A silver-catalyzed transfer hydrogenation of aldehyde in air and water†‡
Mingxin
Liu
a,
Feng
Zhou
a,
Zhenhua
Jia
ab and
Chao-Jun
Li
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bSchool of Pharmaceutical Sciences, Institute of Drug Synthesis and Pharmaceutical Process, Sun Yat-sen University, Guangzhou 510006, P. R. China
First published on 27th January 2014
Abstract
The first example of transfer hydrogenation catalyzed by silver complexes in air and water is reported. Various aromatic and aliphatic aldehydes are reduced by using two ligand systems to give the corresponding alcohols in good to excellent yields, while no reaction is observed with ketones.
The reduction of carbonyl compounds has always been an important index of chemical transformations in both academia and industry.1 Historically, the methods for the reduction of carbonyl compounds are primarily based on boron/aluminum hydride agents and their derivatives.2 Due to the advantages of milder reaction conditions and higher atom efficiency, as well as unique chemo- or stereo-selectivities3 that usually cannot be achieved by other methods, transition-metal catalyzed carbonyl reduction methodologies are intensively studied. Transfer hydrogenation, as an important avenue among transition-metal catalyzed carbonyl reductions, has received much academic attention for its advantages over direct hydrogenations such as the precluding of storage and transportation of hydrogen gas and dangerous high-pressure manipulations.4 Since the first report of the notable Meerwin–Pondorf–Verley (MPV) reduction in 1926,5 and the pioneering work with the introduction of transition-metal catalysis into transfer hydrogenation by Noyori,6 various catalyst systems have been developed to achieve such transformations.7 These catalytic systems generally involve ruthenium [7c], iridium,8 rhodium9 and palladium complexes,10 which are expensive and scarce. To overcome these shortcomings, a few examples using more abundant metal catalysts have been reported. In an early example, Iyer reported the use of more abundant nickel as a successful transition-metal catalyst for transfer hydrogenation.11 More recently, successful transfer hydrogenation catalyzed by iron was introduced by Beller.12 However, most of the existing systems still require an inert atmosphere and anhydrous organic solvents. Thus, catalytic transfer-hydrogenations based on more abundant metals and under eco-friendly conditions are still highly desirable.
Water, among all solvents, has attracted considerable academic attention in transition-metal catalysis.13 Among them, transfer hydrogenations in water have received more and more interest. In 1999, Ogo first reported the use of a bi-nucleus, water-soluble iridium complex to successfully catalyze a transfer hydrogenation of aldehydes and ketones in water.14 In 2005, Xiao reported a highly efficient aqueous-phase asymmetric transfer hydrogenation of ketone catalyzed by rhodium with a water-soluble tosylated Noyori-type ligand.15 Carreira made significant contributions in modifying the iridium system based on the tosylated Noyori-type ligand to achieve a wider range of interesting chemoselectivities in water.16 Most recently, Xiao developed a new iridium complex which is distinct from the previous ones and achieved an extremely high efficiency in transfer hydrogenation in water.17 However, to the best of our knowledge, there seems no example of using a relatively abundant metal as a catalyst to afford transfer hydrogenation in water and under an air atmosphere. Herein, we report a simple, efficient and chemoselective silver-catalyzed transfer hydrogenation of aldehydes into alcohols in air and water for the first time, by using formate as a convenient source of hydrogen (Fig. 1).
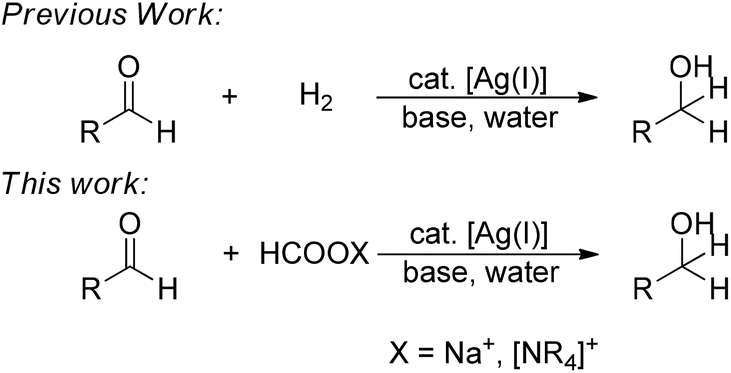 | ||
| Fig. 1 Silver-catalyzed carbonyl reduction strategies. | ||
Previously, based on our earlier work on the silver-catalyzed alkyne–aldehyde additions and aldehyde–alkyne–amine coupling reaction (A3-Reaction),18 we developed a silver-catalyzed catalytic reduction of aldehyde using organosilane as a reducing agent in air and water.19 The reaction exhibited a unique chemoselectivity towards aldehydes with ketones untouched. Most recently, we succeeded in the first silver-catalyzed direct hydrogenation of aldehyde in water (Fig. 1).20 However, while organosilane reduction suffers from low atom-economy,21 the various shortcomings associated with hydrogenation remain.
