NBS-promoted halosulfonylation of terminal alkynes: highly regio- and stereoselective synthesis of (E)-β-halo vinylsulfones†
Yang
Gao
,
Wanqing
Wu
*,
Yubing
Huang
,
Kefan
Huang
and
Huanfeng
Jiang
*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: jianghf@scut.edu.cn; cewuwq@scut.edu.cn; Fax: +86 20-87112906; Tel: +86 20-87112906
First published on 7th March 2014
Abstract
An efficient NBS-promoted method for the synthesis of (E)-β-halo vinylsulfones has been developed. The present protocol went through an environmentally friendly metal-free process to achieve the halosulfonylation of terminal alkynes with high selectivity.
The development of green and efficient methods for the synthesis of valuable molecular skeletons is the central focus of modern organic chemistry and has been intensively pursued by the synthetic community.1 Among them, the difunctionalization of alkynes, which involves the formation of two new vicinal chemical bonds, represents an important contribution to the functionalized alkenes.2 The halosulfonylation of terminal alkynes has drawn considerable attention over the years, particularly in the development of atom-economical and environmentally friendly reaction systems to provide β-halo vinylsulfone products.
On the other hand, β-halo vinylsulfones are well known to be an important class of compounds which are versatile building blocks and valuable intermediates in organic synthesis and medicinal chemistry.3 For example as a core functional group in inhibitors of various enzymatic processes4 and as an important precursor in the synthesis of a series of useful biologically active molecules.5 Due to their importance, various methods have been developed to construct these frameworks.6 Generally, an overwhelming number of these alkyne difunctionalization transformations involve the transition metal-catalyzed (copper, iron, palladium, ruthenium etc.) addition of sulfonyl halogens to the terminal alkynes6a–f (Fig. 1a). Recently, Nakamura6a and Li,6b respectively, reported iron-catalyzed halosulfonylation of terminal alkynes using (p-Tol)3P as a ligand and TBHP as an additive. Despite the synthetic efficiency, some of these metal-catalyzed methods often require air-sensitive and expensive ligands or additives, and produce large amounts of unwanted metal salt by-products, which makes them environmentally unfavourable and results in toxic metal residues in the products. Thus, the development of more efficient and environmentally friendly catalyst systems to achieve the halosulfonylation of terminal alkynes is still highly desirable. Very recently, Lei and coworkers reported an alkyne difunctionalization transformation via a novel metal-free process to form various β-ketosulfone products (Fig. 1b).7 Owing to our growing interest in developing mild and efficient ways for difunctionalization of C–C multiple bonds;8 herein, we disclose a NBS-promoted halosulfonylation of terminal alkynes using commercially available sodium sulfinates as sulfonyl precursors, affording (E)-β-bromo and iodo vinylsulfones with high selectivity (Fig. 1c). This environmentally friendly metal-free transformation has a broad substrate scope and may go through a radical addition process to the terminal alkynes with specific stereoselectivity.
