DOI:
10.1039/C3QO00076A
(Research Article)
Org. Chem. Front., 2014,
1, 395-404
1,1′-Bi-2-naphthol-fluoroacetyl compounds in fluorescent recognition of amines†
Received
22nd December 2013
, Accepted 15th March 2014
First published on 17th March 2014
Abstract
Several 1,1′-bi-2-naphthol (BINOL) derivatives containing fluoroacetyl functions were synthesized, which included (S)-4 with two difluoroacetyl groups at the 3,3′-positions of BINOL, (R)-7 with two trifluoroacetyl groups at the 3,3′-positions of a partially hydrogenated BINOL, and (S)-12 containing two trifluoroacetyl groups at the 6,6′-positions. Compounds (S)-4 and (R)-7 were nonfluorescent but (S)-12 gave intense fluorescence. This indicates that intramolecular hydrogen bondings between the BINOL hydroxyl groups and the 3,3′-dicarbonyl groups of (S)-4 and (R)-7 could have effectively quenched their fluorescence. The fluorescence responses of these compounds towards a variety of amines were studied. It was found that (S)-4 showed greatly enhanced fluorescence in the presence of diamines and also highly enantioselective fluorescent response toward a chiral diamine. However, monoamines could not turn on the fluorescence of this compound. Compound (S)-12 showed greatly increased fluorescence quenching by amines in comparison with BINOL.
Introduction
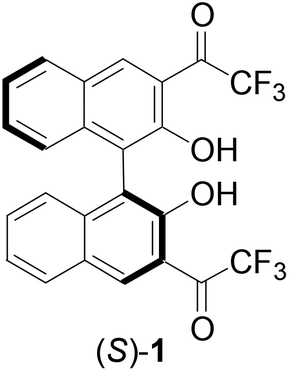
The highly electrophilic property of trifluoromethyl ketones has been utilized in the development of molecular receptors for a variety of nucleophilic species.1–3 Among these studies, the reactions of the trifluoroacetyl groups of these molecules with amines were observed to generate significant changes in spectroscopic signals and allow detection of these substrates.4–7 Recently, we reported that the 1,1′-bi-2-naphthol (BINOL)-based chiral trifluoromethyl ketone (S)-1 exhibited highly enantioselective fluorescent responses toward chiral diamines.8 In addition, this compound also exhibited greatly enhanced fluorescence in the presence of aliphatic diamines but had much smaller or even no fluorescence response toward aromatic diamines and various monoamines. Selective detection of amines is important in environmental, biological and clinical analyses and highly sensitive fluorescent sensors may provide practically useful tools for these areas.9–13 In order to gain additional understanding of the fluorescent recognition of amines by (S)-1, we have synthesized several analogs of (S)-1 by modifying the carbonyl functions or the BINOL units and have compared their fluorescent responses in the presence of amines. These compounds have shown diverse fluorescent responses toward amines and are potentially useful for amine analysis. Herein, these results are reported.
Results and discussion
Synthesis and characterization of the BINOL-fluoroacetyl compounds
We prepared the 3,3′-difluoroacetyl BINOL (S)-4 according to Scheme 1 in order to explore the effect of the reduced electrophilicity of the carbonyl groups on sensing. Protection of the hydroxyl groups of (S)-BINOL with methoxymethyl groups gave compound (S)-2 in 95% yield.14 Treatment of (S)-2 with nBuLi followed by addition of ethyl difluoroacetate gave the 3,3′-diacetyl compound (S)-3 in 41% yield. The methoxymethyl protecting groups of (S)-3 was removed by reaction with trifluoroacetic acid followed by aqueous workup to give the difluoroacetyl compound (S)-4 in 62% yield. The 1H NMR spectrum of (S)-4 in CDCl3 showed a singlet at δ 10.75 for the hydroxyl group of (S)-4, consistent with an intramolecular OH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonding. Its 19F NMR spectrum gave two signals at δ −121.31 (dd, J = 351, 57 Hz) and −119.82 (dd, J = 351, 57 Hz) for the two diastereotopic fluorine atoms in each of the two difluoroacetyl groups. The specific optical rotation of (S)-4 was [α]D = −153.5 (c = 0.21, CHCl3) which is similar to that of (S)-1 previously reported at [α]D = −167.5 (c = 0.355, CHCl3). The difluoroacetyl groups of (S)-4 were expected to be less electrophilic than the trifluoroacetyl groups of (S)-1.
C hydrogen bonding. Its 19F NMR spectrum gave two signals at δ −121.31 (dd, J = 351, 57 Hz) and −119.82 (dd, J = 351, 57 Hz) for the two diastereotopic fluorine atoms in each of the two difluoroacetyl groups. The specific optical rotation of (S)-4 was [α]D = −153.5 (c = 0.21, CHCl3) which is similar to that of (S)-1 previously reported at [α]D = −167.5 (c = 0.355, CHCl3). The difluoroacetyl groups of (S)-4 were expected to be less electrophilic than the trifluoroacetyl groups of (S)-1.
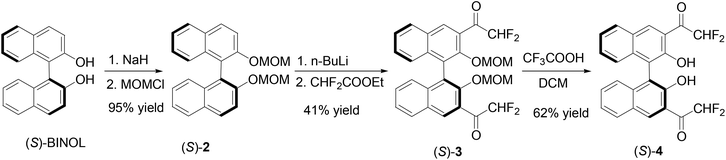 |
| | Scheme 1 Synthesis of 3,3′-di(difluoroacetyl)-BINOL (S)-4. | |
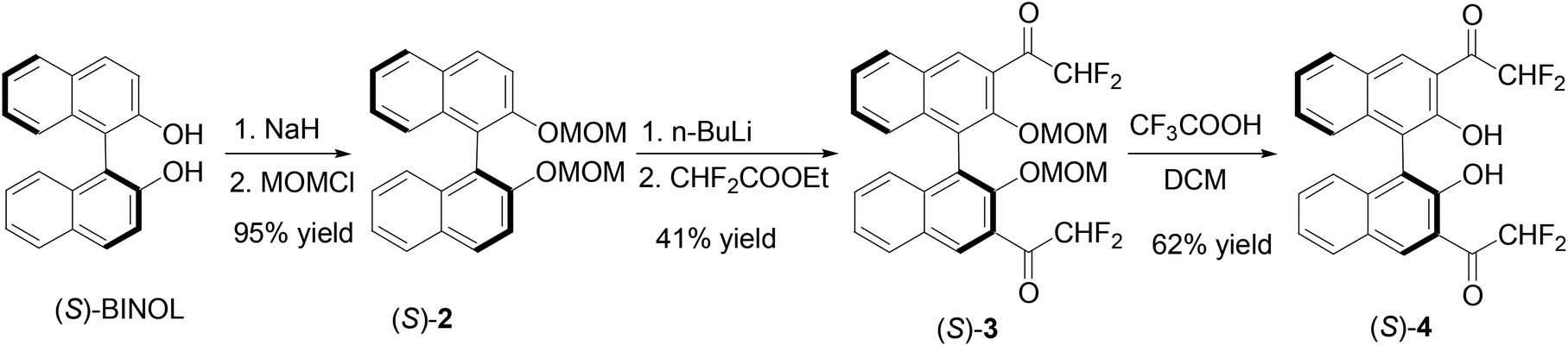
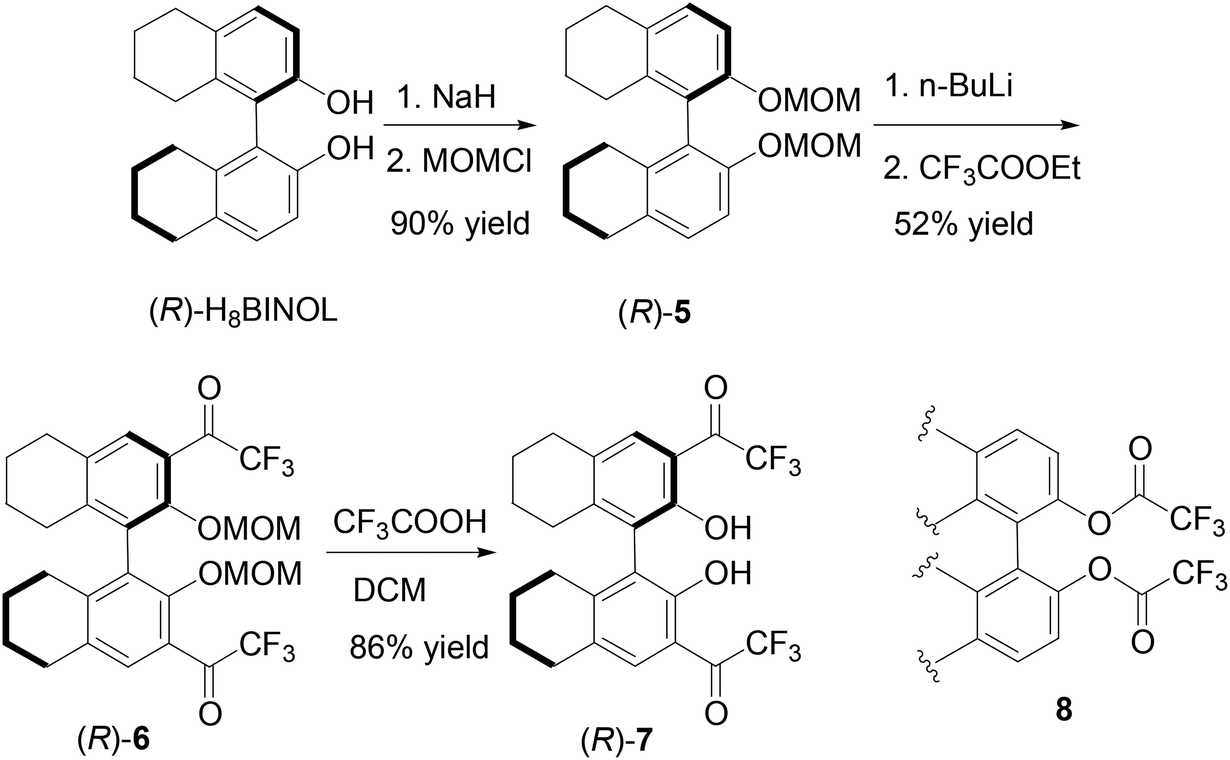
The partially hydrogenated BINOL, H8BINOL, was used to prepare an analog of (S)-1 according to Scheme 2. Protection of the hydroxyl groups of (R)-H8BINOL with methoxymethyl group gave (R)-5 in 90% yield.15 Treatment of (R)-5 with nBuLi followed by addition of ethyl trifluoroacetate gave the 3,3′-diacetyl compound (R)-6 in 52% yield. The methoxymethyl protecting groups of (R)-6 were removed by reaction with trifluoroacetic acid and then aqueous workup to give the H8BINOL analog (R)-7 in 86% yield. The specific optical rotation of (R)-7 was [α]D = −38.5 (c = 0.36, CHCl3). The 1H NMR spectrum of (R)-7 in CDCl3 gave a singlet at δ 11.14 for the hydrogen-bonded hydroxyl protons. The partially hydrogenated BINOL of (R)-7 should make its hydroxyl groups less acidic and its carbonyl groups less electrophilic than those of (S)-1. In addition, the sp3 hybridized carbons in the partially hydrogenated naphthol rings of (R)-7 should also provide a steric environment different from that of (S)-1. A one-step conversion of a fluoroacetate 8 to compounds like (S)-1, (S)-4 or (R)-7 in the presence of a Lewis acid was attempted without success.
