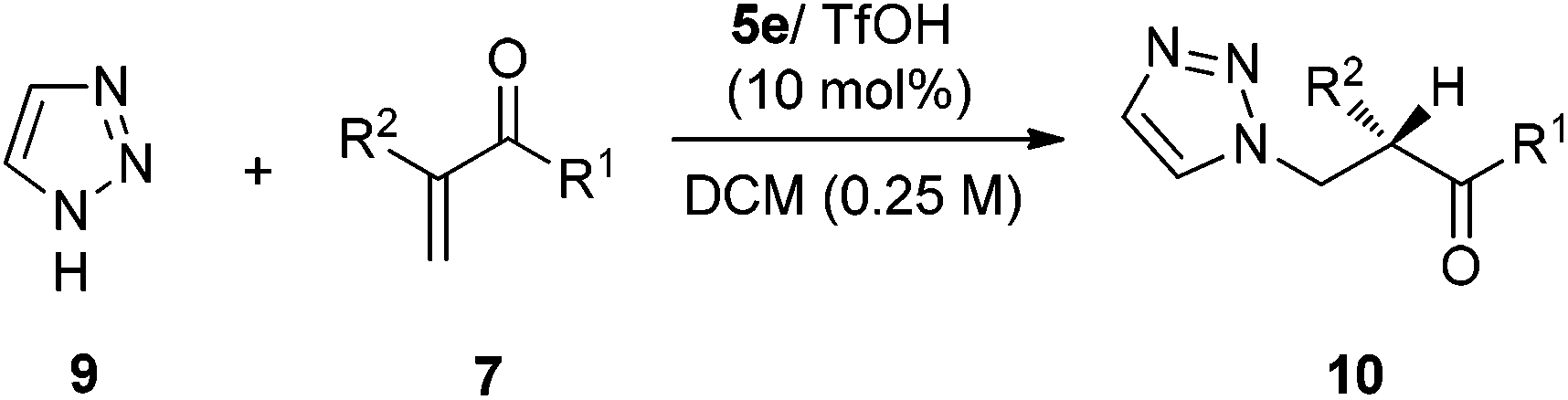
An additional nucleophile that was investigated in this addition–protonation reaction with α-substituted vinyl enones was 5-phenyltetrazole 11. A quick catalyst screening showed that 3a/TfOH was the optimal catalyst, and the use of DCE (1,2-dichloroethane) as the solvent can slightly improve the enantioselectivity. The reactions worked well with the vinyl ketones 7a–d and 7f to provide the enamine protonation products with high ee values and yields (Table 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yieldb (%) | eec (%) | |
| a General conditions: 11 (0.15 mmol), 7 (0.30 mmol), 3a/TfOH (10 mol%) in DCE (0.60 mL). b Yield of isolated yield. c Determined by HPLC analysis. | ||||||
| 1 | Ph | Me | 12a | 24 | 83 | 90 |
| 2 | 4-ClC6H4 | Me | 12b | 24 | 85 | 90 |
| 3 | 4-BrC6H4 | Me | 12c | 32 | 80 | 92 |
| 4 | 4-MeC6H4 | Me | 12d | 24 | 90 | 92 |
| 5 | Ph | Et | 12e | 70 | 80 | 84 |
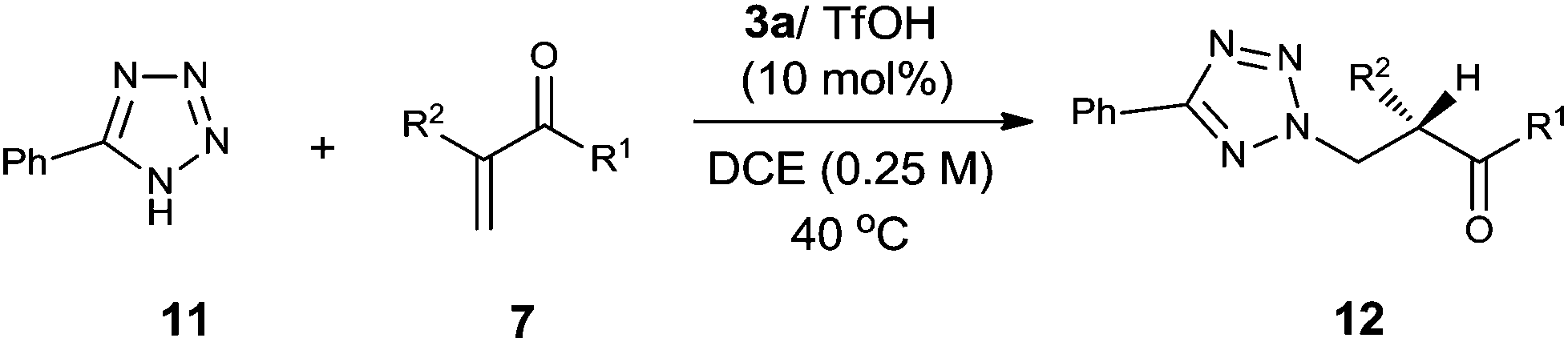
Encouraged by the above results, we continued to examine other azoles, such as 1,2,4-triazole, 6-chloropurine and pyrazole in these aza-Michael addition–protonation reactions using 5b/TfOH as the catalyst. To our delight, all of the reactions worked well to furnish the desired adducts with a good reactivity and enantioselectivity (Scheme 5). The use of readily available imidazoles has also been attempted, with unfortunately no enantioselectivity observed due to uncontrolled background reactions (Scheme 5).