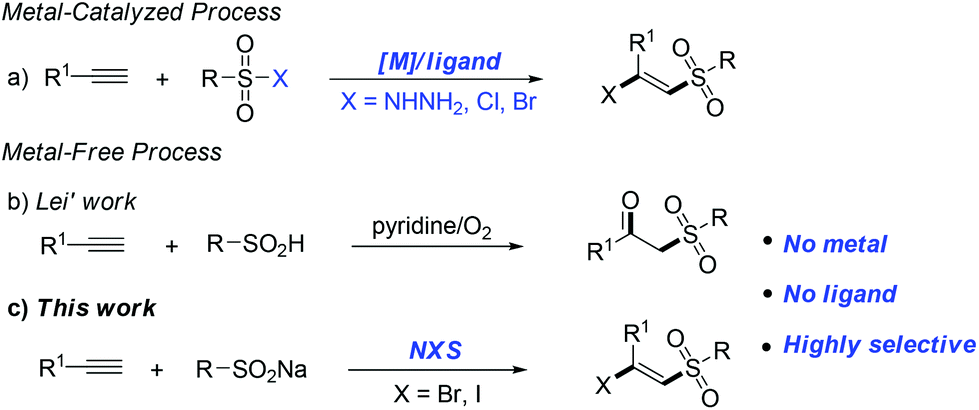 | ||
| Fig. 1 Difunctionalization of the terminal alkyne. | ||
Our initial investigations of this alkyne difunctionalization reaction focused on the addition of sodium sulfinate (1a) to ethynylbenzene (2a) in the presence of NBS in 1,2-dichloroethane (DCE) at 80 °C. To our delight, the desired (E)-β-bromovinylsulfone9 was detected in 53% yield (Table 1, entry 1). The solvent played an important role in the success of the reaction. Polar solvents, such as DMF and DMSO, were found to be totally inefficient for this transformation (entries 2 and 3) and the non-polar solvent toluene was found to be the best solvent (entry 4). Other bromine sources such as LiBr and TBAB were also examined. However, no reaction occurred when LiBr and TBAB were used instead of NBS (entries 5 and 6). This result indicated that NBS might be employed not only as a bromine source, but also as a trigger for this chemical process. In addition, this reaction was performed under a N2 atmosphere and has not much influence on the yield (entry 8). Thus, the optimal reaction conditions were 1a (0.5 mmol), 2a (0.5 mmol), and NBS (0.5 mmol) in 2 mL toluene at 80 °C.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Reaction conditions: unless otherwise noted, all reactions were performed with 1a (0.5 mmol), 2a (0.5 mmol), and catalyst (0.5 mmol) in the indicated solvent (2 mL) at 80 °C for 4 h. n.d. = not detected. b Determined by GC based on 1a. c Performed under N2. | |||
| 1 | NBS | DCE | 53 |
| 2 | NBS | DMF | Trace |
| 3 | NBS | DMSO | Trace |
| 4 | NBS | Toluene | 88 |
| 5 | TBAB | Toluene | n.d. |
| 6 | LiBr | Toluene | n.d. |
| 7 | — | Toluene | n.d. |
| 8c | NBS | Toluene | 85 |
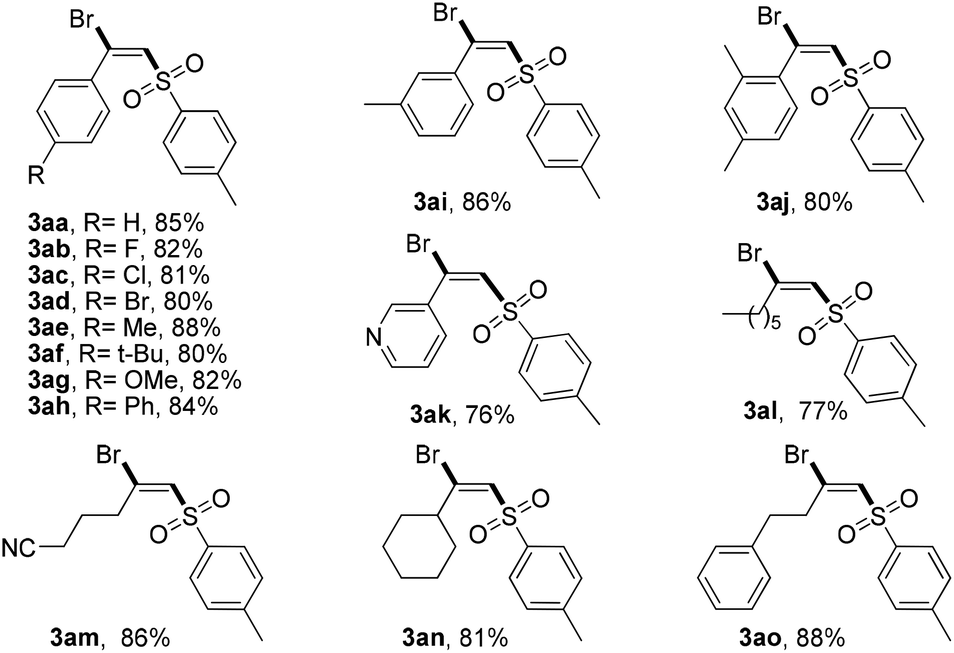
With the optimal reaction conditions in hand, we then examined the scope of this novel transformation (Table 2). Generally, various alkyl- and aryl-substituted terminal alkynes were found to be suitable reaction partners for this difunctionalization process. A series of para-substituted phenylacetylenes including some with electron-donating groups (Me, OMe, t-Bu, and Ph) and some with electron-withdrawing groups (F, Cl, and Br) were well tolerated and converted to the corresponding (E)-β-bromovinylsulfone products 3aa–3ag in good yields (80%–88%). It should be noted that these functional groups could be used for further modification to achieve more complex structures. Then, different substituent positions were tested. ortho- and meta-Methyl substituted phenylacetylene proceeded smoothly to afford the desired products 3ah–3ai in 83% and 85% yields, respectively. Notably, 3-ethynylpyridine also displayed similar reactivity. Pleasingly, alkyl-substituted terminal acetylene can also be successfully transformed into the desired halosulfonylation products 3al–3ao in good yields (70%–82%).
