NHC-catalyzed homoenolate reaction of enals and nitroalkenes: computational study of mechanism, chemoselectivity and stereoselectivity†
Qi
Zhang
a,
Hai-Zhu
Yu
b and
Yao
Fu
*a
aDepartment of Chemistry, University of Science and Technology of China, Hefei, 230026, China. E-mail: fuyao@ustc.edu.cn; Fax: (+86)-551-360-6689; Tel: (+86)-551-360-7476
bDepartment of Polymer Science and Engineering, University of Science and Technology Beijing, Beijing 100083, PR China
First published on 9th May 2014
Abstract
A systematic theoretical study has been carried out on the NHC-catalyzed homoenolate reaction of enals and nitroalkenes. The detailed mechanism of these nitroalkenes participated homoenolate reactions, the chemoselectivity of the homoenolate (vs. Stetter) pathway and the intriguing syn- (vs. anti-) stereoselectivity have been investigated. Calculations show that the homoenolate and Stetter reaction (Path-homo and Path-set) first undergo the same formation process of the Breslow intermediate, which proceeds via a HOAc mediated stepwise mechanism. The subsequent process of Path-homo consists of five steps: nucleophilic attack of the Breslow intermediate (C3 atom) on the nitroalkene, intramolecular proton transfer, HOAc involved protonation of C2, concerted nucleophilic attack of OEt on the carbonyl, and final carbene dissociation. The subsequent process of Path-set consists of three steps: nucleophilic attack of the Breslow intermediate (C1 atom) on the nitroalkene, intramolecular proton transfer and carbene dissociation. Path-homo is more feasible than Path-set. The highly exoergonic nucleophilic attack, the facile intramolecular proton transfer and attack of OEt on the carbonyl in Path-homo result in its feasibility. The stereoselectivity determining step of Path-homo is the attack of the Breslow intermediate on the nitroalkene, in which the steric hindrance and hydrogen bonding determine the syn stereoselectivity.
1. Introduction
N-heterocyclic carbenes (NHCs) catalyzed umpolung reactions have made rapid progress in the past few decades.1 In this context, NHCs are traditionally used as acyl anion equivalents (d1-nucleophile) to participate in Stetter,1c,2 benzoin,3 redox4 and other reactions.5 When the reaction substrates are enals, NHCs can also be used as homoenolate equivalents (d3-nucleophile), to participate in the homoenolate reactions (eqn (1) in Scheme 1).1b,6 In 2004, Bode7a and Glorius8et al. reported pioneering studies on NHC-catalyzed homoenolate reactions. The nucleophilic homoenolate intermediates generated in these processes further reacted with the electrophilic substrates (aldehydes) to synthesize γ-lactones. This breakthrough discovery turns α,β-unsaturated aldehydes into d3-nucleophiles, thus providing a new catalytic method for carbon–carbon bond building. In addition to the Bode7 and Glorius8 groups, Nair,9 Scheidt,10 Liu11 and Rovis12et al. have also made outstanding contributions in homoenolate reactions with a variety of active electrophiles to obtain heterocyclic compounds such as cis-γ-lactam, γ-butyrolactone and cyclopentene. In addition to cyclic compounds, non-cyclic compounds can also be obtained from homoenolate reactions. One of the most important non-cyclization homoenolate reactions involves nitroalkenes as electrophiles.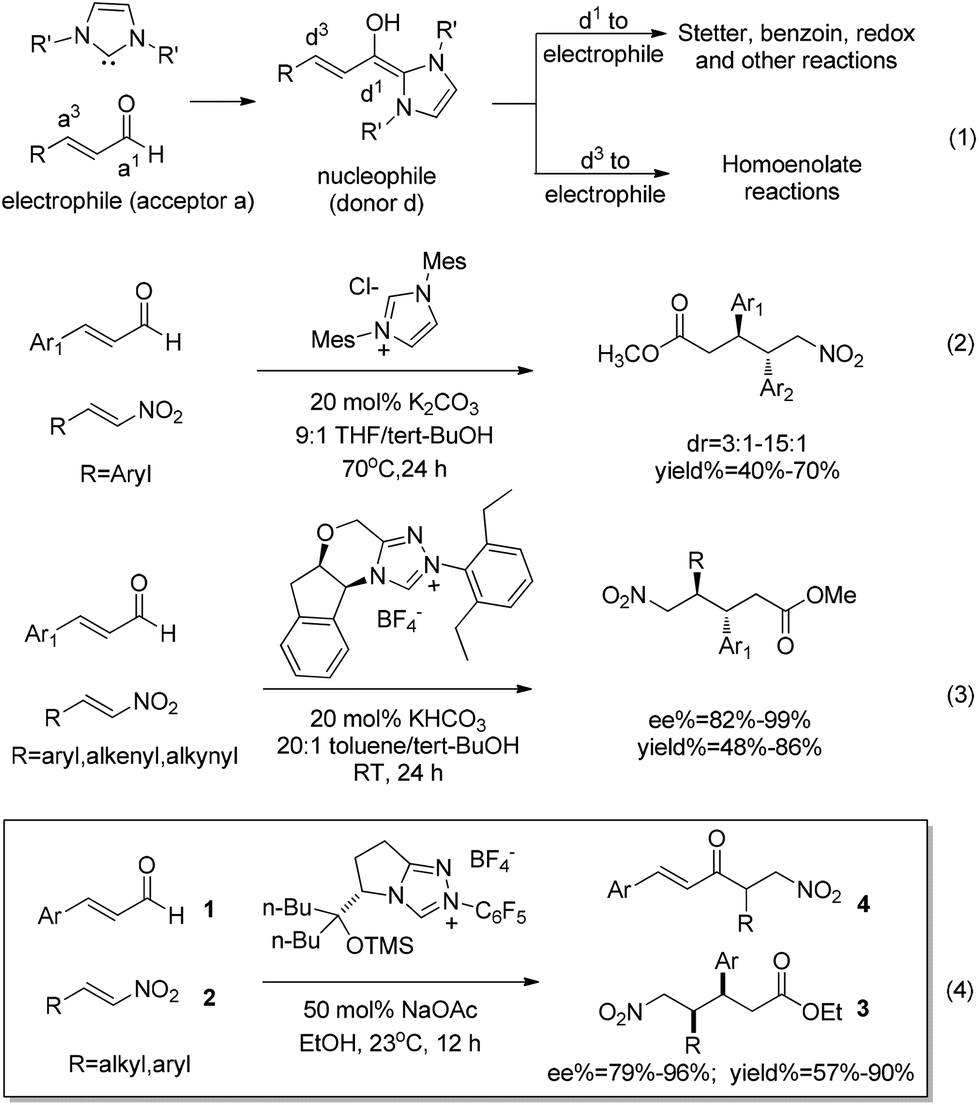 | ||