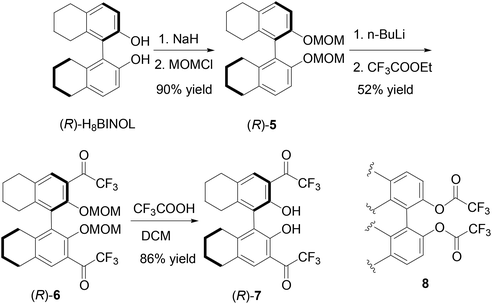 |
| | Scheme 2 Synthesis of 3,3′-di(trifluoroacetyl)-H8BINOL (R)-7. | |
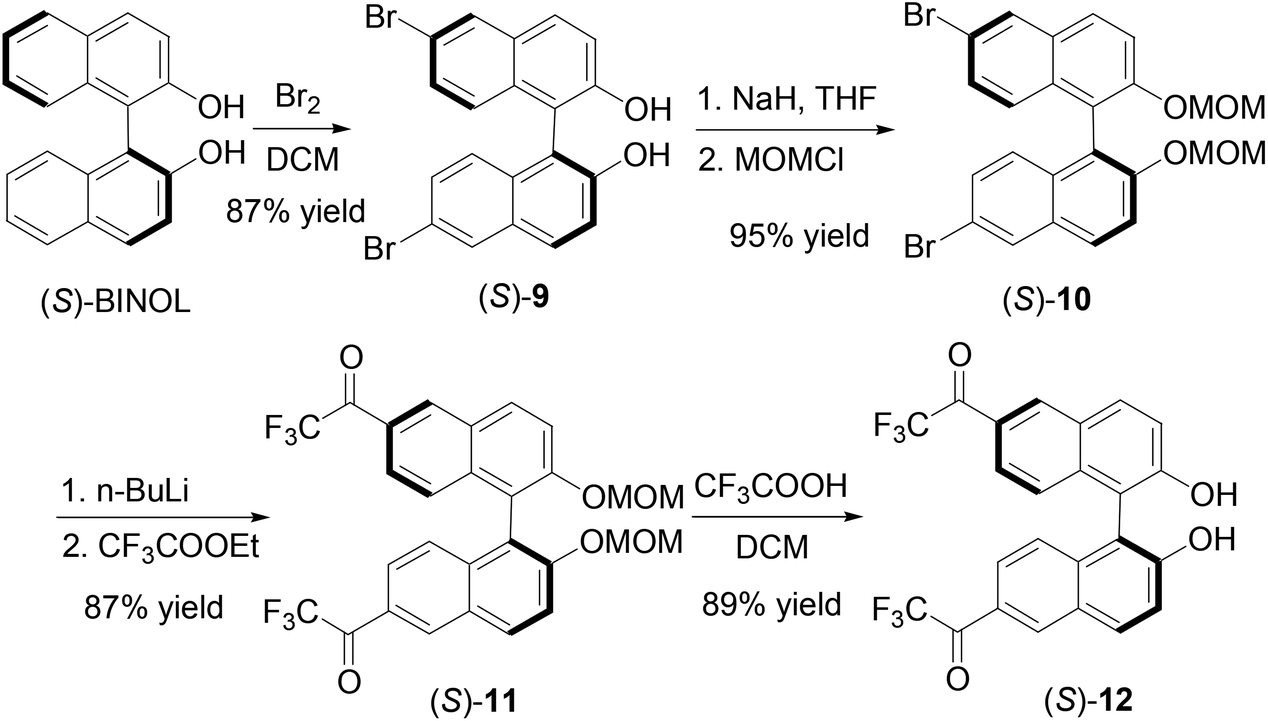
We further introduced trifluoroacetyl group to the 6,6′-positions of BINOL to make compound (S)-12. As shown in Scheme 3, bromination of (S)-BINOL gave the 6,6′-brominated BINOL (S)-9 in 87% yield.16 After protected with the MOM group, the resulting compound (S)-10 was reacted with n-BuLi and ethyl trifluoroacetate to produce (S)-11 in 87% yield. Then trifluoroacetic acid was added to remove the MOM groups to generate (S)-12 in 89% yield. The specific optical rotation of (S)-12 was [α]D = 333.6 (c = 0.16, CHCl3). The 1H NMR spectrum of (S)-12 in CDCl3 showed a singlet at δ 5.33 for the hydroxyl group. This signal is much more upfield than those of the 3,3′-fluoroacetyl-BINOLs (S)-1, (S)-4 and (R)-7 described above because of the absence of the intramolecular OH⋯OC hydrogen bond.
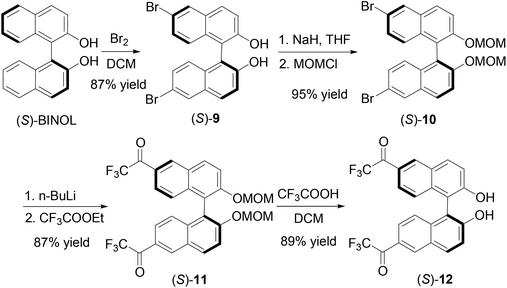 |
| | Scheme 3 Synthesis of 6,6′-di(trifluoroacetyl)-BINOL (S)-12. | |
Comparison of the UV spectra of (S)-4, (R)-7 and (S)-12 with that of (S)-1
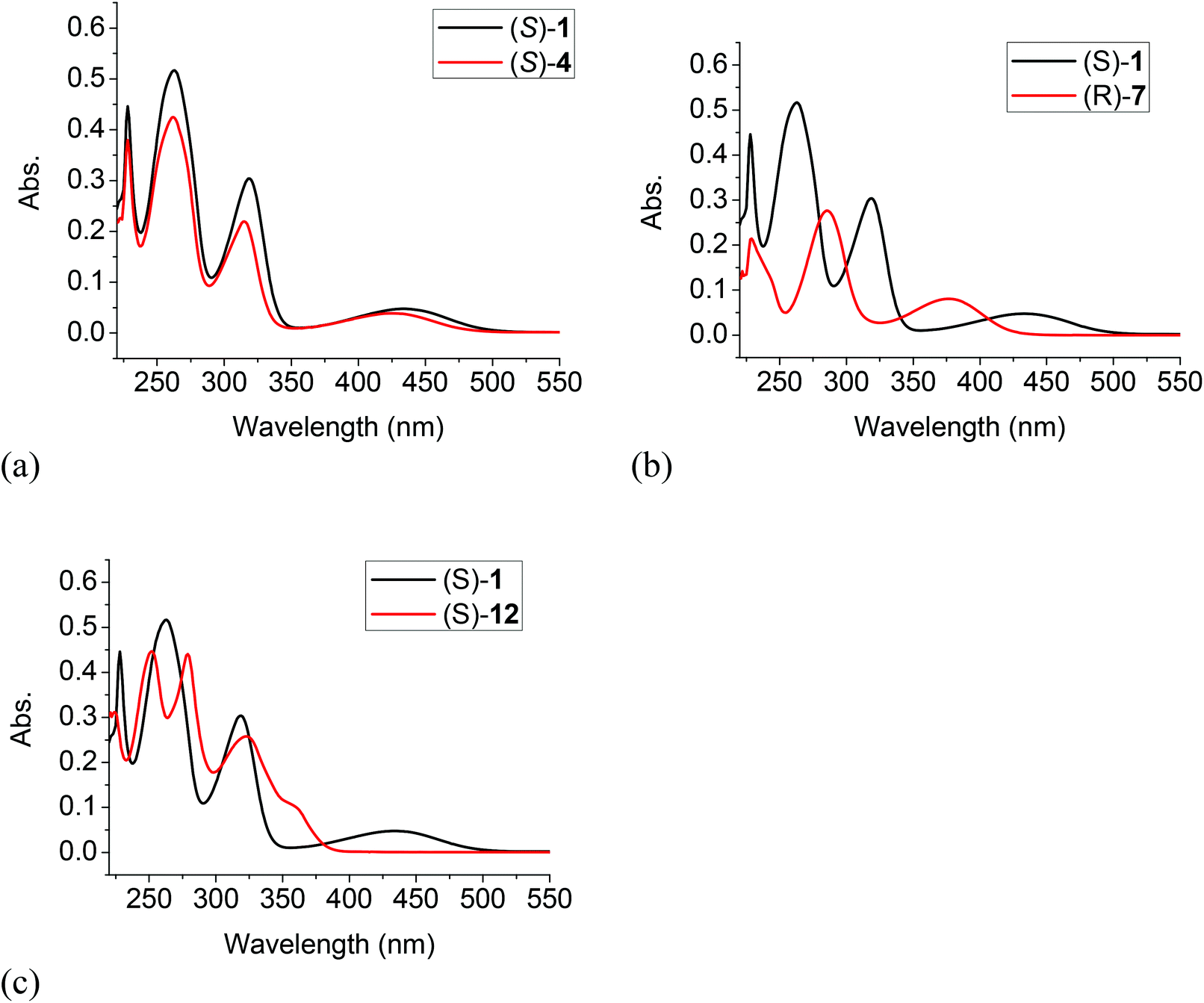
The UV spectra of (S)-4, (R)-7 and (S)-12 in methylene chloride solution were compared with that of (S)-1 in Fig. 1. It shows that the longest wavelength absorption of the difluoroacetyl compound (S)-4 was slightly blue shifted to λmax = 425 nm from that of (S)-1 at λmax = 432 nm since the difluoroacetyl groups of (S)-4 are less efficient electron acceptors in the donor–acceptor conjugated systems of these two compounds. The H8BINOL compound (R)-7 with less extended conjugation due to the partially hydrogenated naphthol rings showed a large blue shift of the longest wavelength absorption to λmax = 377 nm. In the long wavelength absorption of the 6,6′-trifluoroacetyl compound (S)-12, a shoulder was observed at 359 nm which is greatly blue shifted from that of (S)-4. Although both (S)-1 and (S)-12 have the same electron donors and acceptors over a conjugated naphthol ring, without the intramolecular OH⋯OC hydrogen bonding of (S)-1, there was a greater band gap in the absorption of (S)-12. Table 1 summarizes the UV absorption data of these compounds.
