Copper-catalyzed oxidative ipso-carboalkylation of activated alkynes with ethers leading to 3-etherified azaspiro[4.5]trienones†
Wen-Ting
Wei
,
Ren-Jie
Song
,
Xuan-Hui
Ouyang
,
Yang
Li
,
Hai-Bing
Li
and
Jin-Heng
Li
*
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: jhli@hnu.edu.cn; Fax: +86 731 8871 3642; Tel: +86 731 8882 2286
First published on 9th April 2014
Abstract
Cu-catalyzed synthesis of 3-etherified azaspiro[4.5]trienones from N-arylpropiolamides and ethers is presented using TBHP oxidant. This is achieved through C(sp3)–H functionalization, ipso-carbocyclization and dearomatization, and this method represents a new example of alkyne oxidative 1,2-difunctionalization with an ipso-aromatic carbon and a C(sp3)–H bond by simultaneously forming two new carbon–carbon bonds.
The spirocyclohexadienone ring system is a prevalent core structure in numerous natural products, pharmaceuticals and performance materials,1 and it serves as a versatile synthon in organic synthesis.2 The development of efficient and atom-economical methods for the synthesis of spirocyclohexadienones from readily accessible starting materials has therefore recently attracted considerable attention.1–6 Traditionally, the vast majority of efforts focused on the dearomatization of phenols via either the radical ipso-carbocyclization of intermediate A, in situ generated from the corresponding iodides or diazonium salts (Scheme 1a),3,4 or transition-metal-catalyzed intramolecular nucleophilic ipso-carbocyclization of 5-(4-hydroxyaryl)-1-alkenes or 5-(4-hydroxyaryl)-1-alkynes.5 However, these methods only modify the pre-existing frameworks, with no new functional groups being introduced into the system. Recently, the electrophilic ipso-carbocyclization of 4-aryl-1-alkynes with halogen electrophiles has emerged as a powerful and efficient method to construct spirocyclohexadienone rings (Scheme 1b):6 although these transformations could introduce new functional groups into the pre-existing systems, the functional groups were limited to halogen atoms. In light of these reported results, we hypothesized that the presence of an unsaturated bond could serve as a platform to trap radicals leading to a similar version of intermediate A, followed by ipso-carbocyclization and dearomatization affording the expected functionalized spirocyclohexadienone ring system. Herein, we report a new C(sp3)–H functionalization tandem strategy for selective synthesis of 3-etherified azaspiro[4.5]trienone architectures by copper-catalyzed oxidative ipso-carboalkylation of activated alkynes with aromatic ipso-carbon atoms and ethers; this tandem reaction is triggered by the CuCl/TBHP system and involves the use of 4-aryl-1-alkynes as radical acceptors to trap the ether radicals (Scheme 1c).
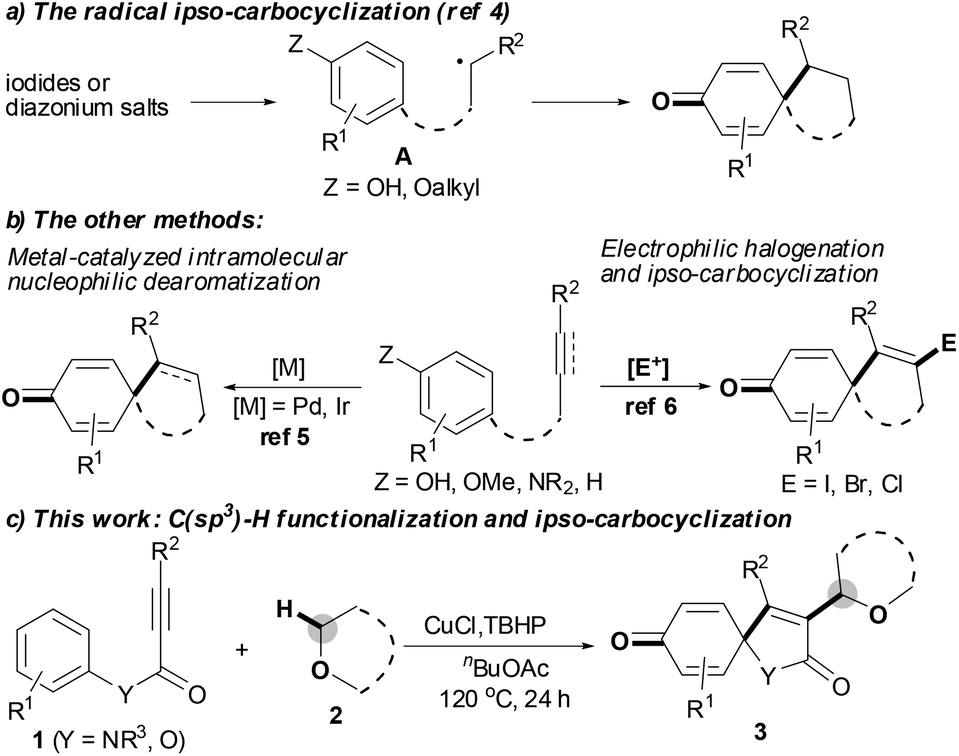 | ||
| Scheme 1 Construction of spirocyclohexadienone rings. | ||
Simple ethers, as important basic chemical feedstocks, are widely used solvents in organic synthesis and industry because of their chemical inertness under many reaction conditions. Nevertheless, the use of transition-metal catalysts affords the opportunity to use them as reactive functionalities. Although ethers, particularly cyclic ethers, frequently occur within the framework of many biologically active molecules,7 transition-metal-catalyzed methods employing simple ethers to construct higher-functionalized ethers through a C–C bond forming process have not been much investigated.8,9 In this field, the methods proceed via a single electron transfer (SET) strategy, and focus on the reaction of the C(sp3)–H bonds adjacent to an oxygen atom (ethers) with other C(sp3)–H bonds,8a–c C(sp2)–H bonds,8d–e aryl organometallic reagents,8f–h alkenes8i–n or terminal alkynes.8o However, methods for the difunctionalization of an alkyne with a C(sp3)–H bond adjacent to an oxygen atom (ethers) and an aromatic ipso-carbon atom to simultaneously form two new carbon–carbon bonds are lacking.