To address these challenges, we began our investigation of catalytic transfer hydrogenation with the silver-diphenylphosphinoferrocene (DPPF) complex (Table 1) based on our previous studies of catalytic hydrogenations, as a catalyst to conduct the transfer hydrogenation of benzaldehyde using sodium formate as the hydrogen source in air and water. Gratifyingly, about 2% 1H NMR yield of the desired product was observed (entry 1). Subsequently, extensive experiments (entries 2–27) were conducted to investigate the effects of silver salts and ligands on the reaction (for complete screening of salts and ligands, please see ESI)†. Changing the anion in silver salt from the non-coordinating PF6 to a weakly coordinating OTf gave the product in 3% 1H-NMR yield (entry 6). Among the halide anions tested, fluoride also gave a ca. 3% yield (entry 2) while chloride only generated <1% yield (entry 3) as observed by 1H NMR of the crude reaction mixture. No desired product was observed with either bromide or iodide anions (entries 3 and 4).
|
|
|||
|---|---|---|---|
| Entry | Silver salt | Ligand | 1H NMR yielda |
| a 1H NMR yields were determined by using mesitylene as the internal standard. b Isolated yield. c Performed without a base. d Carried out without a solvent. e Carried out in ethanol f Carried out in acetonitrile g Carried out in acetone. h Carried out in N,N-dimethylformamide. | |||
| 1 | AgPF6 | L1 | 2% |
| 2 | AgF | L1 | 3% |
| 3 | AgCl | L1 | N.D. |
| 4 | AgBr | L1 | N.D. |
| 5 | AgI | L1 | N.D. |
| 6 | AgOTf | L1 | 3% |
| 7 | AgF | L2 | N.D. |
| 8 | AgF | L3 | N.D. |
| 9 | AgF | L4 | 6% |
| 10 | AgF | L8 | N.D. |
| 11 | AgF | L9 | 61% |
| 12 | AgF | L10 | 66% |
| 13 | AgF | L11 | 50% |
| 14 | AgF | L12 | >99% (92%)b |
| 15 | AgF | L13 | N.D. |
| 16 | AgPF6 | L12 | 38% |
| 17 | AgCl | L12 | 85% |
| 18 | AgBr | L12 | 11% |
| 19 | AgI | L12 | N.D. |
| 20 | // | L12 | N.D. |
| 21 | AgF | // | N.D. |
| 22 | AgF | L12 | 33%c |
| 23 | AgF | L12 | 26%d |
| 24 | AgF | L12 | 80%e |
| 25 | AgF | L12 | 9%f |
| 26 | AgF | L12 | N.D.g |
| 27 | AgF | L12 | N.D.h |