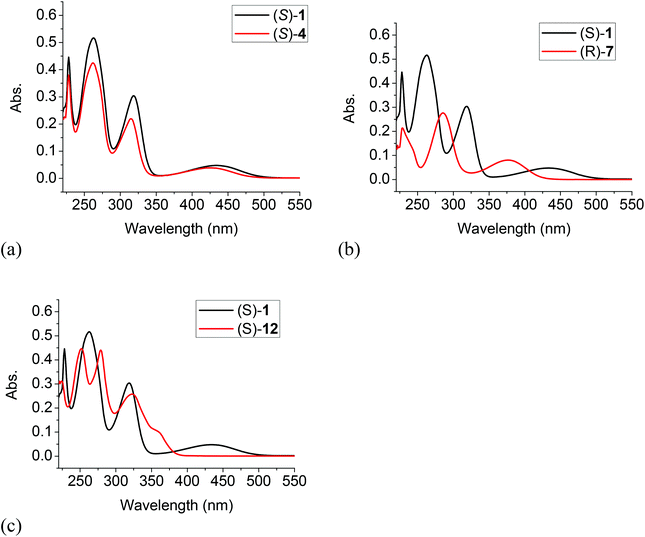 |
| | Fig. 1 Comparison of the UV spectrum of (S)-1 with those of (S)-4 (a), (R)-7 (b) and (S)-12 (c) (1.0 × 10−5 M, CH2Cl2). | |
Table 1 Maximum absorption peaks and extinction coefficients of (S)-1, (S)-4, (R)-7 and (S)-12 (1.0 × 10−5 M in CH2Cl2)
| |
λ
max (ε) |
| (S)-1 |
228 (4.5 × 104), 263 (5.2 × 104), 319 (3.0 × 104), 432 (4.8 × 103) nm |
| (S)-4 |
228 (3.8 × 104), 262 (4.2 × 104), 315 (2.2 × 104), 425 (3.9 × 103) nm |
| (R)-7 |
229 (2.1 × 104), 285 (2.8 × 104), 377 (8.1 × 103) nm |
| (S)-12 |
252 (4.5 × 104), 279 (4.4 × 104), 323 (2.6 × 104), 359 (sh, 1.0 × 104) nm |
Comparison of the fluorescence spectra of (S)-4, (R)-7, and (S)-12 with that of (S)-1
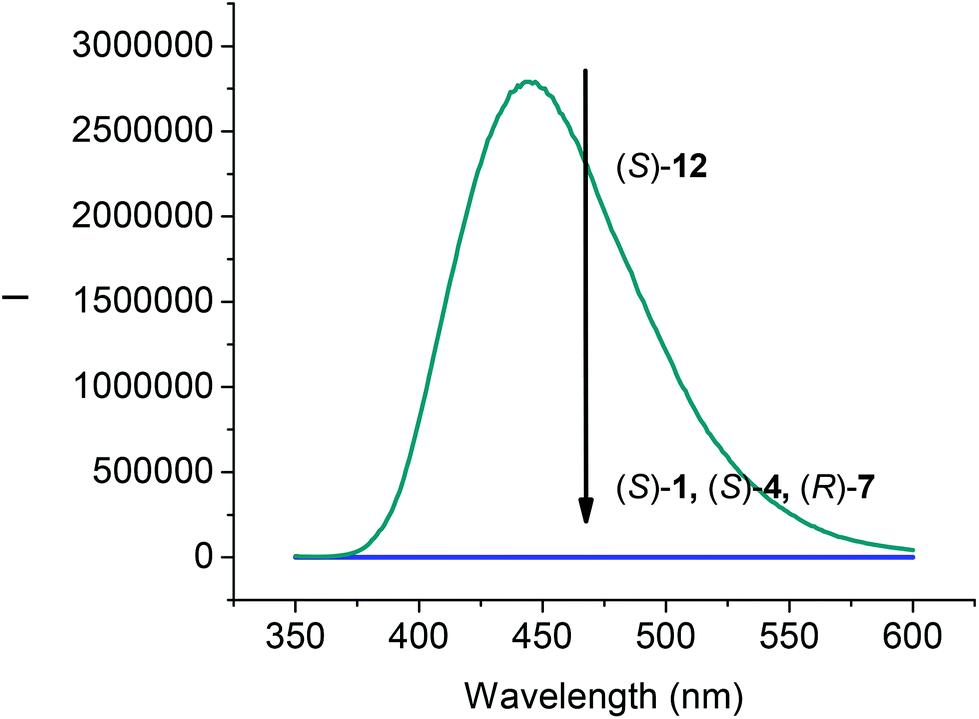
The fluorescence spectra of compounds (S)-4, (R)-7 and (S)-12 in CH2Cl2 at 1.0 × 10−5 M were compared with that of (S)-1. As shown in Fig. 2, all the compounds (S)-1, (S)-4, and (R)-7 that contain the intramolecular OH⋯OC hydrogen bonds were found to be nonemissive. However, the 6,6′-trifluoroacetyl compound (S)-12 showed strong emission at λ = 444 nm. The emission of (S)-12 was significantly red shifted from that of BINOL observed at around 360 nm,17 which could be attributed to the internal charge transfer state arisen from the incorporation of the trifluoroacetyl electron acceptor in (S)-12.
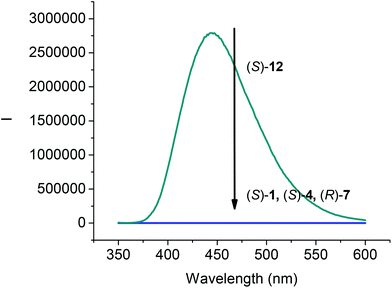 |
| | Fig. 2 Fluorescence Spectra of (S)-1, (S)-4, (R)-7 and (S)-12 (1.0 × 10−5 M) in CH2Cl2. [λexc = 343 nm for (S)-1, (S)-4 and (S)-12; λexc = 290 nm for (R)-7; slit = 2.0/2.0 nm]. | |
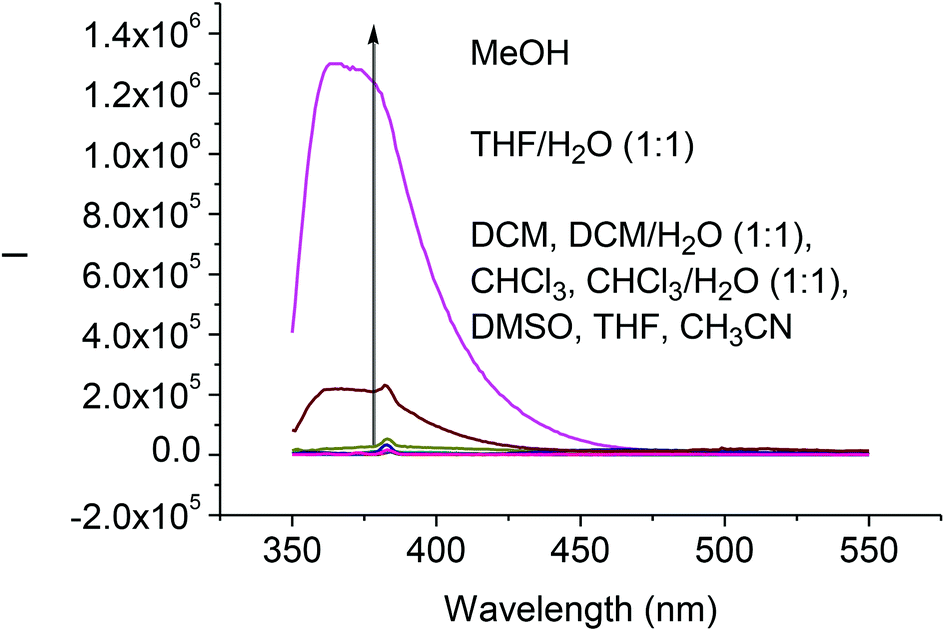
We studied the solvent effect on the fluorescence signals of the difluoroacetyl analog (S)-4. As shown in Fig. 3, addition of water or methanol greatly enhanced the fluorescence of (S)-4. This is similar to what was observed for (S)-1 except that the fluorescence intensity of (S)-4 was much smaller than that of (S)-1 under the same conditions.8b Because of the poor solubility of water in CH2Cl2 and CHCl3, addition of water to these solutions could not enhance the fluorescence of (S)-4. We propose that the fluorescence enhancement of (S)-4 in the presence of water could be attributed to the formation of hydrate upon nucleophilic addition of H2O to the difluoroacetyl groups similar to that proposed for (S)-1.8b
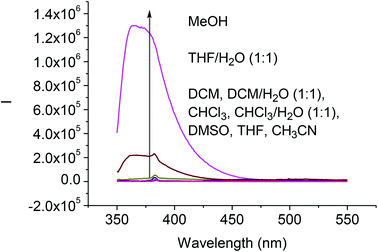 |
| | Fig. 3 Fluorescence spectra of (S)-4 (1.0 × 10−5 M) in various solvents (λexc = 343 nm, slit = 2.0/2.0 nm). | |
Interaction of the BINOL-fluoroacetyl compounds with amines
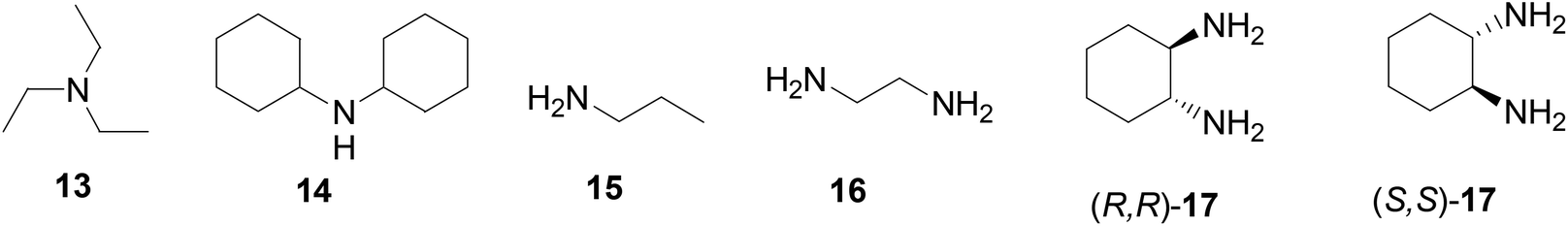
The interactions of the BINOL-fluoroacetyl compounds (S)-4, (R)-7 and (S)-12 (1 × 10−5 M) with several representative amines (Fig. 4) including the tertiary amine 13, the secondary amine 14, the primary amine 15, the 1,2-diamine 16, and the chiral diamine (R,R)- or (S,S)-17 in CH2Cl2 were studied.