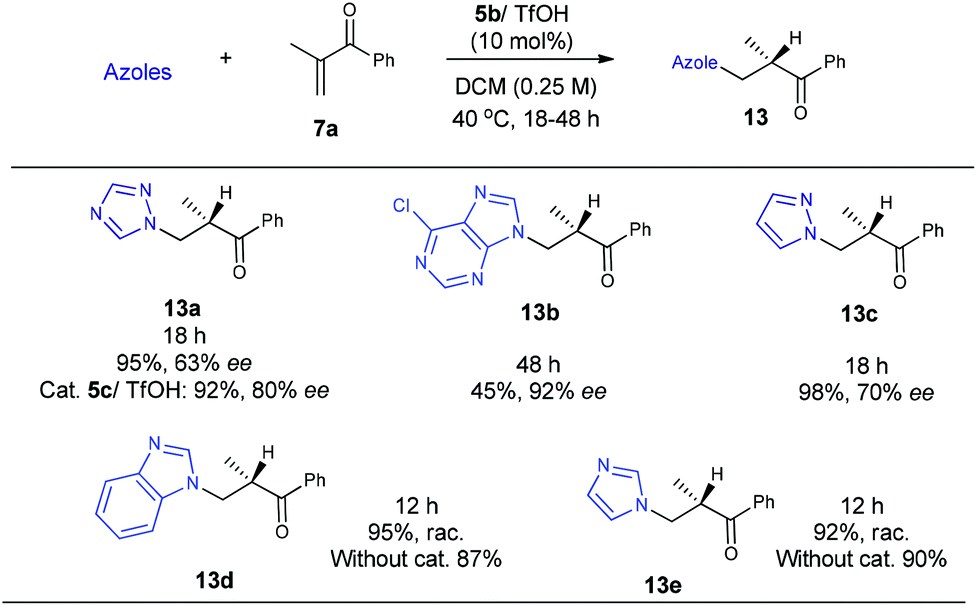 | ||
| Scheme 5 The reactions of vinyl ketone 7a with other azoles. | ||
Collectively, the current studies indicate that the reactions are quite general with respect to azoles. A good enantioselectivity can be readily achieved by screening chiral primary amine catalysts endowed with a common primary-tertiary diamine motif. In this context the sufficient acidity of the azoles to ensure a moderate nucleophilicity seems to be a prerequisite to eliminate possible uncontrolled background reactions (Scheme 5). Provided with the structural diversity of the examined azoles, the fact that a high enantioselectivity can be easily achieved in this intriguing aza-addition–protonation sequence with a common catalytic motif is supportive of a uniform stereocontrol mode. Accordingly, the initially hypothesized Curtin–Hammett control in the C–N bond formation step was next verified by DFT calculations with Gaussian 09.6 We chose the reaction between 5-phenyltetrazole 11 and vinyl ketone 7a as the model reaction and MP2//6-31+G(d)/B3LYP//6-31G(d) as the calculation model.7 The solvation energies were calculated using the Polarizable Continuum Model (CPCM).8 Dielectric constants of 10.125 were used for dichloroethane, and the solute surface was defined with UAHF radii in each case. As expected, protonation on the sterically congested tri-substituted enamines (E- and Z-enamines) are highly facial-selective, which is consistent with our previous observations.5e For example, the energy difference between the R- and S-selective protonation onto the E-enamine (facial selectivity) is very large (ΔΔHa = 3.90 kcal mol−1, ΔΔGa = 4.07 kcal mol−1), indicating that the protonation is a highly enantioselective step and the stereoselectivity is determined prior to this step.