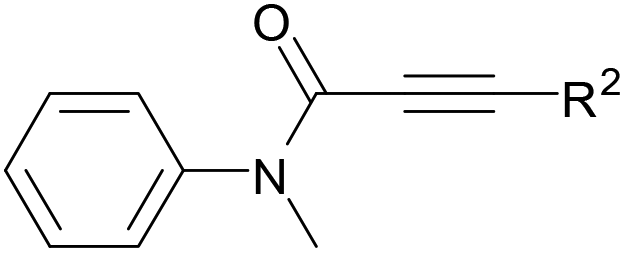
We began our investigation by examining the reaction between N-methyl-N,3-diphenylpropiolamide (1a) and tetrahydrofuran (THF, 2a) (Table 1). Initially, treatment of substrate 1a with THF 2a, FeCl3 and TBHP (tert-butyl hydroperoxide, 5 M in decane) afforded the desired product 3aa in 37% yield (entry 1). Gratifyingly, the Cu catalysts, such as CuCl, CuBr, CuI, CuOAc and CuCl2, were effective to improve the reaction, and CuCl was the most efficient (entries 2–6). In the presence of CuCl and TBHP, product 3aa was obtained in 83% yield (entry 2). It was noted that without Cu catalysts the reaction could take place in the presence of TBHP, albeit with a lower yield (entry 7). In addition, treatment of substrate 1a with THF 2a, TBHP and n-Bu4NI afforded the desired product 3aa in only 26% yield (entry 8). However, the absence of TBHP resulted in no detectable product 3aa (entry 9). Screening the loading of TBHP revealed that the amount of TBHP affected the reaction, and the reaction at 1.2 equiv. TBHP was revealed as the most effective loading (entries 2, 10 and 11). Subsequently, a series of other oxidants, including aqueous TBHP, TAHP (tert-amyl hydroperoxide), CHP (cumene hydroperoxide), DTBP (di-tert-butyl peroxide), DCP (dicumyl peroxide) and BPO (benzoyl peroxide), were tested (entries 12–17). We were surprised to find that only hydroperoxides, aqueous TBHP, TAHP and CHP afforded any results (entries 12–14), and the other oxidants, DTBP, DCP and BPO, were inert (entries 15–17). After the effects of the amount of CuCl and the reaction temperature were examined, it turned out that the best results were obtained in the presence of 5 mol% CuCl at 120 °C (entry 2 and 18–21). Notably, 10 mmol of substrate 1a were successfully converted in good yield (entry 22).
|
|
||||
|---|---|---|---|---|
| Entry | [M] [mol%] | [O] [equiv.] | T [°C] | Isolated yield [%] |
| a Reaction conditions: 1a (0.3 mmol), 2a (1.5 mmol), [Cu], [O], and n-BuOAc (2 mL) under argon atmosphere for 24 h. TBHP (5 M in decane). b n-Bu4NI (0.36 mmol) was added. c >90% of 1a was recovered. d TBHP (70% in water) was added. e 1a (10 mmol, 2.35 g). | ||||
| 1 | FeCl3 (5) | TBHP (1.2) | 120 | 37 |
| 2 | CuCl (5) | TBHP (1.2) | 120 | 83 |
| 3 | CuBr (5) | TBHP (1.2) | 120 | 50 |
| 4 | CuI (5) | TBHP (1.2) | 120 | 54 |
| 5 | CuOAc (5) | TBHP (1.2) | 120 | 59 |
| 6 | CuCl2 (5) | TBHP (1.2) | 120 | 48 |
| 7 | — | TBHP (1.2) | 120 | 41 |
| 8b | — | TBHP (1.2) | 120 | 26 |
| 9c | CuCl (5) | — | 120 | 0 |
| 10 | CuCl (5) | TBHP (1.0) | 120 | 61 |
| 11 | CuCl (5) | TBHP (2.0) | 120 | 83 |
| 12d | CuCl (5) | TBHP (1.2) | 120 | 82 |
| 13 | CuCl (5) | TAHP (1.2) | 120 | 62 |
| 14 | CuCl (5) | CHP (1.2) | 120 | 14 |
| 15c | CuCl (5) | DTBP (1.2) | 120 | 0 |
| 16c | CuCl (5) | DCP (1.2) | 120 | 0 |
| 17c | CuCl (5) | BPO (1.2) | 120 | 0 |
| 18 | CuCl (2) | TBHP (1.2) | 120 | 54 |
| 19 | CuCl (10) | TBHP (1.2) | 120 | 83 |
| 20 | CuCl (5) | TBHP (1.2) | 100 | 51 |
| 21 | CuCl (5) | TBHP (1.2) | 130 | 80 |
| 22e | CuCl (5) | TBHP (1.2) | 120 | 79 |
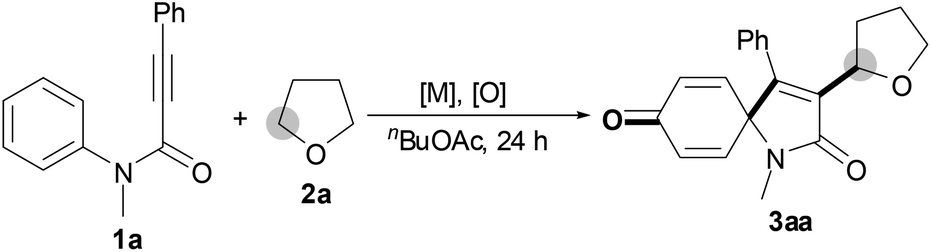
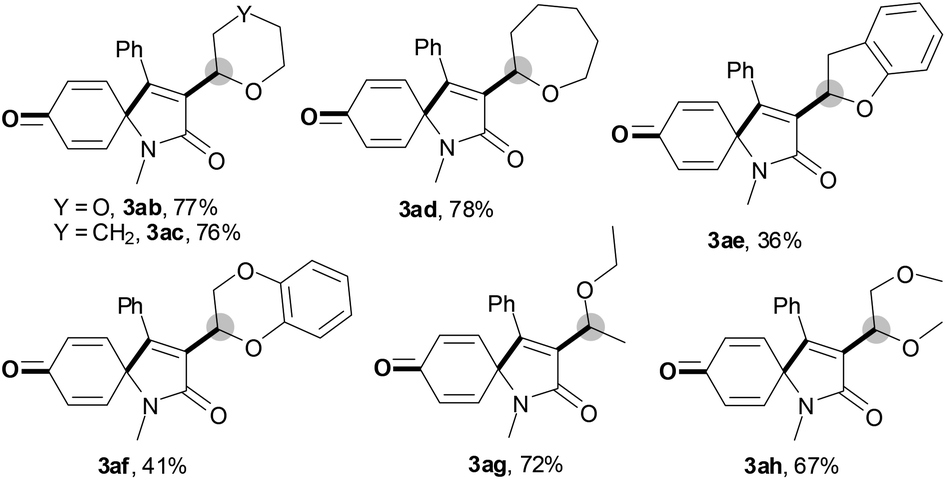
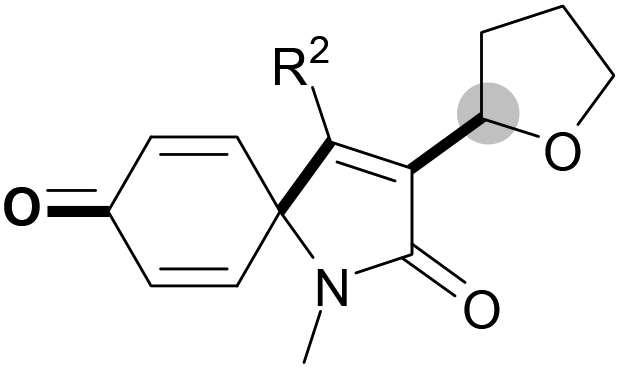
With the optimal conditions in hand, the scope of this Cu-catalyzed tandem reaction of ethers 2 with respect to N-methyl-N,3-diphenylpropiolamide (1a) was firstly investigated (Table 2). The optimal conditions were found to be compatible with a wide range of ethers, such as 1,4-dioxane (2b), tetrahydro-2H-pyran (2c), oxepane (2d), 2,3-dihydrobenzofuran (2e), 2,3-dihydrobenzo[b][1,4]dioxine (2f), diethyl ether (2g) and 1,2-dimethoxyethane (2h). For example, 1,4-dioxane (2b) successfully underwent the reaction with propiolamide 1a, CuCl and TBHP, providing product 3ab in 77% yield. Gratifyingly, the reaction could be expanded to ethers 2c and 2d with different ring sizes (products 3ac and 3ad). Benzoheterocyclic rings are valuable compounds with important chemical, biological, and medicinal properties.10 To our delight, two benzoheterocyclic rings, 2,3-dihydrobenzofuran (2e) or 2,3-dihydrobenzo[b][1,4]dioxine (2f), could be readily introduced into the azaspiro[4.5]trienone structure (products 3ae and 3af). Simple ethers, diethyl ether (2g) and 1,2-dimethoxyethane (2h) were also viable, and their reactions proceeded regioselectively at the CH2 position (products 3ag and 3ah).