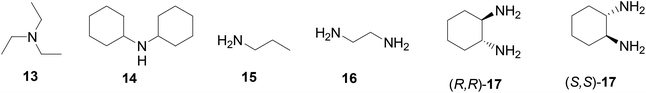 |
| | Fig. 4 Structures of various amines investigated. | |
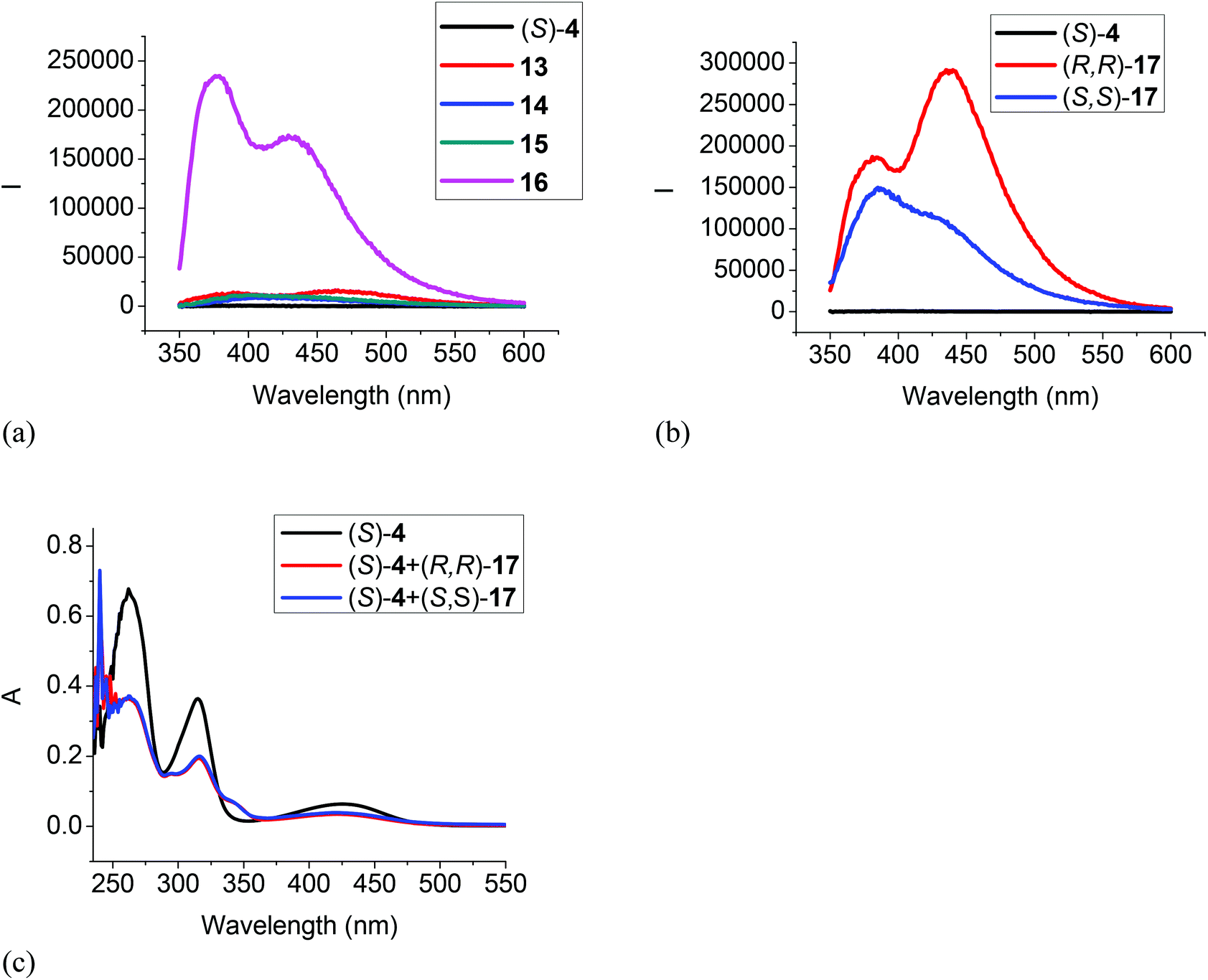
When the difluoroacetyl compound (S)-4 was treated with the monoamines 13–15, only small changes in the fluorescence were observed (Fig. 5a). However, when ethylenediamine 16 was used under the same conditions, large fluorescence enhancement was observed with dual emission signals at λ = 377 and 434 nm. When (S)-4 was treated with the chiral diamine (R,R)-17 (5.0 × 10−3 M), a large fluorescent enhancement was also observed with dual emissions at λ = 385 (λ1) and 438 (λ2) nm with I1/I2 = 0.64 (Fig. 5b). When (S)-4 was treated with (S,S)-17, a similar large fluorescence enhancement at λ1 was also observed, but the fluorescence enhancement at λ2 was much smaller with I1/I2 = 1.47. This demonstrates that (S)-4 can serve as a highly sensitive as well as enantioselective fluorescent sensor for the recognition of the chiral diamine similar to (S)-1.8 It can also be used to distinguish the aliphatic 1,2-diamines from monoamines. Fig. 5c gives the UV absorption responses of (S)-4 toward the enantiomers of the chiral diamine. It shows that when (S)-4 (1.0 × 10−5 M in CH2Cl2) was treated with (R,R)- or (S,S)-17 (5.0 × 10−3 M), there were large absorption decreases at λmax = 262, 315 and 425 nm and a new absorption at λmax = 345 nm, but no enantioselectivity was observed.
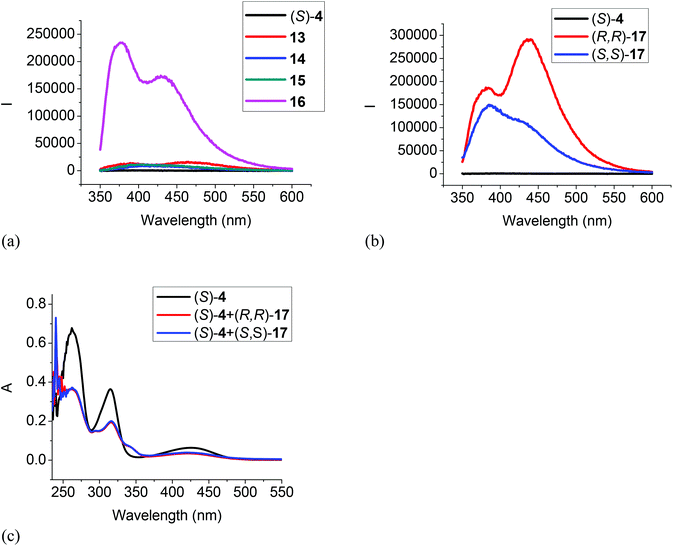 |
| | Fig. 5 Fluorescence spectra of (S)-4 in the presence of amines 13–16 (a), and (R,R)- or (S,S)-17 (b) (λexc = 343 nm, slit = 3.0/3.0 nm). UV spectra of (S)-4 in the presence of (R,R)- or (S,S)-17 (c) [(S)-4: 1.0 × 10−5 M in CH2Cl2; amine: 5.0 × 10−3 M]. | |
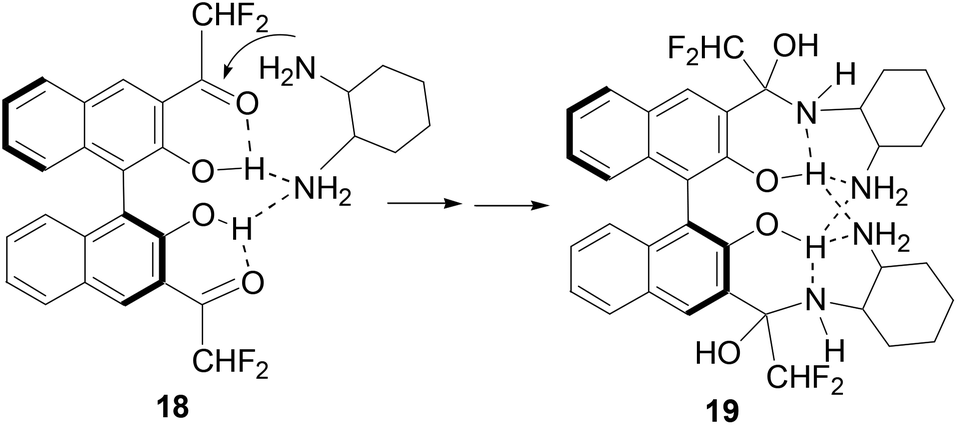
Previously, we observed that (S)-1 reacted with cyclohexane-1,2-diamine to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 adduct.8b On the basis of our study on the reaction of (S)-1 with amines, we propose the following mechanism to account for the reaction and fluorescent responses of (S)-4 with the diamines. As shown in Scheme 4, the intermolecular hydrogen bond between one amine group of a diamine molecule with the hydroxyl groups of (S)-4 in 18 should accelerate the addition of the second amine group to the difluoroacetyl group to generate the highly fluorescent hemiaminal product 19. The fluorescent enhancement is mainly ascribed to the disruption of the original intramolecular OH⋯OC hydrogen bonding. The enantioselectivity in the fluorescent response of (S)-4 could be attributed to the different stability of the diastereomeric isomers of 19 when (S)-4 reacts with the two enantiomers of the diamine 17.