| Scheme 1 | ||
In 2009, the Nair9c group reported an imidazolinium precatalyst-catalyzed homoenolate reaction between aryl nitroalkenes and enals, and obtained anti-δ-nitro esters (eqn (2)). Similarly, Liu et al. recently11 reported a triazolium salts precatalyst-catalyzed homoenolate reaction between aryl/alkenyl/alkynyl nitroalkenes and enals, and obtained anti-δ-nitro ester (eqn (3)). Very recently, the Rovis12a group exploited the homoenolate reaction between alkyl/aryl substituted nitroalkenes and enals, unexpectedly obtaining the syn configuration product 3 accompanied with trace amounts of Stetter product 4 (eqn (4)).
Despite the great progress in non-cyclization homoenolate reactions with the participation of nitroalkene electrophiles, the detailed mechanism of these reactions remains unclear. Taking eqn (4) as an example, Nair9cet al. proposed that the substrate first transforms into Breslow intermediate A; then the C3 atom of A attacks the nitroalkene to obtain B, followed by ethanol mediated protonation, enol–keto tautomerisation, and a final nucleophilic attack of the ethanol on the carbonyl to yield the non-cyclization product E (Scheme 2). The above homoenolate process is favored with respect to the side reaction (Stetter) process. In the Stetter reaction, the C1 atom of intermediate A attacks the nitroalkene, then the intramolecular proton transfer occurs in molecule F to give G, and the formation of the product H is achieved by a carbene dissociation step.13 Nonetheless, two problems remain to be settled in the aforementioned mechanisms. First, the details for the NHC catalyzed formation of Breslow intermediate are under debate. As shown in Scheme 3a, two types of mechanisms, i.e. the mechanisms in absence of conjugated acid and the mechanisms with the aid of conjugated acid might occur. For the first type, the direct 1,2 migration mechanism14 (Path-a), the intermolecular proton transfer mechanism15 (Path-b) between two NHC/benzaldehyde coupled intermediates, and the mechanism involving the carbene ring on NHC undergoing an indirect proton transfer mechanism16 (Path-c) have all been previously proposed computationally. Experimentally, Rovis et al.13a envisioned that the proton is shuttled by the aryl ring on NHC catalyst (Path-d). For the second type, both the concerted proton transfer mechanism involving five-17,18 or seven-membered2b ring transition state (Path-e) and the stepwise proton transfer mechanism19 (Path-f) might occur. Currently, it remains unknown which mechanism (Paths a–f in Scheme 3a) is the most favored. Meanwhile, the subsequent steps from the Breslow intermediate in the homoenolate reaction mechanism are worth examining, because the weak acid (HOEt) mediated protonation20 of intermediate B and the nucleophilic attack of D might be very difficult (Scheme 3b). In this context, we speculate that the intermediate B may undergo an alternative mechanism to generate the homoenolate product.