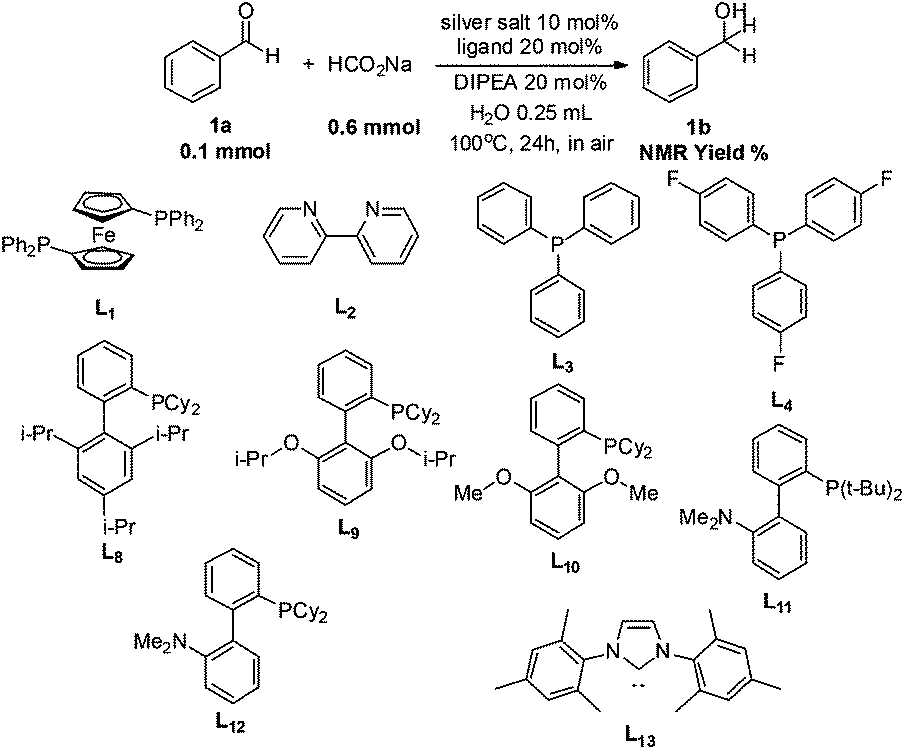
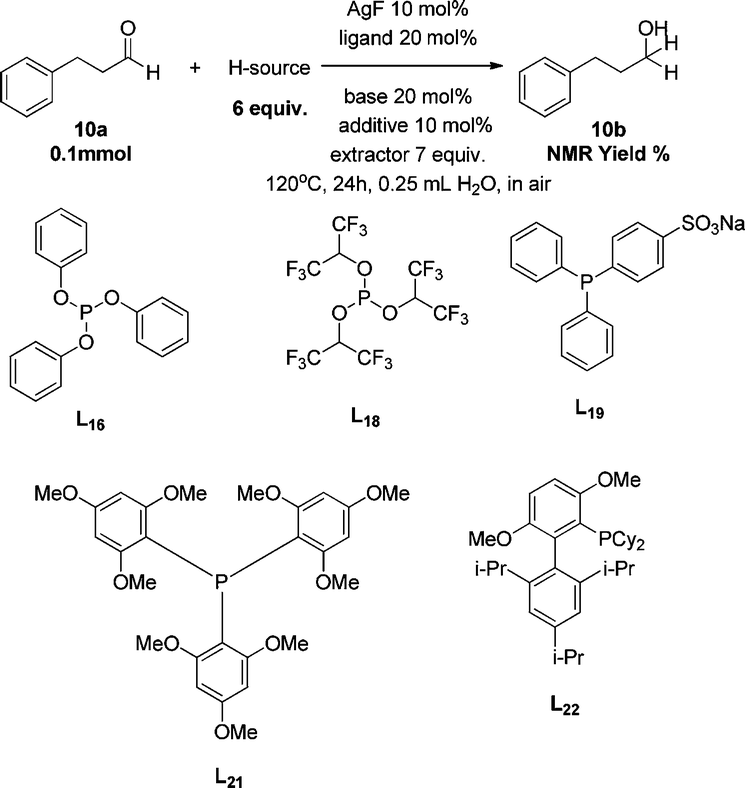
With the identification of the proper silver salt, the optimization was then made to select the preferred ligand. The use of P(p-F-Ph)3 as a ligand provided 6% 1H NMR yield (entry 9) while PPh3 failed to give any desired product (entry 8). Surprisingly, while no product was obtained with Buchwald's biphenyl-type XPhos ligand (entry 10), a very similar Buchwald's biphenyl-ligand RuPhos gave 61% yield of the corresponding transfer hydrogenation product (entry 11), indicating that a second hemi-labile, electron-pair-donor atom on the ligand molecule may be beneficial to the reaction. With this observation, we theorized that a more electron-rich hemi-labile, electron-pair donor might be even more efficient in facilitating the reaction. Indeed, switching the hemi-labile donor of an oxygen atom on RuPhos and SPhos (entry 12) to a nitrogen atom turned out to be a great success. The combination of silver fluoride (AgF) with DavePhos [2′-dicyclohexylphosphino-2-(N′-N′-dimethylamino)-biphenyl] resulted in a nearly quantitative transformation from benzaldehyde to benzyl alcohol (entry 14). Upon direct extraction from the aqueous reaction mixture with chloroform-d, the 1H NMR of the crude reaction mixture was taken, which gave a spectrum perfectly matching that of pure benzyl alcohol with only tiny impurity peaks (due to trace amounts of catalyst and DIPEA). With the optimized ligand in hand, the silver salts were further investigated, which showed a relative catalytic efficiency of AgF > AgCl > AgBr > AgI (entries 17–19). AgPF6 gave a much lower yield than AgF (entry 16). As control experiments, no reaction was observed in the absence of either AgF or the ligand (entries 20 and 21). With the absence of a base, the yield diminished (entry 22). Also, the transformation in organic solvent was carried out (entries 24–27). Except for the very-polar ethanol, all the other solvents seemed to be much less efficient for this transformation. Without solvent, the neat reaction also gave 26% yield (entry 23).