:2 adduct.8b On the basis of our study on the reaction of (S)-1 with amines, we propose the following mechanism to account for the reaction and fluorescent responses of (S)-4 with the diamines. As shown in Scheme 4, the intermolecular hydrogen bond between one amine group of a diamine molecule with the hydroxyl groups of (S)-4 in 18 should accelerate the addition of the second amine group to the difluoroacetyl group to generate the highly fluorescent hemiaminal product 19. The fluorescent enhancement is mainly ascribed to the disruption of the original intramolecular OH⋯OC hydrogen bonding. The enantioselectivity in the fluorescent response of (S)-4 could be attributed to the different stability of the diastereomeric isomers of 19 when (S)-4 reacts with the two enantiomers of the diamine 17.
 |
| | Scheme 4 Proposed mechanism for the reaction of (S)-4 with the chiral diamine 17. | |
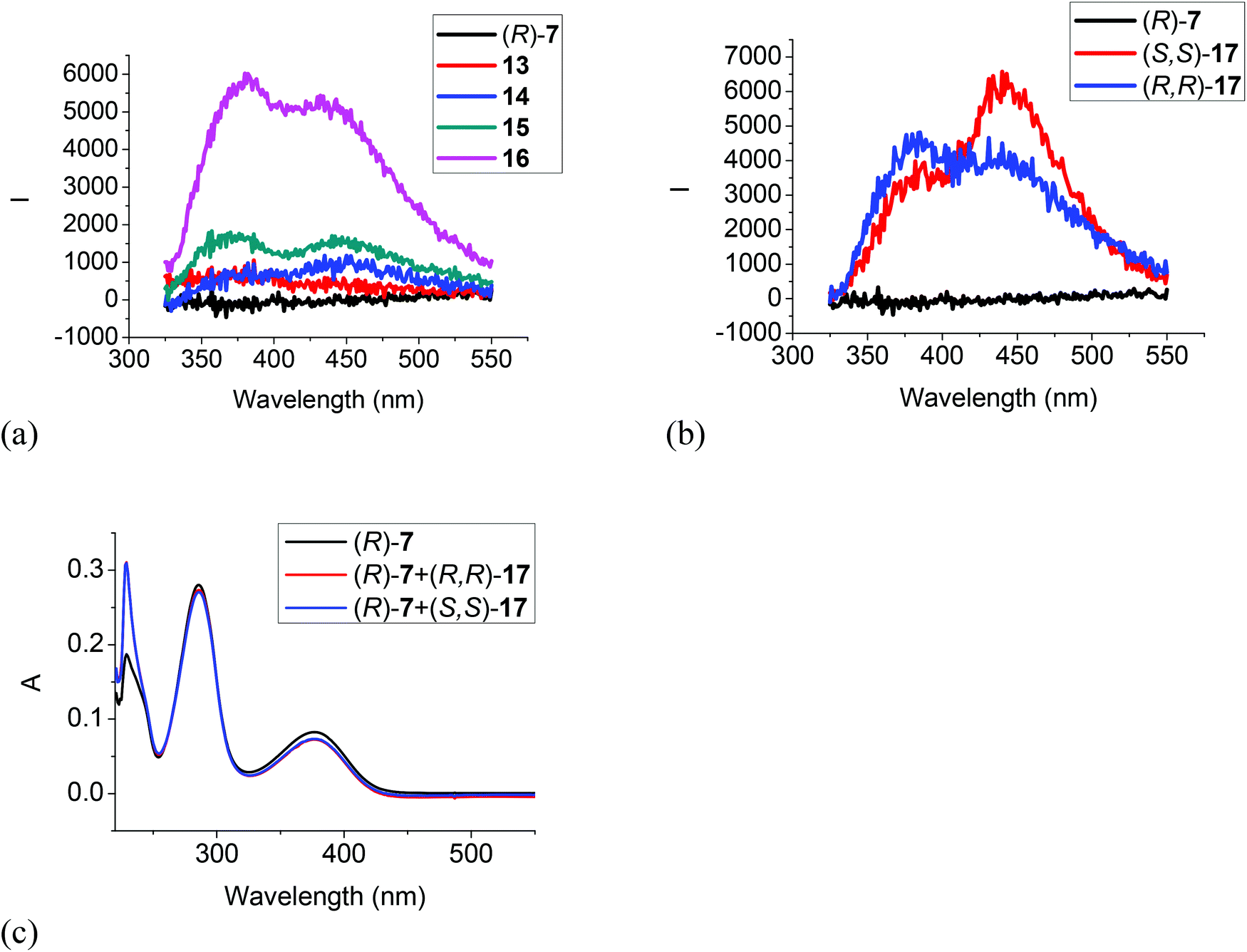
Fig. 6 gives the fluorescence and UV responses of the H8BINOL analogue (R)-7 toward the amines. It shows that monoamines 13, 14 and 15 caused very little change on the fluorescence of this compound (Fig. 6a). Ethylenediamine 16 turned on the fluorescence of (R)-7 with dual emissions at 380 and 435 nm, and the chiral diamine (R,R)- and (S,S)-17 turned on the fluorescence of (R)-7 with dual emissions at 380 nm and 443 nm (Fig. 6a and b). (R)-7 also showed enantioselective response toward (R,R)- and (S,S)-17 but the fluorescent intensity was low. The UV spectrum of (R)-7 in the presence of 500 equiv. of the chiral diamine (R,R)- and (S,S)-17 only gave very small changes (Fig. 6c). These observations demonstrate that the less electrophilic trifluoroacetyl groups of (R)-7 and its less acidic hydroxyl groups due to the partially hydrogenated naphthol rings make it less reactive with the diamines.
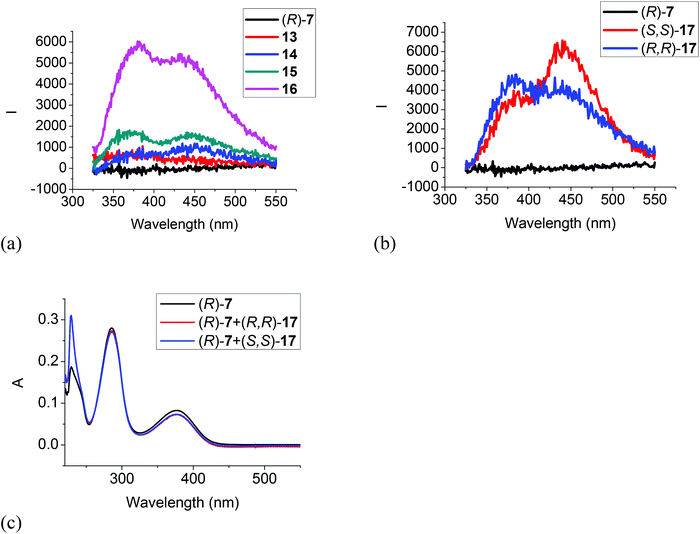 |
| | Fig. 6 Fluorescence spectra of (R)-7 in the presence of amines 13–16 (a), and (R,R)- or (S,S)-17 (b) (amine: 1.0 × 10−3 M) (λexc = 286 nm, slit = 2.0/2.0 nm). UV spectra of (R)-7 in the presence of (R,R)- and (S,S)-17 (5.0 × 10−3 M) (c) [(R)-7: 1.0 × 10−5 M in CH2Cl2]. | |
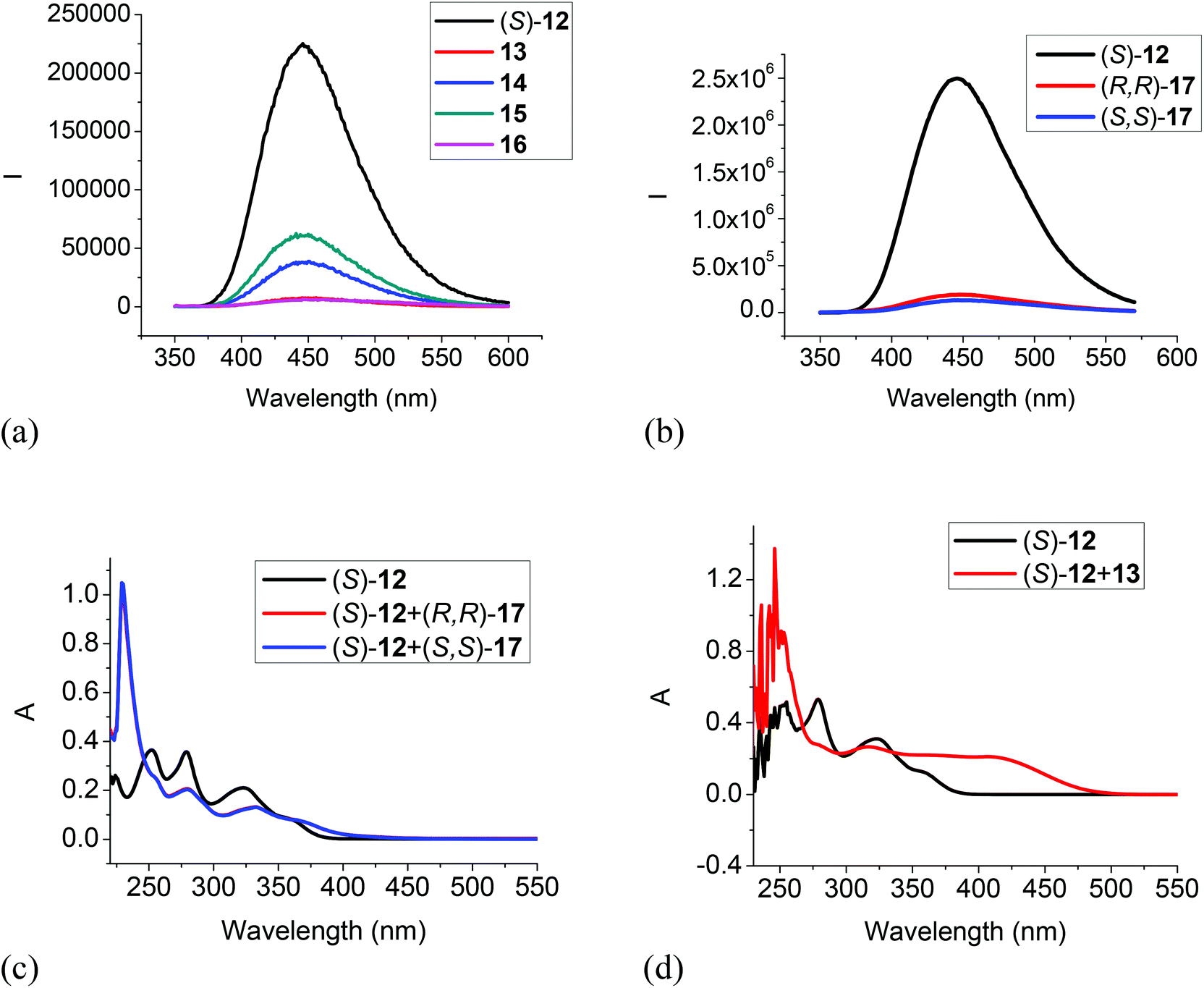
The 6,6′-trifluoroacetyl analog (S)-12 exhibited significant fluorescent quenching in the presence of all of the tested amines, in which chiral diamine 17 quenched over 90% of its fluorescence with almost no enantioselectivity (Fig. 7a and b). The extents of the fluorescence quenching of (S)-12 by the amines in Fig. 7a and b are listed below: 13 quenched 97%; 14 quenched 83%; 15 quenched 73%; 16 quenched 98%; (S,S)-17 quenched 95% and (R,R)-17 quenched 92%. The differences of the fluorescence quenching efficiency should be related to how strongly the amines are interacting with the fluorophore. Since (S)-12 does not have the intramolecular OH⋯OC hydrogen bonds of (S)-1 and (S)-4, it is strongly emissive. It was reported by Iwanek and Mattay that the fluorescence of BINOL can be quenched by amines in various solvents.18 Our observed fluorescence quenching