|
|
|---|
| a Reaction conditions: 1a (0.3 mmol), 2 (1.5 mmol), CuCl (5 mol%), TBHP (1.2 equiv., 5 M in decane), and n-BuOAc (2 mL) at 120 °C under argon atmosphere for 24 h. |
|
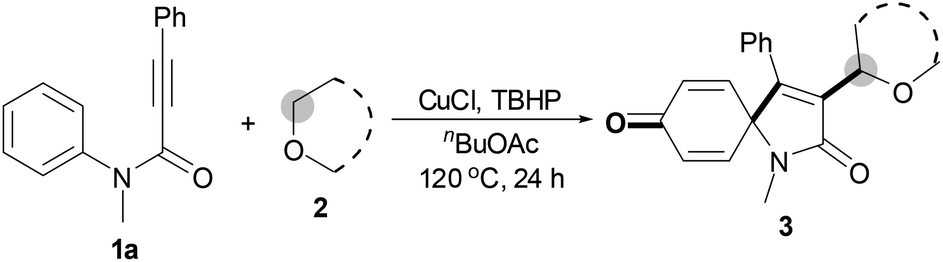
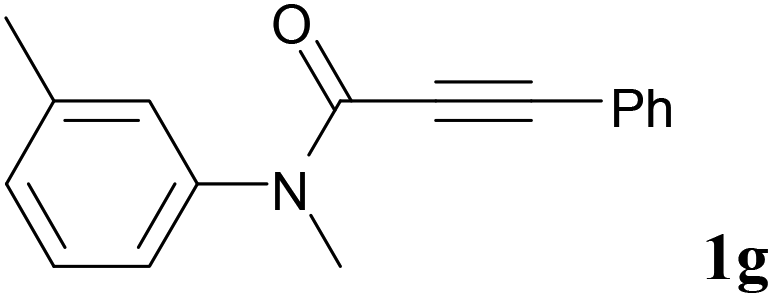
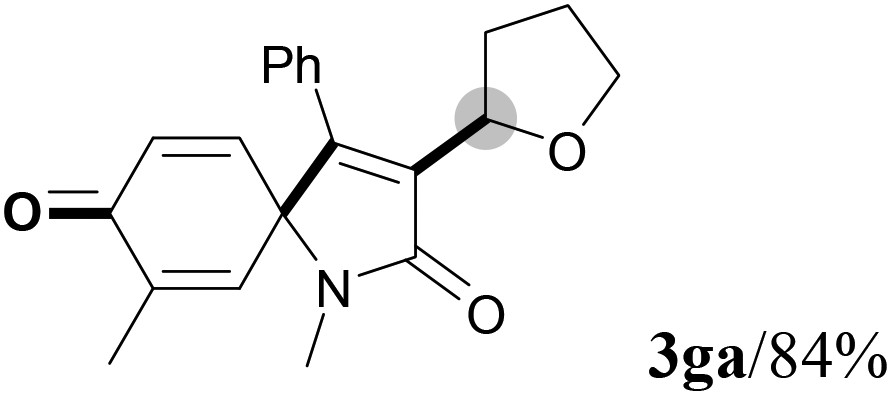
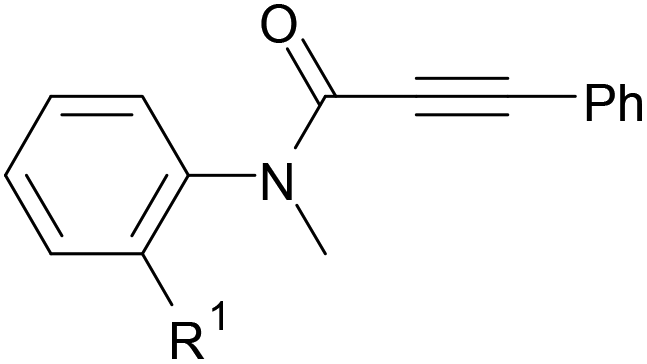
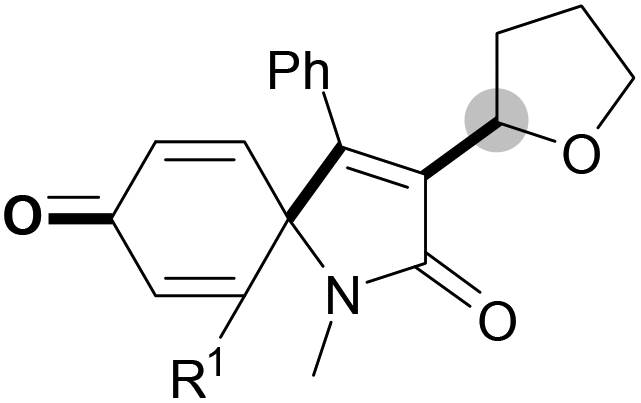
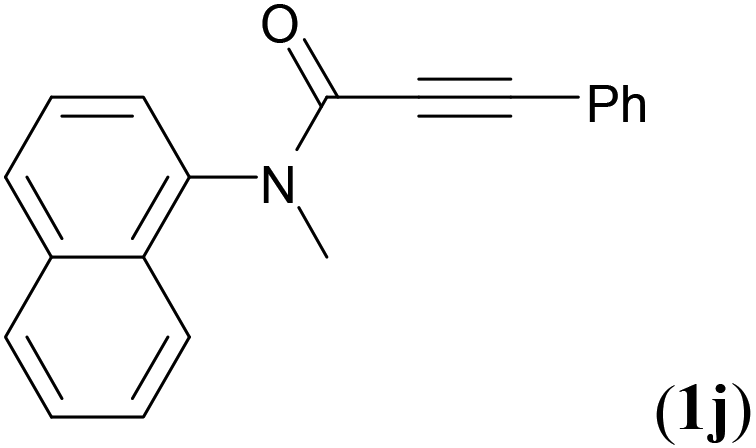
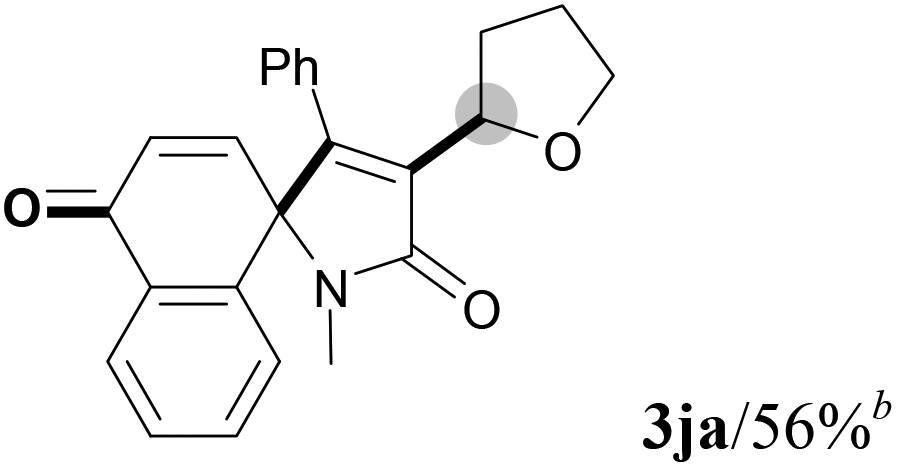
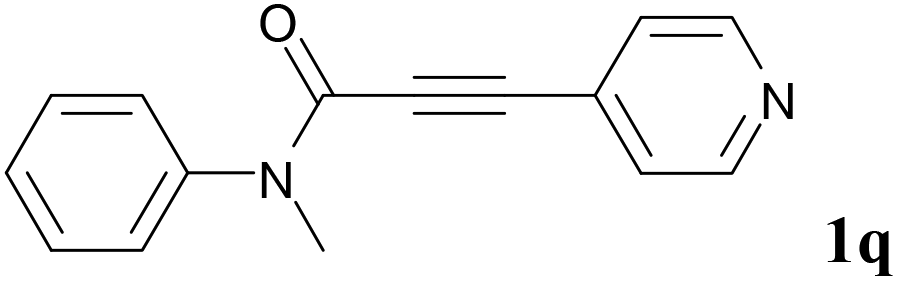
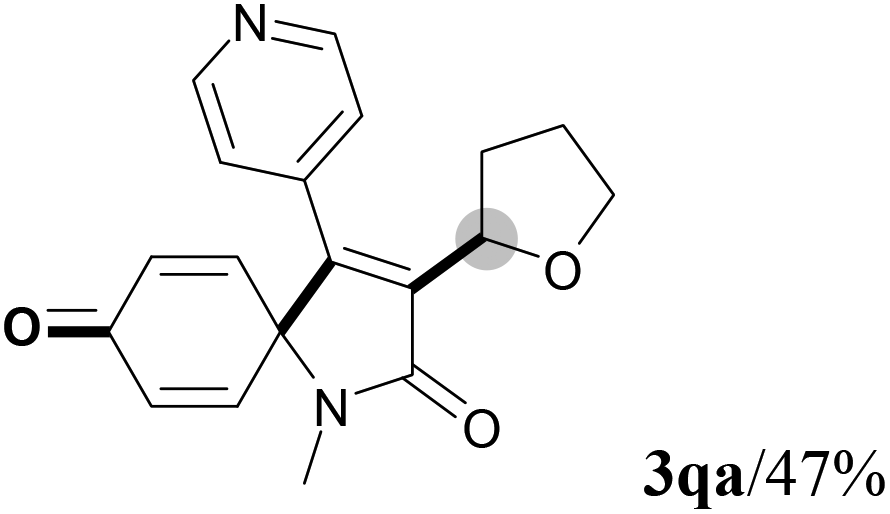
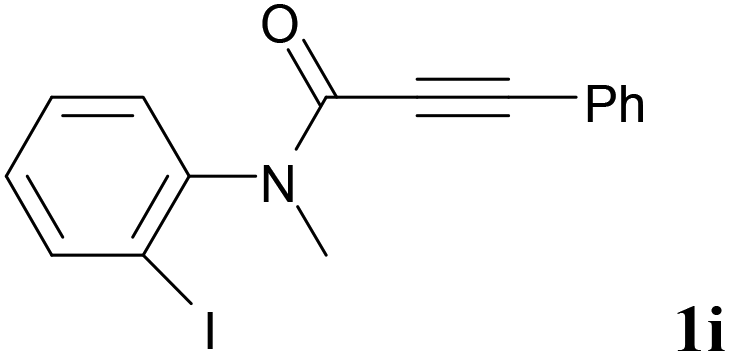
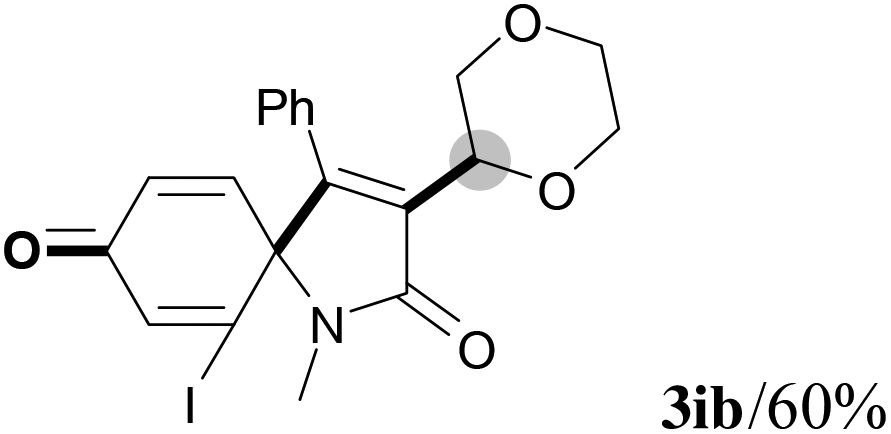
We next set out to examine the possibility of generating 3-etherified azaspiro[4.5]trienones by the Cu-catalyzed reaction of various N-arylpropiolamides 1 in the presence of ethers 2, CuCl and TBHP (Table 3). Changing the N-Me group to a N-Bn, N-(2-I-Bn) or N-allyl group furnished the corresponding products 3ba–da with slightly lower yields, but a N-H group resulted in no reaction (product 3ea). Subsequently, the impact of substituents on the aromatic ring of the N-aryl moiety was investigated (products 3fa–ja). N-Arylpropiolamides with an electron-donating group (o-Me, m-Me or o-MeO) or an electron-withdrawing group (I) on the aromatic ring of the N-aryl moiety were all well-tolerated (products 3fa–ia). Interestingly, the reaction of a naphthalene-derived substrate proceeded smoothly to afford a polycyclic spiro-compound 3ja in moderate yield, albeit with the requirement of a prolonged reaction time. In light of these results, a number of substituents at the terminal alkyne position, including electron-rich or electron-deficient aryl groups and aliphatic groups, were examined (products 3ka–qa). All aryl alkynes were consistent with the optimal conditions, and the order of the substituent reactivity is: electron-rich aryl groups (products 3ka–ma) > electron-deficient aryl groups (products 3na–oa). However, the aliphatic alkyne that was examined showed a lower activity (product 3pa). Gratifyingly, the heteroaryl alkyne that was investigated revealed itself as a suitable substrate (product 3qa). I- or MeO-substituted substrates 1i and 1l reacted with 1,4-dioxane (2b), giving the desired products 3ib and 3lb in moderate yields. It is noteworthy that 3-phenylpropiolate 1r is viable for the tandem reaction with THF 2a in moderate yield (product 3ra).