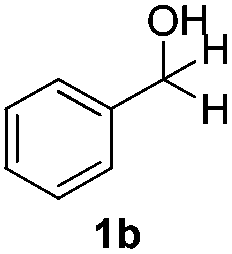
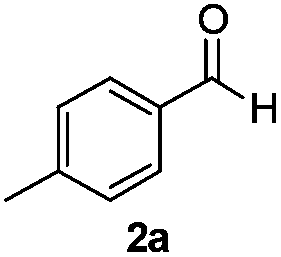
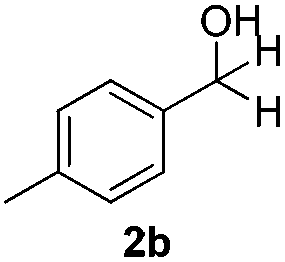
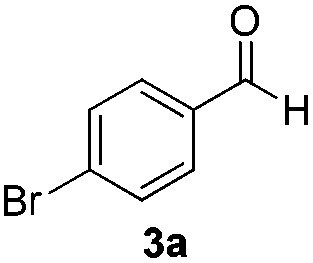
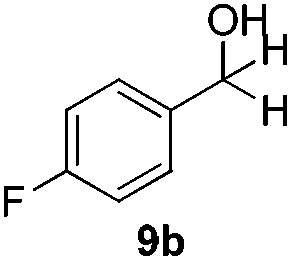
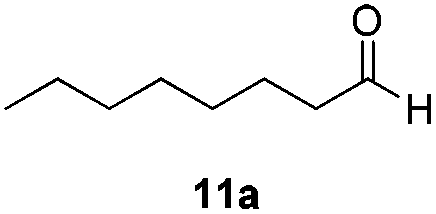
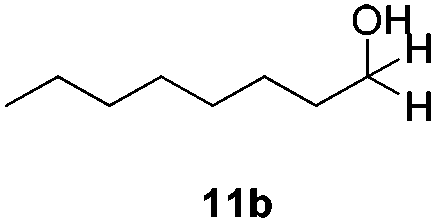
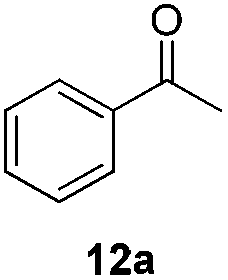
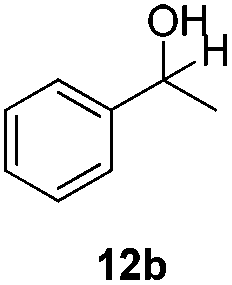
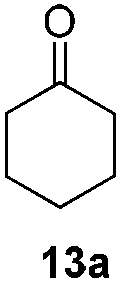
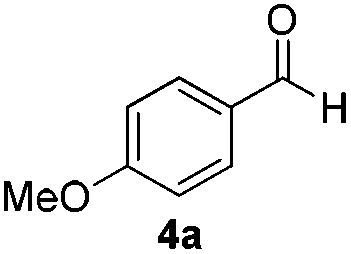
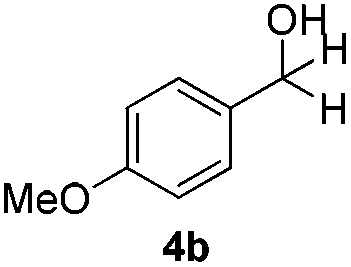
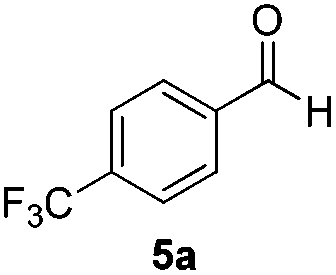
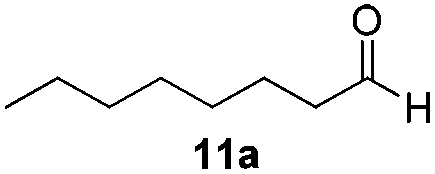
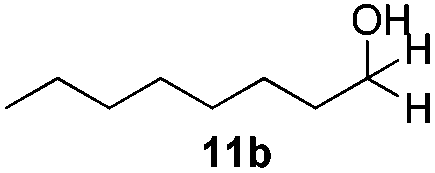
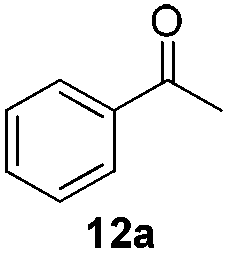
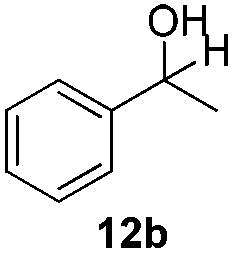
With the optimized conditions in hand, the scope of the reduction was studied with a variety of aldehydes (Table 2). With most aromatic aldehydes, having either an electron-donating or withdrawing substituent, the reduction was efficient and gave the desired products in excellent yields (entries 1–3 and 6). Slightly decreased yields were obtained with the very electron-rich p-anisaldehyde and the very electron-poor α′–α′–α′ p-trifluoro-tolualdehyde (entries 4 and 5). However, this catalytic system appeared to be limited to aromatic aldehydes: while most aromatic aldehydes were reduced in excellent to quantitative yields, only <3% yield or no product was obtained with all the aliphatic aldehydes (entries 10 and 11). Fluorine-substituted aromatic aldehydes also undergo the transformation less efficiently (entries 7–9). At the same time, ketones remained intact under the reaction conditions (entries 12 and 13), similar to our earlier studies on reduction by using organosilanes.19
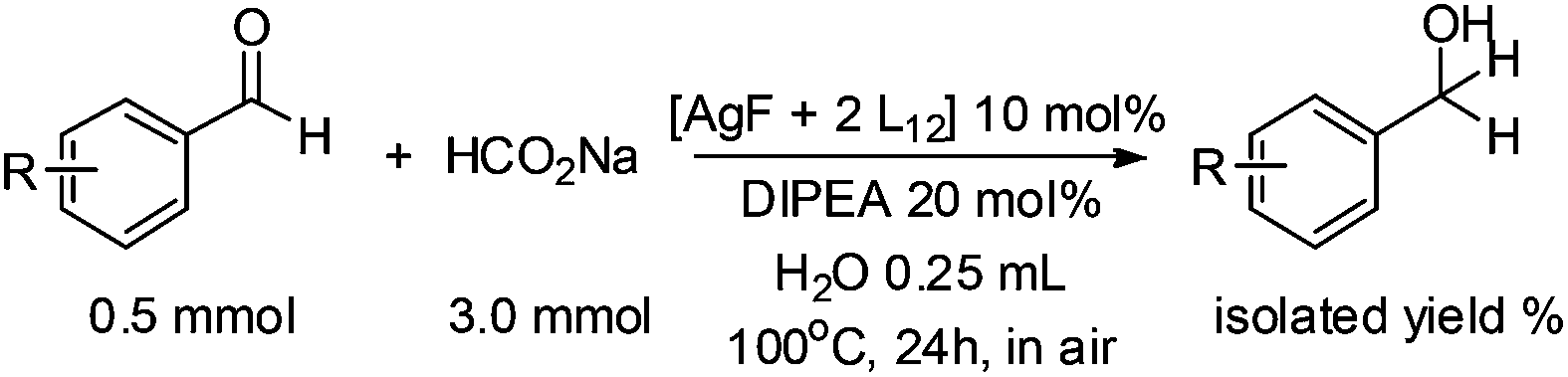
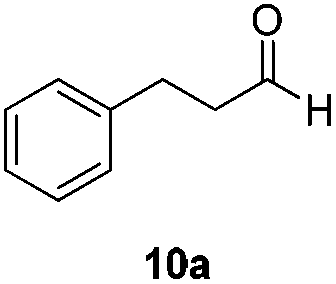
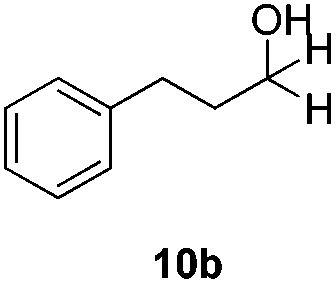
In order to overcome the lack of reactivity with aliphatic aldehydes towards the transfer hydrogenation, another series of condition screenings was engaged by changing the ligands, reaction temperature, and some other additives (Table 3) (for complete screening of salts and ligands, please see ESI)†. Hydrocinnamaldehyde was used as the benchmark for optimizing the conditions. Initially, it was observed that switching the hydrogen source from sodium formate to diisopropyl-ethylammonium formate (DIPEA·HCOOH) slightly raised the product from 3% to 6% (entries 1 