of (S)-12 by the amines appears to be much more efficient than that of BINOL by amines. It was proposed that the fluorescence quenching of BINOL by amines could be attributed to the formation of hydrogen bonded complexes between the BINOL hydroxyl groups and the amine nitrogen as well as the corresponding excited state proton transfer complex. The increased fluorescent quenching of (S)-12 by the amines could be attributed to the increased acidity of the hydroxyl groups of (S)-12 caused by the two electron-withdrawing 6,6′-trifluoroacetyl groups which should increase their interaction with the amine bases. Fig. 7c gives the UV responses of (S)-12 when treated with (S,S)- and (R,R)-17. It shows an absorption increase at 228 nm, decreases at 251, 279 and 323 nm, and a new absorption at 370 nm. No enantioselectivity was observed in the UV responses. Fig. 8d gives the UV responses of (S)-12 when treated with the tertiary amine 13. It shows an absorption increase at 254 nm, decreases at 278 and 323 nm, and a new broad absorption greater than 400 nm. The new long wavelength absorption of (S)-12 indicates the deprotonation of (S)-12 by the stronger base of triethylamine 13.
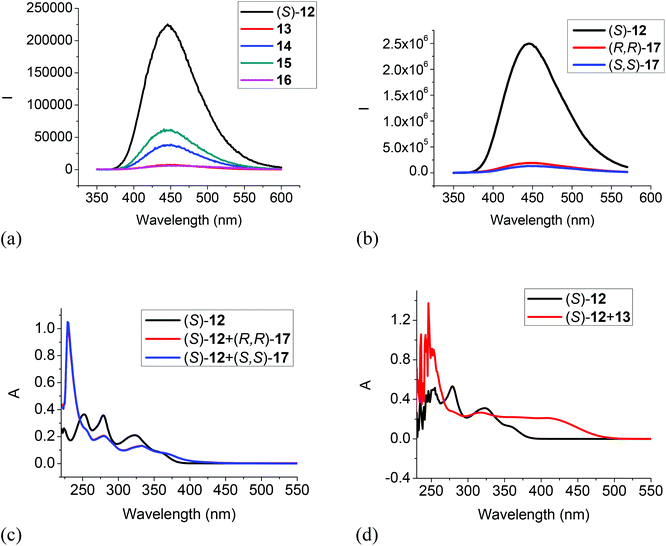 |
| | Fig. 7 Fluorescence spectra of (S)-12 in the presence of amines 13–16 (a), and (R,R)- or (S,S)-17 (b). [λexc = 343 nm, slit = 1.0/1.0 nm for 13–16, and slit = 2.0/2.0 nm for (R,R)- or (S,S)-17]. UV spectra of (S)-12 in the presence of (R,R)- and (S,S)-17 (c) and 13 (d) [(S)-12: 1.0 × 10−5 M in CH2Cl2; amine: 5.0 × 10−3 M]. | |
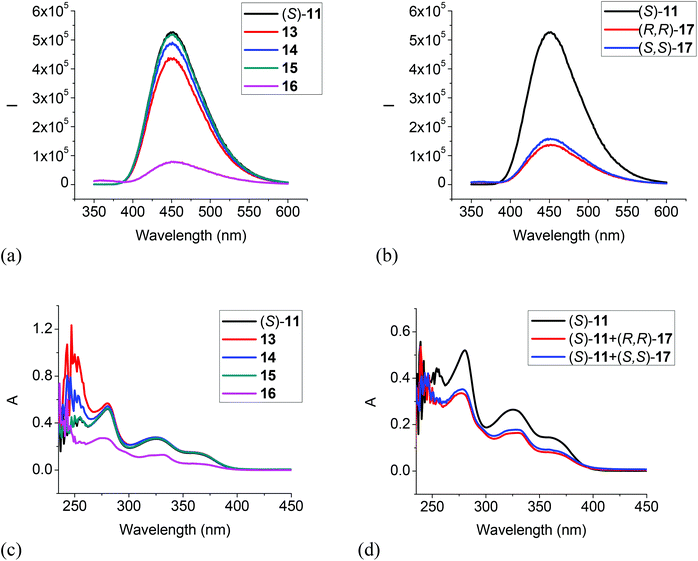 |
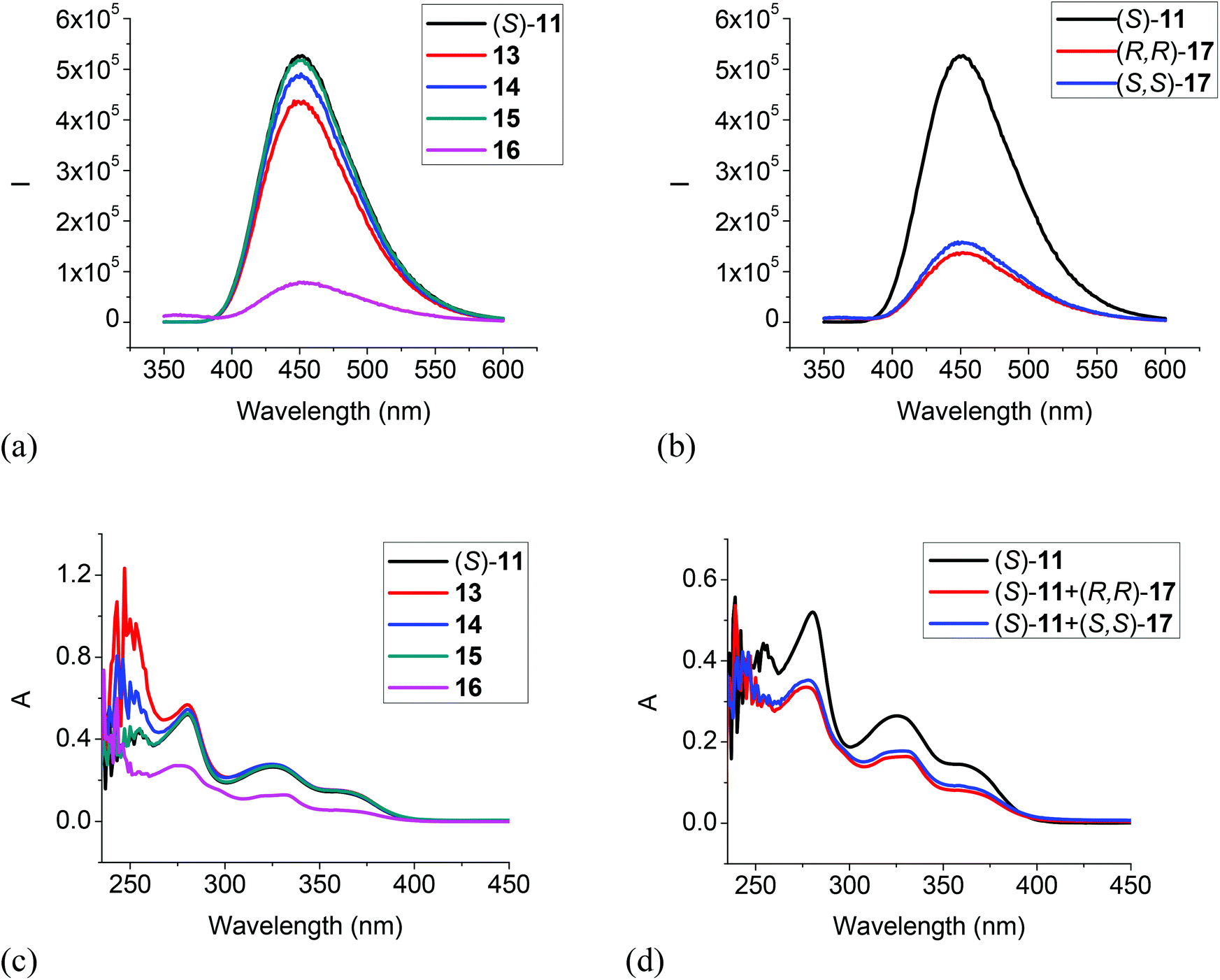
| | Fig. 8 Fluorescence spectra of (S)-11 in the presence of 13–16 (a), and (R,R)- or (S,S)-17 (b). (λexc = 343 nm, slit = 1.0/1.0 nm). UV spectra of (S)-11 in the presence of 13–16 (c), and (R,R)- or (S,S)-17 (d) [(S)-11: 1.0 × 10−5 M in CH2Cl2; amine: 5.0 × 10−3 M]. | |
We also studied the UV and fluorescent responses of the MOM protected compound (S)-11. As Fig. 8a and b show, the monoamines 13, 14 and 15 had very little effect on the fluorescence of (S)-11, but the diamines 16 and 17 could still significantly quench the fluorescence though to a smaller extent than that of (S)-12 by the diamines. The UV spectra in Fig. 8c and d are consistent with the fluorescent responses, that is, the monoamines did not change the UV absorption of (S)-11 but the diamines 16 and 17 significantly decreased the absorption. These observations could be attributed to a more favorable reaction for the diamines with the trifluoroacetyl groups of (S)-11 than the monoamines. This study of the interaction of (S)-11 with the amines also suggests that the fluorescent quenching of (S)-12 by the monoamines should be mainly due to the interaction of the acidic hydroxyl groups with the amine bases, but the diamines 16 and 17 could interact with both the hydroxyl groups and the trifluoroacetyl groups to quench the fluorescence of (S)-12. Thus, the fluorescent quenching of (S)-12 by the diamines is more efficient than that of (S)-11.