|
|
||
|---|---|---|
| Entry | Substrate 1 | Product/isolated yield (%) |
| a Reaction conditions: 1 (0.3 mmol), 2 (1.5 mmol), CuCl (5 mol%), TBHP (1.2 equiv., 5 M in decane) and nBuOAc (2 mL) at 120 °C under argon atmosphere for 24 h. b CuCl (10 mol%) and TBHP (2.4 equiv.) for 48 h. | ||
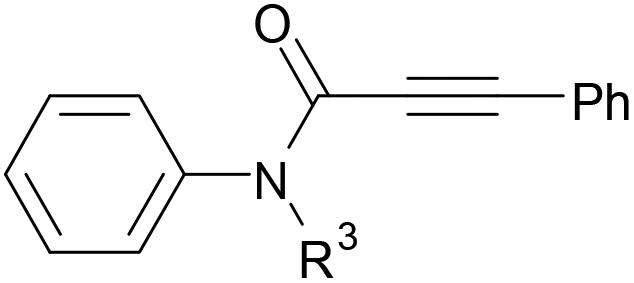
|
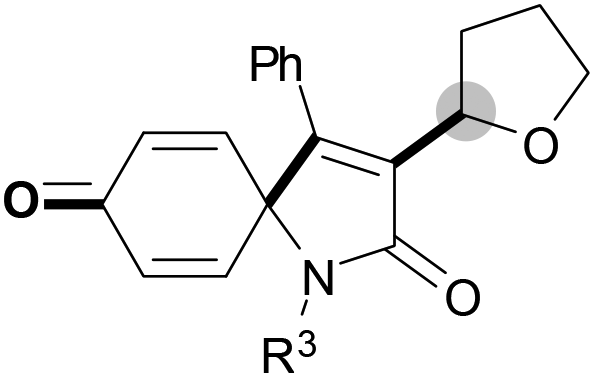
|
|
| 1 | R3 = Bn (1b) | R3 = Bn (3ba)/66% |
| 2 | R3 = 2-I-Bn (1c) | R3 = 2-I-Bn (3ca)/61% |
| 3 | R3 = allyl (1d) | R3 = allyl (3da)/67% |
| 4 | R3 = H (1e) | R3 = H (3ea)/trace |
| 5 |
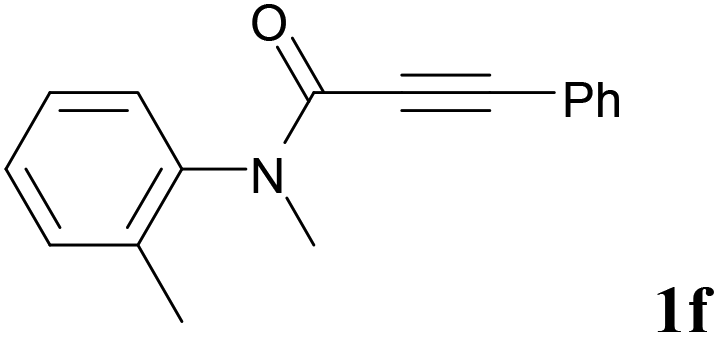
|
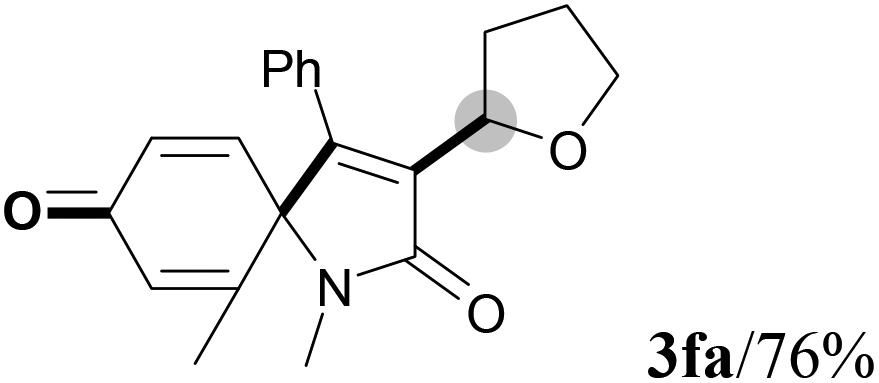
|
| 6 |
|
|
|
|
|
| 7 | R1 = OMe (1h) | R1 = OMe (3ha)/74% |
| 8 | R1 = I (1i) | R1 = I (3ia)/63% |
| 9 |
|
|
|
|
|
| 10 | R2 = 4-MeC6H4 (1k) | R2 = 4-MeC6H4 (3ka)/67% |
| 11 | R2 = 4-MeOC6H4 (1l) | R2 = 4-MeOC6H4 (3la)/70% |
| 12 | R2 = 2-MeOC6H4 (1m) | R2 = 2-MeOC6H4 (3ma)/68% |