and 3). This observation was attributed to the slightly lower basicity of aqueous DIPEA·HCO2H than HCO2Na. Furthermore, the use of CsF instead of DIPEA as a base also improved the yield slightly (entry 4). When the reaction temperature was raised to 120 °C, the NMR yield was boosted to 11% (entry 6). A series of amine bases was also examined for neutralizing the formate (entries 7–10), and DIPEA was still the most efficient one.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | H-source | Base | Ligand | Additive | Extractor | 1H NMR yield |
| a Reactions were carried out at 100 °C. b Reaction was carried out in 1 mL water. | ||||||
| 1 | HCO2Na | DIPEA | L12 | // | // | 3%a |
| 2 | HCO2H | // | L12 | // | // | n.d.a |
| 3 | HCO2H·DIPEA | DIPEA | L12 | // | // | 6%a |
| 4 | HCO2H·DIPEA | CsF | L12 | // | // | 7%a |
| 5 | HCO2H·DIPEA | CsF | L12 | LiF | // | n.d.a |
| 6 | HCO2H·DIPEA | CsF | L12 | // | // | 11% |
| 7 | HCO2H·NH3 | CsF | L12 | // | // | n.d. |
| 8 | HCO2H·1/2TMEDA | CsF | L12 | // | // | n.d. |
| 9 | HCO2H·1/2DABCO | CsF | L12 | // | // | n.d. |
| 10 | HCO2H·DBU | CsF | L12 | // | // | 10% |
| 11 | HCO2H·DIPEA | CsF | L16 | // | // | Trace |
| 12 | HCO2H·DIPEA | CsF | L18 | // | // | n.d. |
| 13 | HCO2H·DIPEA | CsF | L19 | // | // | n.d. |
| 14 | HCO2H·DIPEA | CsF | L21 | // | // | n.d. |
| 15 | HCO2H·DIPEA | CsF | L22 | // | // | 21% |
| 16 | HCO2H·DIPEA | CsF | L10 | // | // | 15% |
| 17 | HCO2H·DIPEA | CsF | L10 | // | DIPEA | 30% |
| 18 | HCO2H·DIPEA | CsF | L10 | // | PhCl | 30% |
| 19 | HCO2H·DIPEA | CsF | L10 | TfOH | PhCl | 55% |
| 20 | HCO2H·DIPEA | CsF | L10 | Benzoic acid | PhCl | 12% |
| 21 | HCO2H·DIPEA | CsF | L10 | CF3CO2H | PhCl | 11% |
| 22 | HCO2H·DIPEA | CsF | L10 | TfOH | PhCl | 75%b |
| 23 | HCO2H·DIPEA | CsF | L22 | TfOH | PhCl | 99% |
| 24 | HCO2H·DIPEA | // | L10 | TfOH | PhCl | 42% |
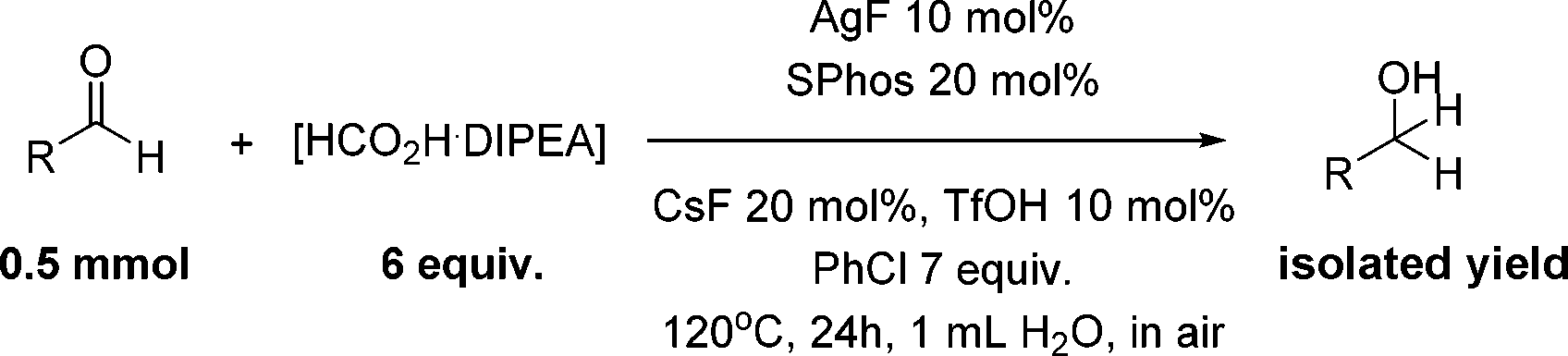
Both electron-rich and electron-poor ligands were then further tested for the aliphatic aldehyde reduction (entries 11–16), which again demonstrated the best results with Buchwald's biphenyl-ligand (BrettPhos and SPhos) (entries 15 and 16) bearing a hemi-labile chelating atom. We also observed that an increased yield was obtained with the use of an excessive amount of DIPEA (entry 17), possibly by providing an organic phase microscopically. Thus, it was found that the excess