Then, the scope of the reaction with respect to sodium sulfinates was also studied (Table 3). Different sodium benzenesulfinates bearing halogen substituents (F, Cl, and Br) could react smoothly with ethynylbenzene (1a) to afford the halosulfonylation products 3ca–3ea in good yields (70%–82%). It is worth mentioning that naphthalene and thiophene substituted sodium sulfinates were also suitable reaction partners for this novel transformation. However, our attempts to employ alkyl-substituted sodium sulfinates as substrates turned out to be unfruitful, which might be caused by the instability of the alkyl-substituted sulfone radicals.
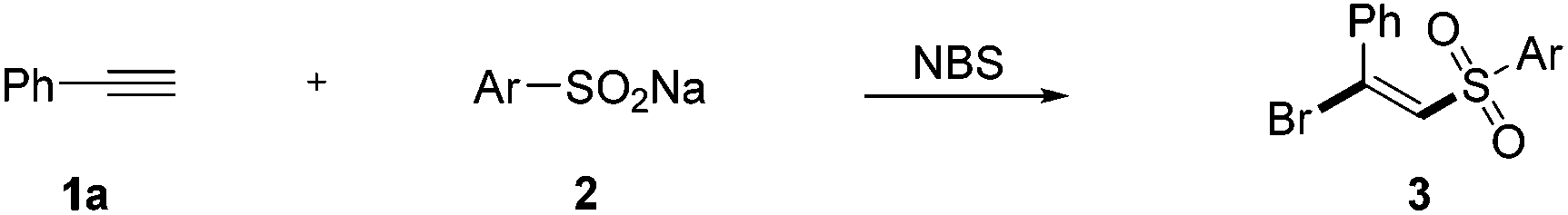
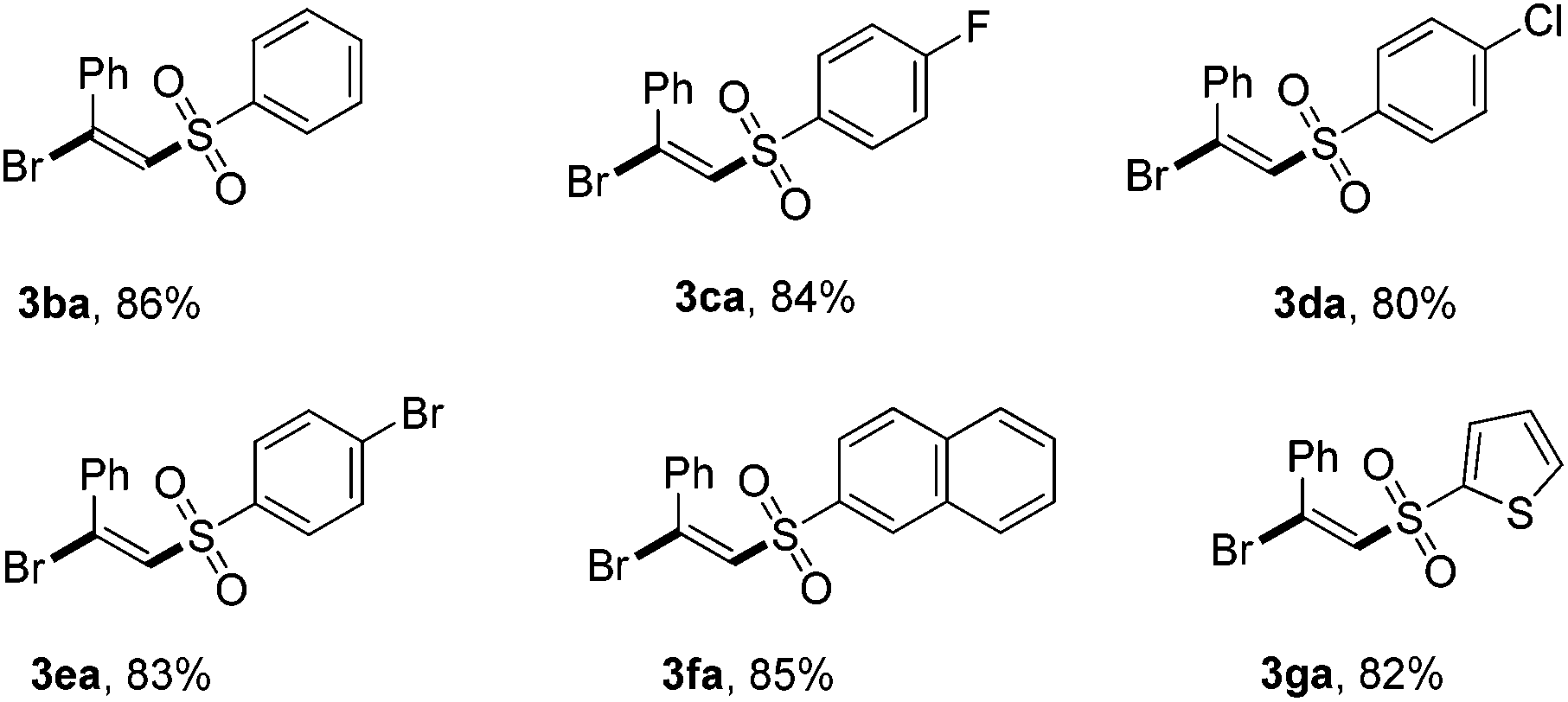
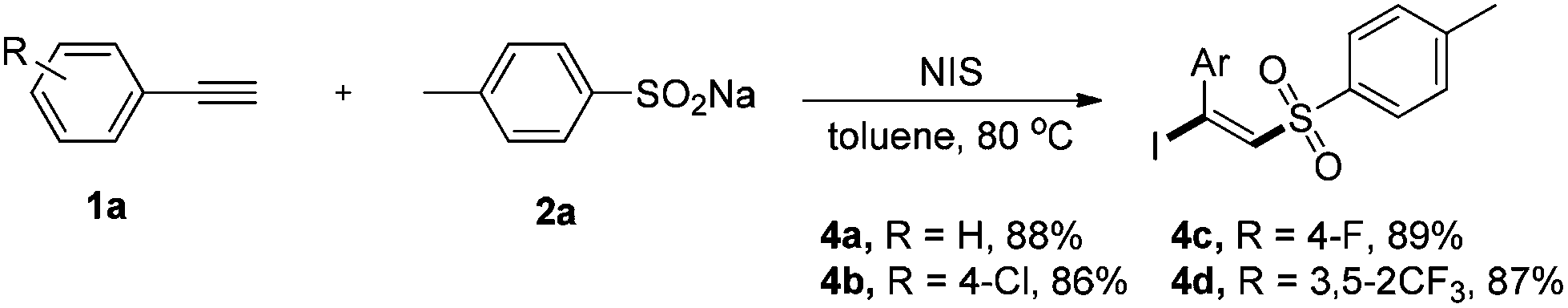
To further highlight the versatility of this alkyne difunctionalization strategy, we applied this method to the synthesis of (E)-β-iodovinylsulfones and the expected products 4a–4d were obtained in good yields using NIS instead of NBS. However, NCS was found to be totally inefficient for this transformation.