| 13 | R2 = 4-IC6H4 (1n) | R2 = 4-IC6H4 (3na)/66% |
| 14 | R2 = 4-CNC6H4 (1o) | R2 = 4-CNC6H4 (3oa)/62% |
| 15 | R2 = n-C5H11 (1p) | R2 = n-C5H11 (3pa)/trace |
| 16 |
|
|
| 17 |
|
|
| 18 |
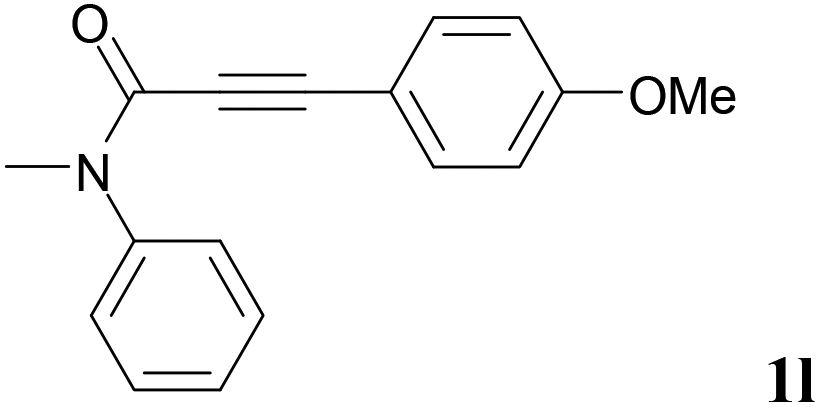
|
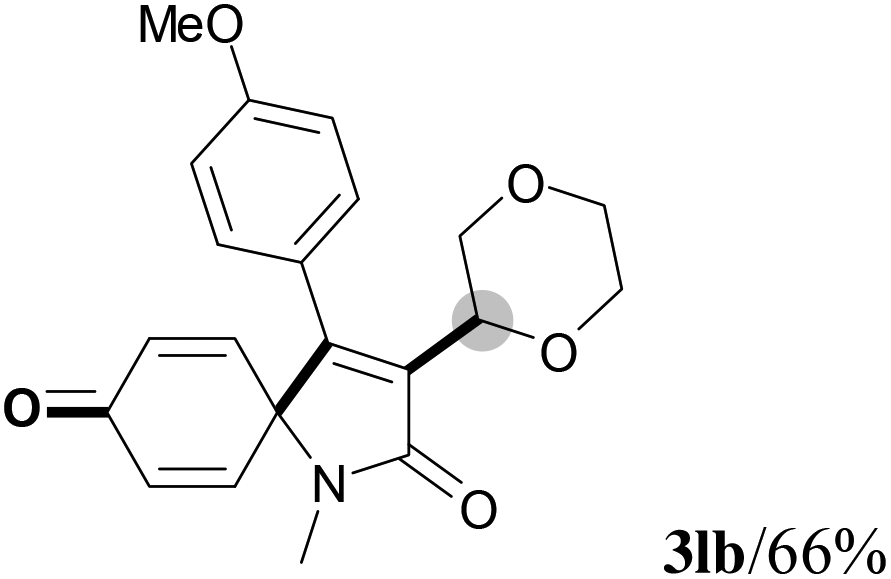
|
| 19 |
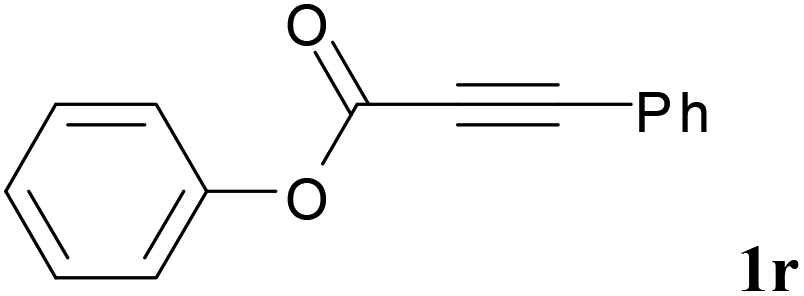
|
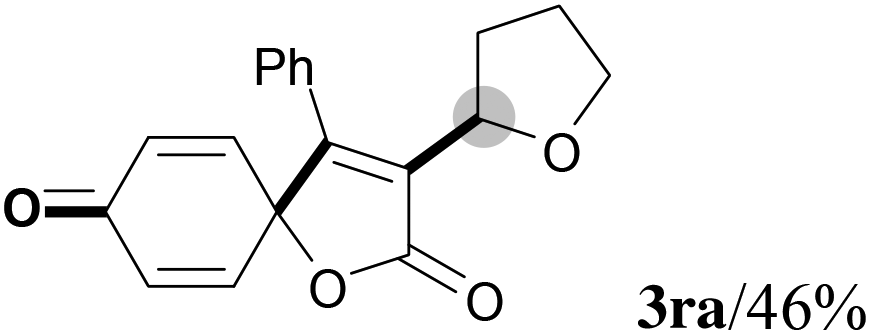
|
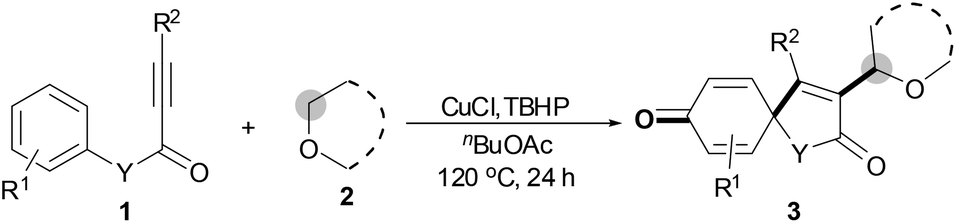
To our surprise, p-MeO- or p-F-substituted N-arylpropiolamides 1s and 1t gave the corresponding spiro[4,5]trienone 3aa in 68% and 66% yields, respectively, by releasing the para-substituents (eqn (1); Scheme 2). However, p-Me-substituted substrate 1u afforded hydroxylated product 3ua in 36% yield (eqn (2)). Notably, no 18O atom was incorporated in the corresponding spiro[4,5]trienones 3 when the reaction of 1a, 1s, 1t or 1u with THF 2a was performed in the presence of 2 equiv. H218O, suggesting that the carbonyl and hydroxyl oxygen atom does not come from water.