DIPEA could be replaced by hydrophobic chlorobenzene (entry 18). Surprisingly, we noticed that the use of a freshly distilled starting aldehyde decreased the reaction yield compared to an aged one, which was attributed to the small amount of acid caused by oxidation of aldehyde. A series of acids was then tested (entries 19–21) as reaction additives and trifluorosulfonic acid appeared to be the most effective (entry 19). After amplifying the amount of solvent, 75% NMR yield was obtained by using SPhos ligand (entry 22), whereas a nearly quantitative yield was achieved with the use of the more expensive BrettPhos (entry 23). The reaction that proceeds without cesium fluoride gave a decreased yield of 42% (entry 24).
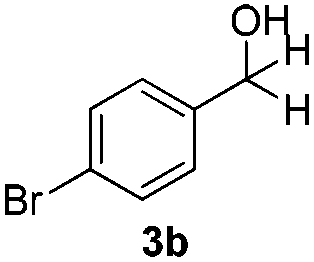
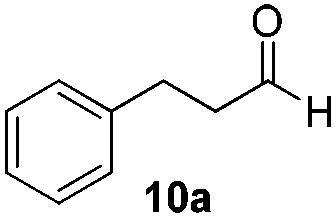
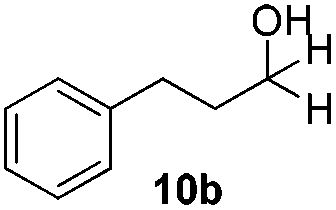
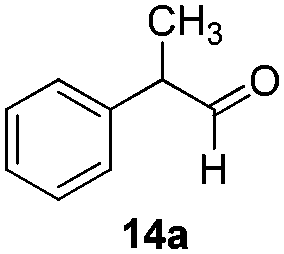
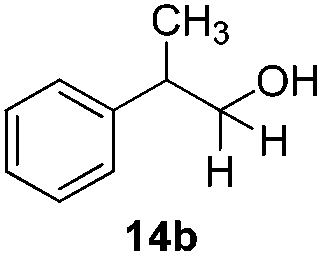
With the conditions optimized, transfer hydrogenations of both aromatic and aliphatic aldehydes were examined (Table 4) with the slightly less effective but relatively cheaper SPhos ligand. As expected, the new conditions still gave a nearly quantitative NMR yield with most aromatic aldehydes (entries 1–3 and 5–9), except for the very electron rich p-anisaldehyde which only gave 51% NMR yield (entry 4). Fluorine-substituted benzaldehydes also give excellent to nearly quantitative yield, regardless of the o-, m-, p-substituted regioisomers (entries 7–9). On the other hand, simple aliphatic n-octanal gave 76% isolated yield of the corresponding alcohol (entry 11). The α-substituted phenylpropionaldehyde seems to give a lower yield of 43% (entry 13). This could be due to the steric effect that the methyl group at α-position hindered the carbonyl from being attacked. With a double bond adjacent to the carbonyl, 82% isolated yield of the corresponding alcohol was obtained with cinnamaldehyde (entry 14). With an isolated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond present, the reaction diminished (entries 15–17). This might be due to the competing stronger coordination of Ag(I) complex towards CC bond compared to CO and conjugated CO bonds, which stopped the reaction. However, in the presence of CC bond, the hydrogenation of CC was not observed. Still, ketones seem to be inert towards our reaction conditions (entry 12).