The Curtin–Hammett control in the C–N formation step was then explored. The C–N bond formation of 5-phenyltetrazole 11 with E-s-trans iminium ion is slightly lower in energy than that with its equilibrated E-s-cis isomer (ΔΔHa = 1.97 kcal mol−1, ΔΔGa = 0.86 kcal mol−1) which corresponds to a diastereoisomeric excess of 95% favouring the E-enamine. Provided with the stereospecific protonation, this gives an enantiomeric excess of 95% for the final protonation product S-12c (Scheme 6), which is consistent with the 90% ee experimentally observed for S-12c.9
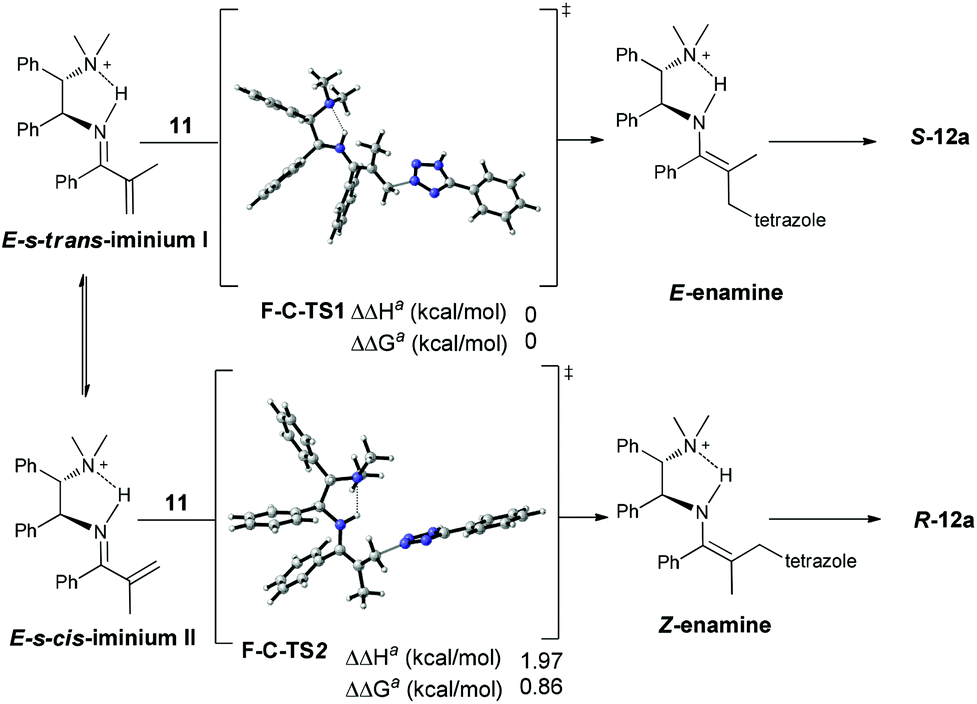 | ||
| Scheme 6 The transition states for the reaction between 5-phenyltetrazole 11 and vinyl ketone 7a. | ||
Finally, to demonstrate the synthetic utility of the catalyst system, a larger scale synthesis of 8e using 5b/TfOH as the catalyst was performed, and the reactions furnished the desired product with similar results (Scheme 7). Further transformation has also been explored. The Baeyer–Villiger oxidation of 8e proceeded with m-CPBA successfully afforded the β-benzotriazolyl ester 14, which is difficult to synthesize using enolate chemistry, in 78% yield and 85% ee (Scheme 7).