 | ||
| Scheme 2 Control experiments. | ||
The results in Table 1 were carefully reviewed, and we found that only hydroperoxides displayed catalytic activity for the current reaction (entries 2, 12–14 vs. entries 15–17). In light of this, we deduced that the oxygen atom of the newly formed carbonyl group comes from the hydroperoxide.11 The results in eqn (3) show that a stoichiometric amount of radical inhibitor (1.2 equiv. TEMPO) resulted in no conversion of substrate 1a; moreover, THF 2a was converted into 2,2,6,6-tetramethyl-1-(tetrahydrofuran-2-yloxy)piperidine 4. These results imply that the tandem reaction includes a radical process. A large kinetic isotope effect was discovered using a mixture of THF 2a and THF-D8 [D8]-2a (kH/kD = 3.0), indicating that the C(sp3)–H bond cleavage of THF constitutes a rate-limiting step (eqn (4)).8
Consequently, we proposed a working mechanism as outlined in Scheme 3 on the basis of the present results and the literature reports.8,9 Initially, alkyl radical B is formed from THF (1a) by a single electron transfer from TBHP with the aid of the active Cu+ species.8,9 Subsequently, alkylation of alkyne 2a with alkyl radical B affords vinyl radical intermediate C, followed by selective ipso-carbocyclization to give intermediate D.4,5 Intermediate D reacts with Cu2+(OH) to yield intermediate E and regenerates the active Cu+ species.11 Finally, oxidation of intermediate E by TBHP/[Cu] produces the desired spiro[4,5]trienone 3aa.12
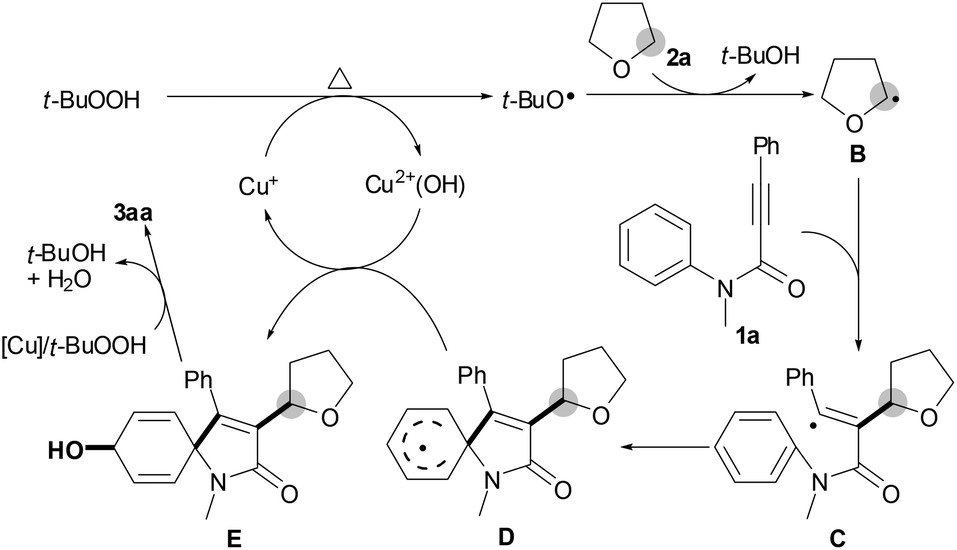 | ||
| Scheme 3 Possible mechanism. | ||
In summary, we have developed a new Cu-catalyzed synthesis of 3-etherified azaspiro[4.5]trienones from N-arylpropiolamides and ethers through oxidative C(sp3)–H functionalization, ipso-carbocyclization and dearomatization cascade. Mechanistic studies suggested that a radical process is involved, and the oxygen atom of the newly formed carbonyl group comes from the hydroperoxide. Importantly, this tandem method makes the construction of higher-functionalized ethers from simple ethers easy by a C(sp3)–H oxidative functionalization strategy, and represents the first example of alkyne oxidative 1,2-difunctionalization to simultaneously form two carbon–carbon bonds and one carbon–oxygen double bond. Studies on applications of this oxidative difunctionalization method in organic synthesis are currently underway in our laboratory.
This research was supported by the Hunan Provincial Natural Science Foundation of China (no. 13JJ2018), the Natural Science Foundation of China (no. 21172060), and the Specialized Research Fund for the Doctoral Program of Higher Education (no. 20120161110041).
Notes and references
- (a) C. H. Heathcock, S. L. Graham, M. C. Pirrung, F. Plavac and C. T. White, The Total Synthesis of Natural Products, ed. J. Apsimon, Wiley-Interscience, New York, 1983, vol. 5, p. 264 Search PubMed; (b) K. Yoneda, E. Yamagata, T. Nakanishi, T. Nagashima, I. Kawasaki, T. Yoshida, H. Mori and I. Miura, Phytochemistry, 1984, 23, 2068 CrossRef CAS; (c) A. S. Chawla and A. H. Jackson, Nat. Prod. Rep., 1989, 6, 55 RSC; (d) Z. Jin, Nat. Prod. Rep., 2005, 22, 111 RSC; (e) E. M. Antunes, B. R. Copp, M. T. Davies-Coleman and T. Samaai, Nat. Prod. Rep., 2005, 22, 62 RSC; (f) A. S. Chawla and V. K. Kapoor, in The Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, 1995, vol. 9, p. 86 Search PubMed; (g) Y. Tsuda and T. Sano, in The Alkaloids, ed. G. A. Cordell, Academic Press, San Diego, 1996, vol. 48, p. 249 Search PubMed; (h) Y.-S. Cai, Y.-W. Guo and K. Krohn, Nat. Prod. Rep., 2010, 27, 1840 RSC; (i) E. Gravel and E. Poupon, Nat. Prod. Rep., 2010, 27, 32 RSC.
- For reviews: (a) S. T. Roche and J. A. Porco, Jr., Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (b) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed.
- For reviews on the oxidative spirocyclization of phenol derivatives: (a) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef PubMed; (b) T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC; (c) S. Quideau, L. Pouységu and D. Deffieux, Synlett, 2008, 467 CrossRef CAS PubMed; (d) M. A. Ciufolini, N. A. Braun, S. Canesi, M. Ousmer, J. Chang and D. Chai, Synthesis, 2007, 3759 CrossRef CAS PubMed; (e) S. Rodríguez and P. Wipf, Synthesis, 2004, 2767 Search PubMed; (f) D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS PubMed; (g) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed.
- For representative papers on the synthesis of spirocyclohexadienones via radical ipso-carbocyclization: (a) F. G.-L. de Turiso and D. P. Curran, Org. Lett., 2005, 7, 151 CrossRef PubMed; (b) T. Lanza, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and G. Zanardi, Angew. Chem., Int. Ed., 2008, 47, 9439 CrossRef CAS PubMed; (c) T. Dohi, A. Maruyama, M. Yoshimura, K. Morimoto, H. Tohma and Y. Kita, Angew. Chem., Int. Ed., 2005, 44, 6193 CrossRef CAS PubMed; (d) Y. Dohi, Y. Minamitsuji, A. Maruyama, S. Hirose and Y. Kita, Org. Lett., 2008, 10, 3559 CrossRef PubMed.
- For representative papers on metal-catalyzed synthesis of spirocyclohexadienones: (a) F. C. Pigge, J. J. Coniglio and R. Dalvi, J. Am. Chem. Soc., 2006, 128, 3498 CrossRef CAS PubMed; (b) S. Chiba, L. Zhang and J.-Y. Lee, J. Am. Chem. Soc., 2010, 132, 7266 CrossRef CAS PubMed; (c) Y. L. Tnay, C. Chen, Y. Y. Chua, L. Zhang and S. Chiba, Org. Lett., 2012, 14, 3550 CrossRef CAS PubMed; (d) T. Nemoto, Y. Ishige, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020 CrossRef CAS PubMed; (e) Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455 CrossRef CAS PubMed; (f) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS PubMed; (g) M. Yoshida, T. Nemoto, Z. Zhao, Y. Ishige and Y. Hamada, Tetrahedron: Asymmetry, 2012, 23, 859 CrossRef CAS PubMed; (h) T. Nemoto, Z. Zhao, T. Yokosaka, Y. Suzuki, R. Wu and Y. Hamada, Angew. Chem., Int. Ed., 2013, 52, 2217 CrossRef CAS PubMed.