Summary
We have synthesized and studied several BINOL-fluoroacetyl compounds for the fluorescent recognition of amines. The difluoroacetyl compound (S)-4 showed large fluorescence enhancement in the presence of diamines and highly enantioselective responses were observed toward a chiral diamine. However, the primary, secondary and tertiary monoamines cannot turn on the fluorescence of this compound. When the carbonyl groups are moved from the 3,3′-positions of the BINOL unit of (S)-1, (S)-4 and (R)-7 to the 6,6′-positions of (S)-12, greatly enhanced fluorescence was observed. This demonstrates that the intramolecular H⋯OC hydrogen bonding in the 3,3′-dicarbonyl compounds of the BINOL derivatives should be responsible for their fluorescent quenching. (S)-12 showed efficient fluorescent quenching in the presence of the mono and diamines. Thus, the 6,6′-substituted analog gave the opposite fluorescent responses of the 3,3′-substituted compounds. The H8BINOL derivative (R)-7 showed much weaker fluorescent responses. This work has demonstrated that the steric and electronic properties of the carbonyl groups of the BINOL-based compound can greatly influence their fluorescent responses in amine recognition.
Experimental section
General data
THF was distilled over sodium and benzophenone under nitrogen atmosphere. Methylene chloride and diethyl ether were dried by passing through activated alumina columns under nitrogen. Solvents were stored over 4 Å molecular sieves.
Melting points were uncorrected and obtained on a Mel-Temp II capillary melting point apparatus. Optical rotations were measured on a Jasco P-2000 digital polarimeter. NMR spectra were recorded on a Varian-300 MHz or Bruker-600 MHz spectrometer. Chemical shifts for 1H NMR spectra were reported in parts per million relative to a singlet at 7.26 ppm for deuterated chloroform. Chemical shifts for 13C NMR were reported relative to the centerline of a triplet at 77.16 ppm for deuterated chloroform. The 19F NMR spectra were reported in units of part per million (ppm) relative to trifluoroacetic acid (δ −76.55 ppm) as an external reference. High resolution mass spectra were obtained from the University of Illinois at Urbana-Champaign (UIUC) Mass Spectrometry Facility. UV-Vis spectra were recorded on a Hewlett-Packard 8452A diode-array spectrophotometer. Steady-state fluorescence emission spectra were recorded on a Horiba FluoroMax-4 spectrofluorometer.
Preparation of samples for fluorescence measurement
Materials.
The molecular receptors were purified by column chromatography followed by recrystallization and then stored in a refrigerator. All of the solvents were either HPLC or spectroscopic grade. The stock solutions of the receptors were freshly prepared for each measurement. A 0.01 M stock solution of an amine substrate in methylene chloride was freshly prepared. For the fluorescent study, a receptor solution was mixed with the amine solution at room temperature in a 5 mL volumetric flask and diluted to the desired concentration. The resulting solution was allowed to stand at room temperature for 0.5 h before the fluorescent measurement and all the fluorescence spectra were taken within 2 h.
Synthesis and characterization of compounds
Synthesis and characterization of (S)-3.
Under nitrogen, 2,2′-bis(methoxymethoxy)-1,1′-binaphthalene, (S)-2 (3.0 mmol, 1.12 g), was dissolved in diethyl ether (36 mL). The solution was cooled to 0 °C, and n-BuLi (10.5 mmol, 2.5 M in hexane, 4.2 mL) was added dropwise. The reaction mixture was stirred for 3 h at room temperature and cooled to 0 °C, and then ethyl difluoroacetate (12.0 mmol, 1.3 mL) was added slowly. The reaction mixture was allowed to warm to room temperature and stirred for 2 h to afford a cream-like mixture. A saturated aqueous NH4Cl solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (1/2) to afford compound (S)-3 as a yellow oil in 41% yield (1.23 mmol, 652 mg). 1H NMR (300 MHz, CDCl3) δ 2.90 (s, 6H), 4.60 (d, J = 5.7 Hz, 2H), 4.66 (d, J = 5.7 Hz, 2H), 6.69 (t, J = 54 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.44 (m, 2H), 7.54 (m, 2H), 8.05 (d, J = 8.1 Hz, 2H), 8.44 (s, 2H). 19F NMR (282 MHz, CDCl3) δ −127.93 (dd, J = 321, 54 Hz, 2F), −126.57 (dd, J = 321, 54 Hz, 2F). 13C NMR (75 MHz, CDCl3) δ 57.0, 100.2, 110.0 (t, J = 249 Hz), 125.4, 125.9, 126.6, 129.4, 129.7, 129.9, 130.0, 133.1, 136.0, 151.0, 190.6 (t, J = 25.5 Hz). HRMS Calcd for C28H22O6F4Na (MNa+): 553.1250, Found: 553.1259. [α]D = 31.9 (c = 0.19, CHCl3).
Synthesis and characterization of (S)-4.
After compound (S)-3 (0.94 mmol, 500 mg) was dissolved in a minimum amount of CH2Cl2, trifluoroacetic acid (1.0 mL) was added slowly, and the mixture was stirred at room temperature for 1 h. A saturated aqueous NaHCO3 solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (1/1) to afford compound (S)-4 as a light yellow solid in 62% yield (0.58 mmol, 258 mg). 1H NMR (600 MHz, CDCl3) δ 6.53 (t, J = 53.4 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 7.40–7.46 (m, 4H), 8.00 (d, J = 7.8 Hz, 2H), 8.76 (s, 2H), 10.75 (s, 2H). 19F NMR (282 MHz, CDCl3) δ −121.31 (dd, J = 351, 57 Hz, 2F), −119.82 (dd, J = 351, 57 Hz, 2F). 13C NMR (150 MHz, CDCl3) δ 111.3 (t, J = 252 Hz), 116.6, 117.6, 124.7, 124.9, 127.2, 130.8, 131.6, 135.4 (t, J = 4.5 Hz), 138.1, 154.8, 192.2 (t, J = 25.5 Hz). HRMS Calcd for C24H15O4F4 (MH+): 443.0906, Found: 443.0907. m.p. 271 °C. [α]D = −153.5 (c = 0.21, CHCl3).
Synthesis and characterization of (R)-6.
(1) Synthesis of (R)-5. Under nitrogen, (R)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl-2,2′-diol (H8BINOL) (25 mmol, 7.4 g) was dissolved in THF (300 mL). The solution was cooled to 0 °C, and NaH (62.8 mmol, 60% in mineral oil, 2.52 g) was added in small portions. The reaction mixture was stirred for 30 min and then chloromethyl methyl ether (62.8 mmol, 4.8 mL) was added slowly. The reaction mixture was allowed to warm to room temperature and stirred for overnight. Water was added slowly to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–ethyl acetate (15/1) to afford compound (R)-5 as a white solid in 90% yield (22.5 mmol, 8.6 g). 1H NMR (300 MHz, CDCl3) δ 1.65–1.76 (m, 8H), 2.07–2.17 (m, 2H), 2.27–2.37 (m, 2H), 2.78 (t, J = 6.0 Hz, 4H), 3.30 (s, 6H), 4.97 (d, J = 6.6 Hz, 2H), 5.03 (d, J = 6.6 Hz, 2H), 6.99 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H). (2) Synthesis of (R)-6. Under nitrogen, (R)-2,2′-bis(methoxymethoxy)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl, (R)-5 (4.4 mmol, 1.7 g), was dissolved in diethyl ether (50 mL). The solution was cooled to 0 °C, and n-BuLi (17.8 mmol, 2.5 M in hexane, 7.1 mL) was added dropwise. The reaction mixture was stirred for 4 h at room temperature and cooled to 0 °C, and then ethyl trifluoroacetate (20.0 mmol, 2.4 mL) was added slowly. The reaction mixture was allowed to warm to room temperature and stirred for 2.5 h to afford a cream-like mixture. A saturated aqueous NH4Cl solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (1/3) to afford compound (R)-6 as a yellow oil in 52% yield (2.3 mmol, 1.31 g). 1H NMR (300 MHz, CDCl3) δ 1.65–1.82 (m, 8H), 2.12–2.22 (m, 2H), 2.47–2.57 (m, 2H), 2.83–2.87 (m, 10H), 4.79 (d, J = 6.6 Hz, 2H), 4.83 (d, J = 6.6 Hz, 2H), 7.49 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 22.5, 28.5, 29.6, 56.2, 100.8, 116.4 (q, J = 290 Hz), 123.2, 130.47, 130.51, 132.2, 133.9, 145.4, 153.5, 181.4 (q, J = 34.5 Hz). HRMS Calcd for C28H28O6F6Na (MNa+): 597.1688, Found: 197.1678. [α]D = 43.34 (c = 0.39, CHCl3).