 | ||
| Fig. 2 The synthetic utility of the (E)-β-halo vinylsulfones. | ||
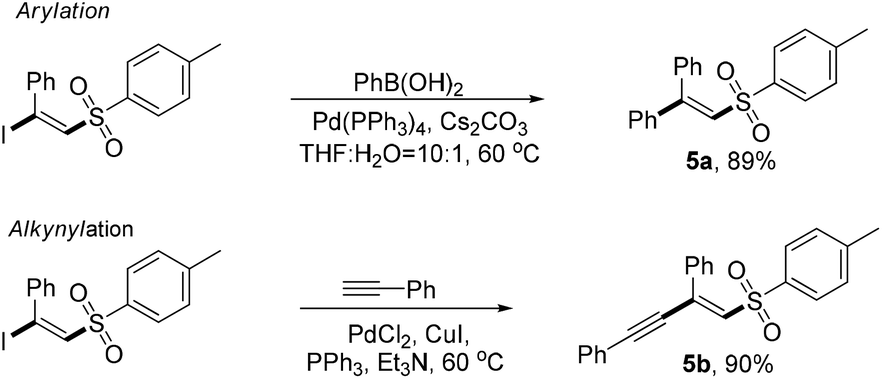
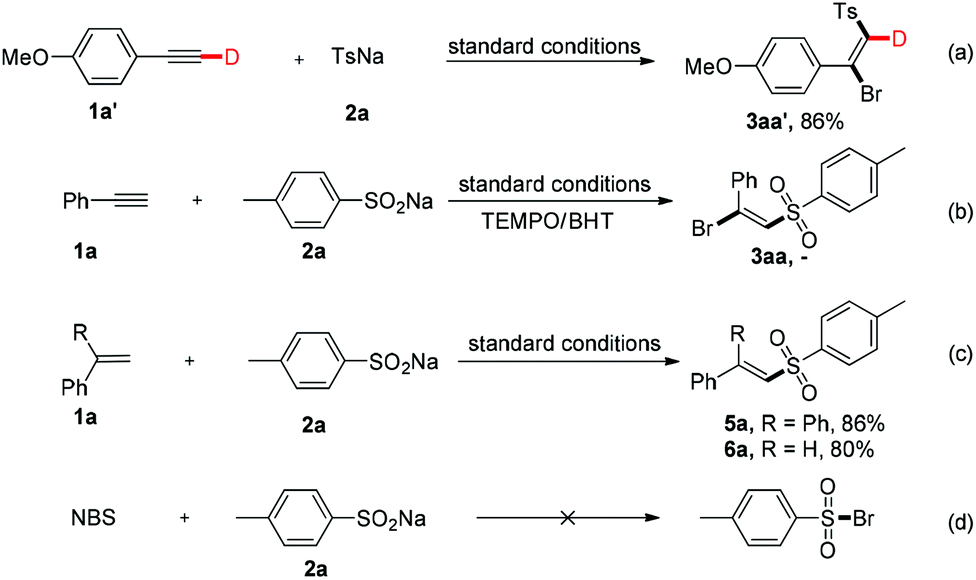
To gain insight into the mechanism of the chemical process, several control experiments were conducted (Fig. 3). First, a deuterated experiment was carried out to distinguish the hydrogen position of the terminal alkyne. As depicted in eqn (a), the deuterium product 3aa′ was obtained exclusively in 86% isolated yield and the deuterium atom (98% examined by 1H NMR spectroscopy) was still present. Next, when the radical scavengers, TEMPO and BHT, were employed in the reaction system, both could inhibit this halosulfonylation process, indicating that a radical pathway should be involved [eqn (b)]. To find more direct evidence of the radical process, the styrene compounds were used to capture the sulfone radicals and the vinyl sulfone products 5a and 5b could be obtained in 86% and 80% yields, respectively. These observations suggested that the NBS-promoted alkyne halosulfonylation might go through a sulfone radical process. Furthermore, no sulfonyl bromide could be detected when 2a was treated with NBS, thereby making this radical process different from the reported metal-promoted halosulfonylation processes6a–f [eqn (d)].