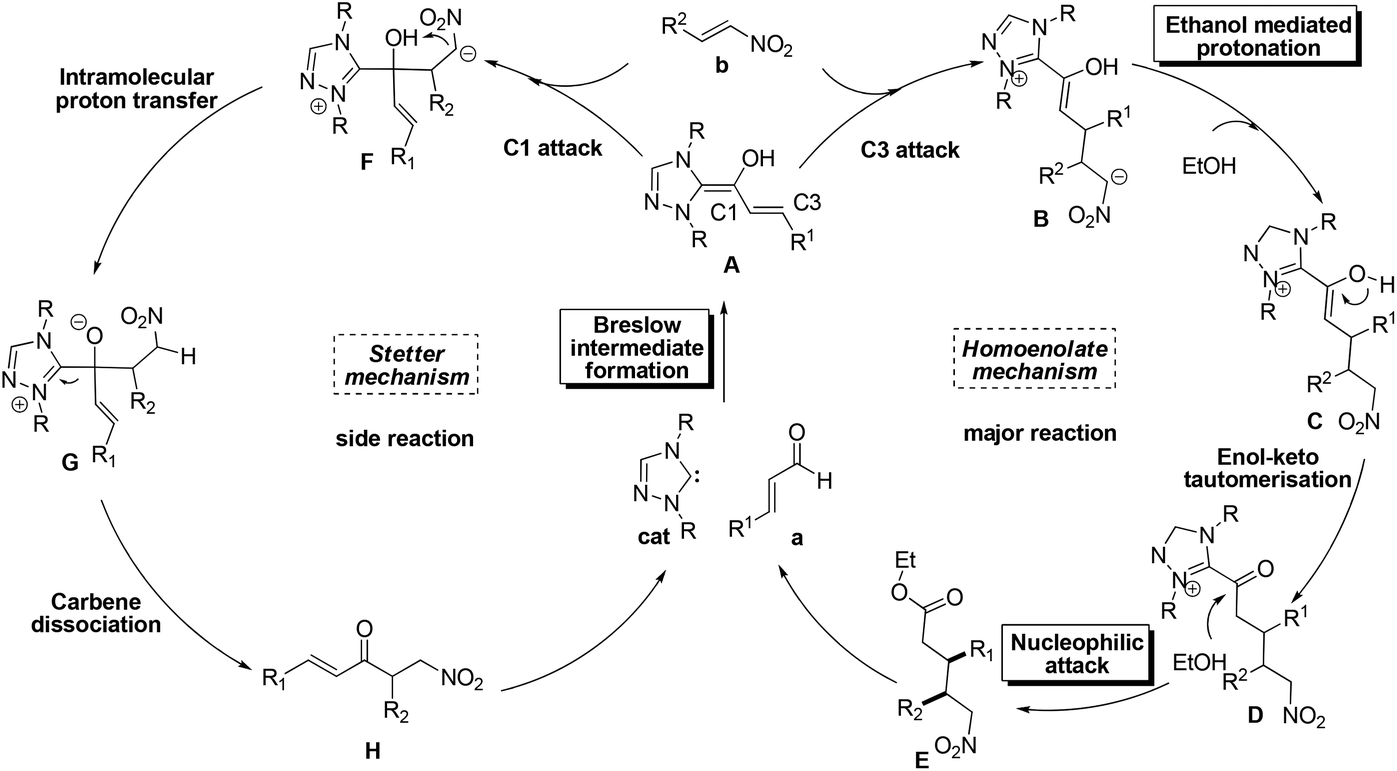 | ||
| Scheme 2 Proposed mechanism of homoenolate and Stetter reaction. | ||
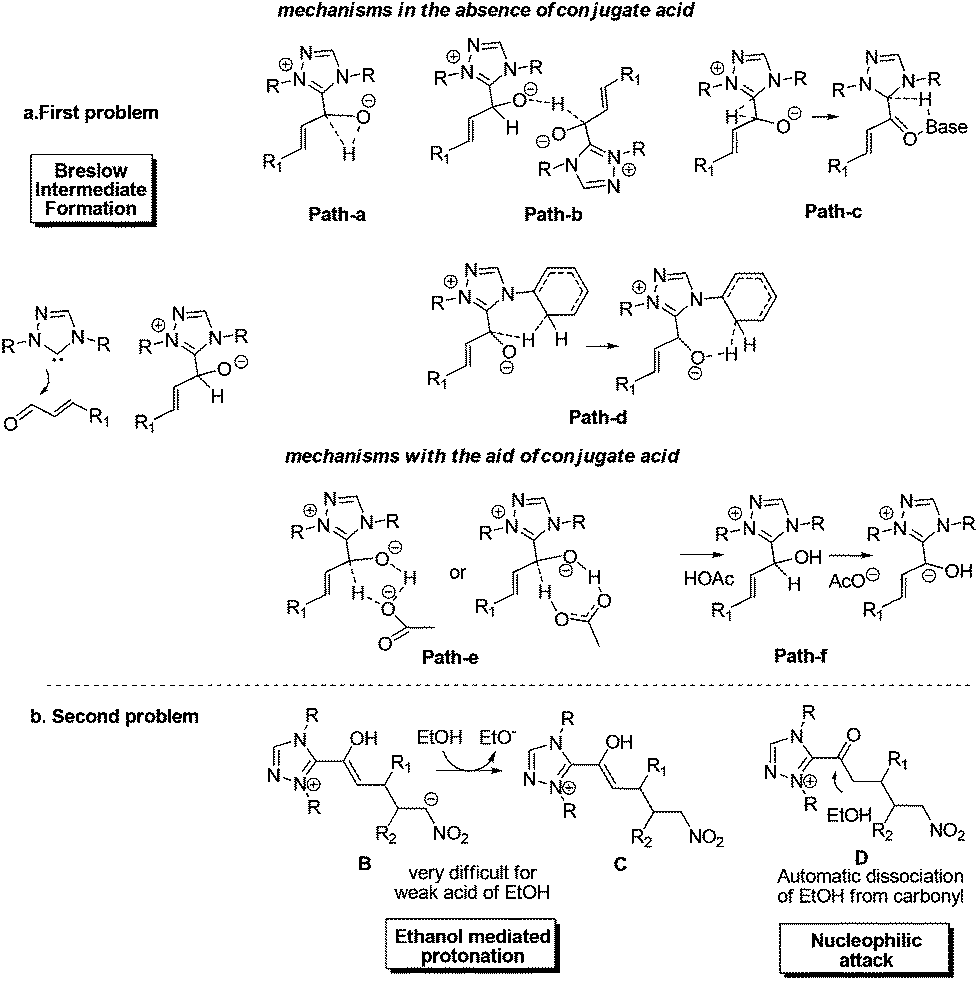 | ||
| Scheme 3 Two mechanistic questions to be settled. | ||
In order to solve the above mechanistic problems, we carried out theoretical calculations on the mechanism of the nitroalkene participated homoenolate reaction. Meanwhile, the origin of the chemoselectivity of the homoenolate (vs. Stetter) reaction and the stereoselectivity of the intriguing syn- (vs. anti-) homoenolate reaction shown in eqn (4) has also been investigated. According to our calculations, the Breslow intermediate is generated through Path-f (stepwise proton transfer involving a conjugate acid), rather than any of Paths a–e. As for the subsequent steps in the homoenolate reaction (Path-homo), the Breslow intermediate first undergoes the nucleophilic attack of the C3 atom on the nitroalkene, and the formed intermediate (B, Scheme 2) then experiences an intramolecular protonation by the hydroxyl group (instead of the ethanol mediated intermolecular protonation). After the subsequent enol–keto tautomerisation, the formed intermediate (D) is attacked by an OEt− anion derived from HOEt (rather than a direct attack by HOEt). Finally, the product is obtained after the dissociation of carbene. The proposed mechanism is slightly different from that shown in Scheme 2 in the steps involving the intramolecular protonation and the attack of OEt− on the carbonyl. The proposed mechanism has been verified by the good consistency between the calculation results and the experiments on the observed chemo- and stereoselectivity. Analyzing the reason for