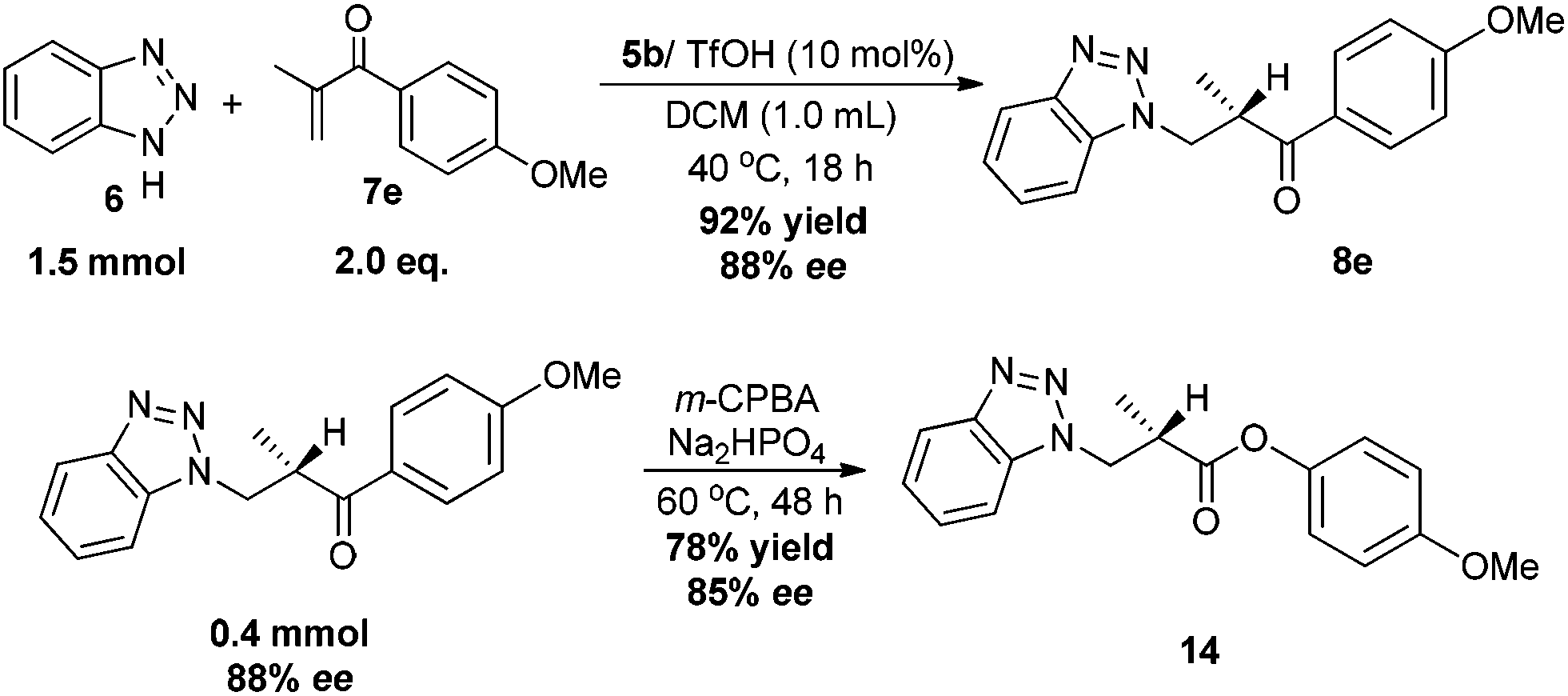 | ||
| Scheme 7 The gram scale reaction of 6 and 7e and the subsequent synthetic manipulation. | ||
In summary, we have developed the first example of conjugate addition–protonation reactions with azoles in iminium catalysis of α-branched vinyl ketones. The reactions can be broadly applied to a range of azoles with good yields and high enantioselectivities. This study also further verified the Curtin–Hammett control in dictating the stereoselectivity of the stereogenic protonation step. Given the largely unexplored iminium catalysis with α-branched enones, more successes to pursue the addition–protonation in new reaction development are anticipated. Importantly, the mechanism-oriented new catalyst design for the reactions with α-branched acroleins is also warranted.
We thank the Natural Science Foundation of China (21025208, 21202171) and the National Basic Research Program of China (2011CB808600) for financial support.
Notes and references
-
(a)
A. R. Katritzky and A. F. Pozharskii, Handbook of Heterocyclic Chemistry, Pergamon, Oxford, 2nd edn, 2002 Search PubMed
; (b) J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 4th edn, 2000 Search PubMed
- For recent review on organocatalytic asymmetric Aza-Michael Additions, see:
(a) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS PubMed
- For recent review, see:
(a) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed
- For reviews on enantioselective protonation, see:
(a)
A. Yanagisawa and H. Yamamoto, in Comprehensive Asymmetric Catalysis Vol. III, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Heidelberg, 1999, p. 1295 Search PubMed
- For an account, see:
(a) L. Zhang and S. Luo, Synlett., 2012, 1575 Search PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, (Revision A.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed
-
(a) M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503 CrossRef CAS
-
(a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS
- The absolute configuration was determined by analogy with compound 8c. A derivative of 8c was characterized by X-ray crystallography. See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of 1H NMR and 13C NMR spectra and HPLC traces. CCDC number 965694. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00027c |
| This journal is © the Partner Organisations 2014 |