C bond present, the reaction diminished (entries 15–17). This might be due to the competing stronger coordination of Ag(I) complex towards CC bond compared to CO and conjugated CO bonds, which stopped the reaction. However, in the presence of CC bond, the hydrogenation of CC was not observed. Still, ketones seem to be inert towards our reaction conditions (entry 12).

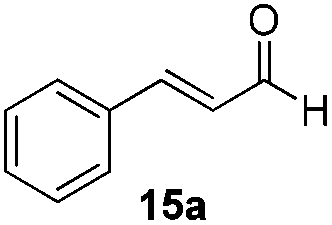
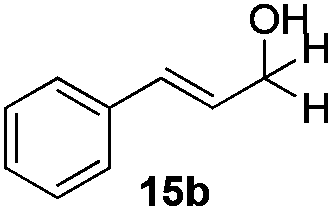
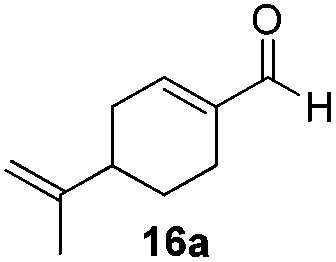
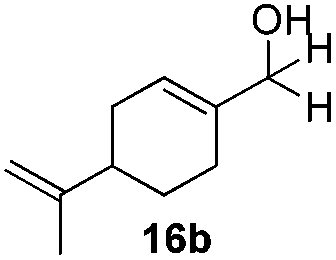
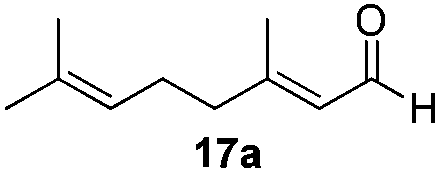
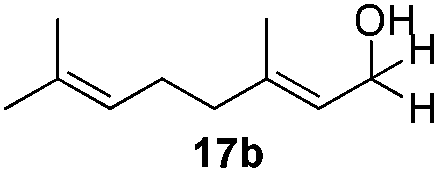
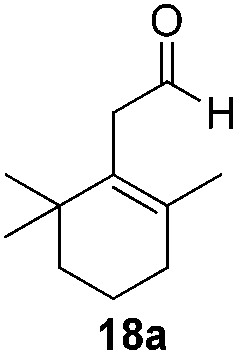
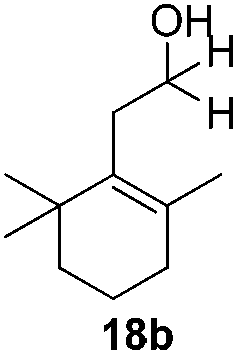
Conclusions
The first silver-catalyzed transfer hydrogenation has been discovered in air and water. Various aromatic and aliphatic aldehydes were reduced efficiently by using formate as a safe and convenient hydrogen source. The use of AgF-DavePhos leads to a selective reduction of aromatic aldehydes, whereas both aromatic and aliphatic aldehydes are reduced efficiently with AgF-BrettPhos and AgF-SPhos catalysts. The scope, mechanism and synthetic applications of this novel catalytic transfer hydrogen are under further investigation in our laboratory.Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.J.L.), the CFI, FQRNT Center for Green Chemistry and Catalysis and NSERC for support of our research. Z.J. thanks the Oversea Study Program of the Guangzhou Elite Project for financial support.Notes and references
- H. C. Brown and P. V. Ramachandran, in Reductions in Organic Synthesis, ed. A. F. Abdel-Magid, American Chemical Society, 1996, p. 1–30 Search PubMed.
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Structure and Mechanisms, Kluwer Academic, New York, 4th edn, 2000 Search PubMed; (b) J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 3rd edn, 1985 Search PubMed.
- X. Wu and J. Xiao, in Metal-Catalyzed Reactions in Water, ed. P. H. Dixneuf and V. Cadierno, Wiley-VCH Verlag, Weinheim, 2003, pp. 173–242 Search PubMed.
- A. Robertson, T. Matsumoto and S. Ogo, Dalton Trans., 2011, 40, 10304 RSC.