 | ||
| Fig. 3 Control experiments. | ||
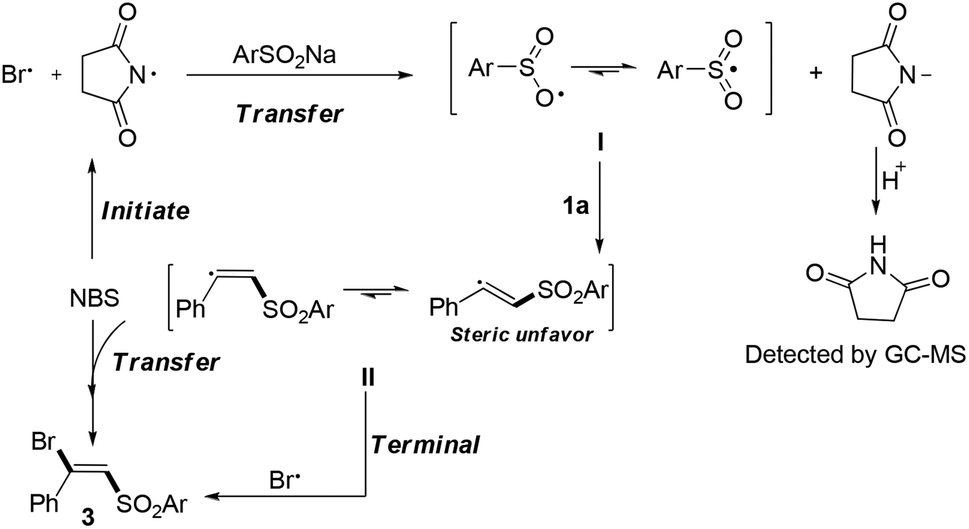
Based on the experimental results and previous reports,6a–g a plausible mechanism for this transformation is proposed in Scheme 1. The reaction was initially triggered by NBS to form the sulfone radical11 which may go through the cracking of the N–Br bond.12 Subsequently, the radical addition of I to the terminal alkyne formed the vinyl sulfone radical II,6,7 which would transfer the radical to NBS and afford the difunctionalization products.
 | ||
| Scheme 1 Possible reaction mechanism. | ||
In conclusion, an efficient NBS-promoted method for the synthesis of (E)-β-halo vinylsulfones has been developed. The present protocol went through an environmentally friendly metal-free process to achieve the halosulfonylation of terminal alkynes with high selectivity. NBS plays a dual role as both a trigger and a bromine source in this chemical process. The mild reaction conditions, easily available starting materials and high selectivity make the present halosulfonylation protocol rather attractive and applicable. In addition, the resulting halo vinylsulfone products can be further modified efficiently, which may find their potential applications in organic synthesis and medicinal chemistry.
The authors thank the National Natural Science Foundation of China (21172076 and 21202046), the National Basic Research Program of China (973 Program) (2011CB808600), the Guangdong Natural Science Foundation (10351064101000000 and S2012040007088), and the Fundamental Research Funds for the Central Universities (2014ZP0004 and 2014ZZ0046) for financial support.
Notes and references
- (a) J. L. Tucker, Org. Process Res. Dev., 2006, 10, 315 CrossRef CAS; (b) B. M. Trost, Science, 1983, 219, 245 CAS; (c) B. M. Trost, Science, 1991, 254, 1471 CAS; (d) M. C. Cann and T. A. Dickneider, J. Chem. Educ., 2004, 81, 977 CrossRef CAS; (e) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (f) R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS; (g) B. M. Trost, Acc. Chem. Res., 2002, 35, 695 CrossRef CAS PubMed; (h) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley-VCH, Weinheim, 6th edn, 2006, pp. 999–1250 Search PubMed.
- (a) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793 CrossRef CAS PubMed; (b) R. Ettari, E. Nizi, M. E. D. Francesco, M.-A. Dude, G. Pradel, R. Vicik, T. Schirmeister, N. Micale, S. Grasso and M. Zappala, J. Med. Chem., 2008, 51, 988 CrossRef CAS PubMed; (c) I. Forristal, J. Sulfur Chem., 2005, 26, 163 CrossRef CAS.