Synthesis and characterization of (R)-7.
After compound (R)-6 (237.8 mg, 0.41 mmol) was dissolved in a minimum amount of CH2Cl2, trifluoroacetic acid (1.0 mL) was added slowly, and the mixture was stirred at room temperature for 10 min. A saturated aqueous NaHCO3 solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (2/1) to afford compound (R)-7 as a yellow solid in 86% yield (0.35 mmol, 172 mg). 1H NMR (300 MHz, CDCl3) δ 1.71–1.88 (m, 8H), 2.25–2.35 (m, 2H), 2.50–2.60 (m, 2H), 2.88 (m, 4H), 7.67 (s, 2H), 11.1 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 22.4, 22.6, 28.3, 29.4, 112.2, 116.8 (q, J = 288 Hz), 124.4, 129.9, 130.6 (q, J = 3.0 Hz), 150.0, 159.4, 184.1 (q, J = 34.5 Hz). HRMS Calcd for C24H21O4F6 (MH+): 487.1344, Found: 487.1338. m.p. 209 °C. [α]D = −38.5 (c = 0.36, CHCl3).
Synthesis and characterization of (S)-9.
(S)-BINOL (14.7 mmol, 4.2 g) was dissolved in CH2Cl2 (150 mL) and the solution was cooled to −78 °C. Br2 (37.8 mmol, 2 mL) was added dropwise. After the reaction mixture was stirred for 2.5 h, it was warmed up to room temperature and stirred for additional 30 min. 10% NaHSO3 solution (100 mL) was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–ethyl acetate (4/1) to afford compound (S)-9 as a white solid in 87% yield (12.8 mmol, 5.68 g). The NMR data of (S)-9 matched those of the literature.19
Synthesis and characterization of (S)-10.
Under nitrogen, (S)-9 (8.0 mmol, 3.55 g) was dissolved in THF (150 mL). The solution was cooled to 0 °C, and NaH (20 mmol, 60% in mineral oil, 0.8 g) was added in small portions. The reaction mixture was stirred for 30 min and then chloromethyl methyl ether (20 mmol, 1.5 mL) was added slowly. The reaction mixture was allowed to warm to room temperature and stirred for 1 h. Water was added slowly to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (three 30 mL aliquots). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–ethyl acetate (10/1) to afford compound (S)-10 as a white solid in 95% yield (7.6 mmol, 4.04 g). The NMR data of (S)-10 matched those reported.16
Synthesis and characterization of (S)-11.
Under nitrogen, (S)-10 (1.0 mmol, 532 mg) was dissolved in THF (15 mL). The solution was cooled to −78 °C, and n-BuLi (4.4 mmol, 2.5 M in hexane, 1.8 mL) was added dropwise. The reaction mixture was stirred for additional 3 h at the same temperature, and then ethyl trifluoroacetate (6.6 mmol, 0.8 mL) was added slowly. The reaction mixture was stirred at −78 °C for 30 min and then at rt for 30 min. A saturated aqueous NH4Cl solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (1/2) to afford compound (S)-11 as a light yellow oil in 87% yield (0.87 mmol, 493 mg). 1H NMR (300 MHz, CDCl3) δ 3.24 (s, 6H), 5.12 (d, J = 7.2 Hz, 2H), 5.21 (d, J = 7.2 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 7.77 (d, J = 9.0 Hz, 2H), 7.88 (d, J = 9.0 Hz, 2H), 8.20 (d, J = 9.0 Hz, 2H), 8.72 (s, 2H). 19F NMR (282 MHz, CDCl3) δ −71.63. 13C NMR (75 MHz, CDCl3) δ 56.2, 94.5, 117.0 (t, J = 290 Hz), 117.6, 119.8, 125.1, 125.6, 126.1, 128.4, 132.7, 133.7, 137.4, 156.2, 180.2 (t, J = 34.5 Hz). HRMS Calcd for C28H20O6F6Na (MNa+): 589.1062, Found: 589.1063. [α]D = 36.35 (c = 0.305, CHCl3).
Synthesis and characterization of (S)-12.
After compound (S)-11 (320 mg, 0.56 mmol) was dissolved in a minimum amount of CH2Cl2, trifluoroacetic acid (1 mL) was added slowly, and the mixture was stirred at room temperature for 2 h. A saturated aqueous Na2CO3 solution was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine, and dried over Na2SO4. After evaporation of the solvent, the residue was purified by column chromatography on silica gel eluted with hexane–methylene chloride (2/1) to afford compound (S)-12 as a light yellow solid in 89% yield (0.50 mmol, 238 mg). 1H NMR (300 MHz, CDCl3) δ 5.33 (s, 2H), 7.19 (d, J = 8.7 Hz, 2H), 7.53 (d, J = 9.0 Hz, 2H), 7.92 (d, J = 9.0 Hz, 2H), 8.21 (d, J = 9.0 Hz, 2H), 8.69 (s, 2H). 19F NMR (282 MHz, CDCl3) δ −71.78. 13C NMR (150 MHz, DMSO-d6) δ 115.0, 116.8 (t, J = 291 Hz), 120.2, 123.3, 124.4, 125.3, 126.7, 132.5, 133.6, 137.7, 179.1 (t, J = 33 Hz). [α]D = 333.6 (c = 0.16, CHCl3). m.p. 244 °C. HRMS Calcd for C24H13O4F6 (MH+): 479.0718, Found: 479.0714.
Acknowledgements
Partial supports of this work from the US National Science Foundation (CHE-0717995 and CHE-1047104) are gratefully acknowledged.
References
- H. B. Herman and G. A. Rechnitz, Science, 1974, 184, 1074–1075 CAS.
- M. E. Meyerhoff, E. Pretsch, D. H. Welti and W. Simon, Anal. Chem., 1987, 59, 144–150 CrossRef CAS.
-
(a) K. Wang, K. Seiler, J.-P. Haug, B. Lehmann, S. W. K. Hartman and W. Simon, Anal. Chem., 1991, 63, 970–974 CrossRef CAS;
(b) K. Seiler, K. Wang, M. Kuratli and W. Simon, Anal. Chim. Acta, 1991, 244, 151–160 CrossRef CAS.
- G. J. Mohr, N. Tirelli, C. Lohse and U. E. Spichiger-Keller, Adv. Mater., 1998, 10, 1353–1357 CrossRef CAS.
- E. Mertz and S. C. Zimmerman, J. Am. Chem. Soc., 2003, 125, 3424–3425 CrossRef CAS PubMed.
-
(a) S.-i. Sasaki, Y. Kotegawa and H. Tamiaki, Tetrahedron Lett., 2006, 47, 4849–4852 CrossRef CAS;
(b) S. Sasaki, G. Monma, D. Citterio, K. Yamada and K. Suzuki, Chimia, 2005, 59, 204–208 CrossRef CAS.
- D. Ryu, E. Park, D.-S. Kim, S. Yan, J. Y. Lee, B.-Y. Chang and K. H. Ahn, J. Am. Chem. Soc., 2008, 130, 2394–2395 CrossRef CAS PubMed.
-
(a) S. Yu, W. Plunkett, M. Kim and L. Pu, J. Am. Chem. Soc., 2012, 134, 20282–20285 CrossRef CAS PubMed;
(b) S. Yu, W. Plunkett, M. Kim, E. Wu, M. Sabat and L. Pu, J. Org. Chem., 2013, 78, 12671–12680 CrossRef CAS PubMed.
- E. K. Feuster and T. E. Glass, J. Am. Chem. Soc., 2003, 125, 16174–16175 CrossRef CAS PubMed.
- F. Tanaka, N. Mase and C. F. Barbas, Chem. Commun., 2004, 1762–1763 RSC.
- S. Oguri, A. Mizusawa, M. Kamada and M. Kohori, Anal. Chim. Acta, 2006, 558, 326–331 CrossRef CAS.
- J. Kumpf and U. H. F. Bunz, Chem. – Eur. J., 2012, 18, 8921–8924 CrossRef CAS PubMed.
- For reviews on fluorescent recognition of amines, see:
(a) G. J. Mohr, Anal. Bioanal. Chem., 2006, 386, 1201–1214 CrossRef CAS PubMed;
(b) G. J. Mohr, Sens. Actuators, B, 2005, 107, 2–13 CrossRef CAS.
- P. J. Cox, W. Wang and V. Snieckus, Tetrahedron Lett., 1992, 33, 2253–2256 CrossRef CAS.
- A. M. DeBerardinis, M. Turlington, J. Ko, L. Sole and L. Pu, J. Org. Chem., 2010, 75, 2836–2850 CrossRef CAS PubMed.
- Q. S. Hu, X. F. Zheng and L. Pu, J. Org. Chem., 1996, 61, 5200–5201 CrossRef CAS.
- H.-C. Zhang and L. Pu, Macromolecules, 2004, 37, 2695–2702 CrossRef CAS.
- W. Iwanek and J. Mattay, J. Photochem. Photobiol., A, 1992, 67, 209–226 CrossRef CAS.
- G. D. Y. Sogah and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 3035–3042 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the new compounds and additional optical spectra. See DOI: 10.1039/c3qo00076a |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.