- (a) D. Klomp, U. Hanefeld and J. A. Peters, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, vol. 1 Search PubMed; (b) W. Ponndorf, Angew. Chem., 1926, 39, 138 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- (a) Ruthenium in Organic Synthesis, ed. M. Kitamura, R. Noyori and S.-I. Murahashi, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) J. M. Brunel, Tetrahedron, 2007, 63, 3899 CrossRef CAS PubMed; (c) C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750 CrossRef CAS PubMed; (d) F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379 CrossRef CAS PubMed; (e) M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818 CrossRef CAS.
- (a) K. Ikariya and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 RSC; (b) W. Barrata, K. Siega and P. Rigo, Adv. Synth. Catal., 2007, 349, 1633 CrossRef; (c) B. R. James and R. H. Morris, J. Chem. Soc., Chem. Commun., 1978, 929 RSC; (d) X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718 CrossRef CAS PubMed; (e) J. Li, Y. Zhang, D. Han, G. Jia, J. Gao, L. Zhong and C. Li, Green Chem., 2008, 10, 608 RSC; (f) Y. Himeda, N. Onozawa-Komatsuzaki, S. Miyazawa, H. Sugihara, T. Hirose and K. Kasuga, Chem.–Eur. J., 2008, 14, 11076 CrossRef CAS PubMed.
- T. Mizugaki, Y. Kanayama, K. Ebitani and K. Kaneda, J. Org. Chem., 1998, 63, 2378 CrossRef CAS.
- M. Oba, K. Kojima, E. Endo, H. Sano and K. Nishiyama, Green Chem. Lett. Rev., 2013, 6, 233 CrossRef CAS.
- (a) S. Iyer and J. P. Varghese, J. Chem. Soc., Chem. Commun., 1995, 465 RSC; (b) S. Iyer and A. K. Sattar, Synth. Commun., 1998, 28, 1721 CrossRef CAS.
- G. Wienhöfer, F. Westerhaus, K. Junge and M. Beller, J. Org. Chem., 2013, 744, 156 CrossRef PubMed.
- (a) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 5, 68 RSC; (b) C.-J. Li and T.-H. Chan, Comprehensive Organic Reactions in Aqueous Media, Wiley, New York, 2007 Search PubMed; (c) Metal-Catalyzed Reactions in Water, ed. P. H. Dixneuf and V. Cadierno, Wiley VCH, Weinheim, 2012 Search PubMed.
- (a) S. Ogo, N. Makihara and Y. Watanabe, Organometallics, 1999, 18, 5470 CrossRef CAS; (b) S. Ogo, N. Makihara, Y. Kaneko and Y. Watanabe, Organometallics, 2001, 20, 4903 CrossRef CAS; (c) T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 4149 CrossRef CAS PubMed; (d) S. Ogo, K. Uehara, T. Abura and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 3020 CrossRef CAS PubMed.
- (a) X. Wu, D. Vinci, T. Ikariyab and J. Xiao, Chem. Commun., 2005, 4447 RSC; (b) X. Wu, X. Li, F. King and J. Xiao, Angew. Chem., Int. Ed., 2005, 44, 3407 CrossRef CAS PubMed; (c) X. Wu and J. Xiao, Chem. Commun., 2007, 2449 RSC; (d) X. Wu, X. Li, A. Zanotti-Gerosa, A. Pettman, J. Liu, A. Mills and J. Xiao, Chem.–Eur. J., 2008, 14, 2209 CrossRef CAS PubMed.
- (a) O. Soltani, M. Ariger and E. Carreira, Org. Lett., 2009, 11, 4196 CrossRef CAS PubMed; (b) O. Soltani, M. Ariger, H. Vázquez-Villa and E. Carreira, Org. Lett., 2010, 12, 2893 CrossRef CAS PubMed; (c) H. Vázquez-Villa, S. Reber, M. Ariger and E. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979 CrossRef PubMed; (d) M. Ariger and E. Carreira, Org. Lett., 2012, 14, 4522 CrossRef CAS PubMed.
- Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629 RSC.
- (a) C. Wei, Z. Li and C.-J. Li, Org. Lett., 2003, 5, 4473 CrossRef CAS PubMed; (b) Z. Li, C. Wei, L. Chen, R. S. Varma and C.-J. Li, Tetrahedron Lett., 2004, 45, 2443 CrossRef CAS PubMed; (c) X. Yao and C.-J. Li, Org. Lett., 2005, 7, 4395 CrossRef CAS PubMed; (d) Z. Jia, X. Li, A. S. C. Chan and C.-J. Li, Synlett, 2012, 2758 CAS.
- Z. Jia, M. Liu, X. Li, A. S. C. Chan and C.-J. Li, Synlett, 2013, 2049 CAS.
- Z. Jia, F. Zhou, M. Liu, X. Li, A. S. C. Chan and C.-J. Li, Angew. Chem., Int. Ed., 2013, 52, 11871 CrossRef CAS PubMed.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00063j |
| ‡ This article is part of the celebration of the 40th Anniversary of the Mukaiyama Reaction. |
| This journal is © the Partner Organisations 2014 |