- (a) L. Ni, X. S. Zheng, P. K. Somers, L. K. Hoong, R. R. Hill, E. M. Marino, K.-L. Suen, U. Saxena and C. Q. Meng, Bioorg. Med. Chem. Lett., 2003, 13, 745 CrossRef CAS; (b) J. T. Palmer, D. Rasnick, J. L. Klaus and D. Bromme, J. Med. Chem., 1995, 38, 3193 CrossRef CAS.
- (a) V. Aranapakam, G. T. Grosu, J. M. Davis, B. Hu, J. Ellingboe, J. L. Baker, J. S. Skotnicki, A. Zask, J. F. DiJoseph, A. Sung, M. A. Sharr, L. M. Killar, T. Walter, G. Jin and R. Cowling, J. Med. Chem., 2003, 46, 2361 CrossRef CAS PubMed; (b) J. N. Desrosiers and A. B. Charette, Angew. Chem., Int. Ed., 2007, 46, 5955 CrossRef CAS PubMed; (c) M. N. Noshi, A. El-Awa, E. Torres and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 11242 CrossRef CAS PubMed; (d) L. Mu, K. Drandarov, W. H. Bisson, A. Schibig, C. Wirz, P. A. Schubiger and G. Westera, Eur. J. Med. Chem., 2006, 41, 640 CrossRef CAS PubMed.
- (a) X. Zeng, L. Ilies and E. Nakamura, Org. Lett., 2012, 14, 954 CrossRef CAS PubMed; (b) X. Li, X. Shi, M. Fang and X. Xu, J. Org. Chem., 2013, 78, 9499 CrossRef CAS PubMed; (c) Y. Amiel, J. Org. Chem., 1971, 36, 3697 CrossRef CAS; (d) H. Mataunoto, T. Nakano, K. Ohkawa and Y. Nagai, Chem. Lett., 1978, 363 Search PubMed; (e) S. R. Dubbaka and P. Vogel, Chem.–Eur. J., 2005, 11, 2633 CrossRef CAS PubMed; (f) X. Huang, D. Duan and W. Zheng, J. Org. Chem., 2003, 68, 1958 CrossRef CAS PubMed; (g) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed; (h) W. E. Truce and G. C. Wolf, J. Org. Chem., 1971, 36, 1727 CrossRef CAS.
- (a) Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. Lei, J. Am. Chem. Soc., 2013, 135, 11481 CrossRef CAS PubMed; (b) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed.
- (a) Y. Li, X. Liu, D. Ma, B. Liu and H. Jiang, Adv. Synth. Catal., 2012, 354, 2683 CrossRef CAS; (b) Z. Chen, J. Li, H. Jiang, S. Zhu, Y. Li and C. Qi, Org. Lett., 2010, 12, 3262 CrossRef CAS PubMed; (c) Z. Chen, H. Jiang, Y. Li and C. Qi, Chem. Commun., 2010, 46, 8049 RSC.
- The structures of the products were confirmed by comparing with the standard NMR spectra. See the ESI† for details.
- (a) K. Sonogashira, Y. Tohda and N. Hagiwara, Tetrahedron Lett., 1975, 50, 4467 CrossRef; (b) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866 RSC.
- (a) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (b) X. Li, X. Xu, P. Hu, X. Xiao and C. Zhou, J. Org. Chem., 2013, 78, 7343 CrossRef CAS PubMed; (c) X. Li, X. Xu and Y. Tang, Org. Biomol. Chem., 2013, 11, 1739 RSC; (d) X. Li, X. Xu and X. Shi, Tetrahedron Lett., 2013, 54, 3071 CrossRef CAS PubMed.
- (a) N. O. Calloway, Chem. Rev., 1935, 17, 327 CrossRef CAS; (b) W. Baik, H. J. Lee, J. M. Jang, S. Koo and B. H. Kim, J. Org. Chem., 2000, 65, 108 CrossRef CAS PubMed; (c) Y. Kitayama, M. Yorizane, H. Minami and M. Okubo, Macromolecules, 2012, 45, 2286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, copies of 1H and 13C NMR spectra for selected compounds. See DOI: 10.1039/c3qo00075c |
| This journal is © the Partner Organisations 2014 |