- For other representative papers on the synthesis of spirocyclohexadienones. Halocyclization: (a) T. R. Appel, N. A. M. Yehia, U. Baumeister, H. Hartung, R. Kluge, D. Ströhl and E. Fanghänel, Eur. J. Org. Chem., 2003, 47 CrossRef CAS; (b) X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 12230 CrossRef CAS PubMed; (c) B.-X. Tang, D.-J. Tang, S. Tang, Q.-F. Yu, Y.-H. Zhang, Y. Liang, P. Zhong and J.-H. Li, Org. Lett., 2008, 10, 1063 CrossRef CAS PubMed; (d) Q. Yin and S.-L. You, Org. Lett., 2012, 14, 3526 CrossRef CAS PubMed; (e) B.-X. Tang, Y.-H. Zhang, R.-J. Song, D.-J. Tang, G.-B. Deng, Z.-Q. Wang, Y.-X. Xie, Y.-Z. Xia and J.-H. Li, J. Org. Chem., 2012, 77, 2837 CrossRef CAS PubMed, and references cited therein; For a review: (f) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937 CrossRef CAS PubMed.
- (a) Polyether Antibiotics: Naturally Occurring Acid Ionophores, ed. J. W. Westley, Marcel Dekker, New York, 1982 Search PubMed; (b) D. E. Levy and C. Tang, The Chemistry of C-Glycosides, Pergamon, Oxford, 1st edn, 1995 Search PubMed; (c) W. Heitmann, G. Strehlke and D. Mayer, Ethers, Aliphatic in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed; (d) F. Q. Alali, X. X. Liu and J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504 CrossRef CAS PubMed; (e) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407 CrossRef CAS PubMed; (f) M. Saleem, H. J. Kim, M. S. Ali and Y. S. Lee, Nat. Prod. Rep., 2005, 22, 696 RSC; (g) M. Sasaki and H. Fuwa, Synlett, 2004, 1851 CrossRef CAS PubMed; (h) L. F. Tietze and N. Rackelmann, Pure Appl. Chem., 2004, 76, 1967 CrossRef CAS; (i) E. Lee, Pure Appl. Chem., 1996, 68, 631 CrossRef CAS; (j) G. M. Nicholas and A. J. Phillips, Nat. Prod. Rep., 2005, 22, 144 RSC.
- For the reactions with other C(sp3)–H bonds: (a) Y. Zhang and C.-J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949 CrossRef CAS PubMed; (b) Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS PubMed; (c) Y. Xie, M. Yu and Y. Zhang, Synthesis, 2011, 2803 CAS; The C(sp2)–H bonds: (d) T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016 CrossRef CAS PubMed; (e) X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848 CrossRef CAS PubMed; Aryl organometallic reagents: (f) P. P. Singh, S. Gudup, H. Aruri, U. Singh, S. Ambala, M. Yadav, S. D. Sawant and R. A. Vishwakarma, Org. Biomol. Chem., 2012, 10, 1587 RSC; (g) P. P. Singh, S. Gudup, S. Ambala, U. Singh, S. Dadhwal, B. Singh, S. D. Sawant and R. A. Vishwakarma, Chem. Commun., 2011, 47, 5852 RSC; (h) D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453 CrossRef CAS PubMed; Alkenes: (i) K. Cao, Y.-J. Jiang, S.-Y. Zhang, C.-A. Fan, Y.-Q. Tu and Y.-J. Pan, Tetrahedron Lett., 2008, 49, 4652 CrossRef CAS PubMed; (j) K. Cheng, L.-H. Huang and Y.-H. Zhang, Org. Lett., 2009, 11, 2908 CrossRef CAS PubMed; (k) J. Y. Kim, J. C. Park, H. Song and K. H. Park, Bull. Korean Chem. Soc., 2010, 31, 3509 CrossRef CAS; (l) H. Sun, Y. Zhang, F. Guo, Z. Zha and Z. Wang, J. Org. Chem., 2012, 77, 3563 CrossRef CAS PubMed; (m) R. L. Jacobs and G. G. Ecke, J. Org. Chem., 1963, 28, 3036 CrossRef CAS; (n) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; Alkynes through hydroallylation with ethers: (o) L. Huang, K. Cheng, B. Yao, J. Zhao and Y. Zhang, Synthesis, 2009, 3504 CAS.
- For selected papers on other methods employing simple ethers to construct higher-functionalized ethers: (a) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (b) H. S. A. Inoue and K. Oshima, Synlett, 1999, 1582 CrossRef PubMed; (c) A. Ishida, D. Sugita, Y. Itoh and S. Takamuku, J. Am. Chem. Soc., 1995, 117, 11687 CrossRef CAS; (d) T. Yoshimitsu, T. Makino and H. Nagaoka, J. Org. Chem., 2003, 68, 7548 CrossRef CAS PubMed; (e) H. M. L. Davies, T. Hansen and M. R. Churchill, J. Am. Chem. Soc., 2000, 122, 3063 CrossRef CAS; (f) J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef CAS; (g) J. Xiang and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 11986 CrossRef CAS; (h) J. Xiang, W. Jiang, J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 4123 CrossRef CAS.
- (a) J. Jankun, S. H. Selman, R. Swiercz and E. Skrzypcak-Jankun, Nature, 1997, 387, 561 CrossRef CAS PubMed; (b) A. D. Cington, Chem. Soc. Rev., 1997, 26, 111 RSC; (c) B. A. Keay and P. W. Dibble, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Elsevier, Oxford, U.K., 1996, vol. 2, p. 395 Search PubMed; (d) X.-L. Hou, Z. Yang and H. N. C. Wong, Prog. Heterocycl.Chem., 2002, 14, 139 CAS.
- For a paper on the use of TBHP to introduce an oxygen atom into the products: H. Jiang, H. Huang, H. Cao and C. Qi, Org. Lett., 2010, 12, 5561 CrossRef CAS PubMed.
- For selected recent papers on Cu-catalyzed oxidation of alcohols: (a) Y. Zhu, B. Zhao and Y. Shi, Org. Lett., 2013, 15, 992 CrossRef CAS PubMed; (b) X. Liu, Q. Xia, Y. Zhang, C. Chen and W. Chen, J. Org. Chem., 2013, 78, 8531 CrossRef CAS PubMed , and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00006d |
| This journal is © the Partner Organisations 2014 |