selectivity, we found that the two facile steps (the intramolecular protonation and attack of −OEt on the carbonyl) in Path-homo result in the feasibility of the homoenolate reaction over the Stetter reaction. Meanwhile, the stereoselective determining step of Path-homo is the attack of the Breslow intermediate (A) on the nitroalkene. In this step, the steric hindrance between the ethyl of the nitroalkene and the alkyl substituent on the carbene ring, and the hydrogen bonding between the nitro of the nitroalkene and the hydroxyl group, determine the stereoselectivity.
2. Computational methods and model reaction
2.1 Computational methods
The Gaussian09 suite of programmes21 was used for the calculations in this study. The B3LYP13b,c,15,17,18,22,23 method combined with the 6-31+G(d) basis set was used for unrestricted geometry optimization in ethanol solvent (consistent with Rovis's experiments,12a with SMD24,25 model) on all structures. To get the thermodynamic corrections of Gibbs free energy and verify the stationary points to be local minima or saddle points, we conducted frequency analysis at the same level with optimization. For all transition states, we performed the intrinsic reaction coordinate (IRC) analysis to confirm that they connect the correct reactants and products on the potential energy surfaces.26 For compounds that may have different conformations, various structures were calculated and the lowest energy conformation is used in the following discussions.M0627,28/6-31+G(d,p) method with the SMD24,25 model was used for the solution phase single-point energy calculations of all these stationary points (ethanol is used as solvent). All energetics involved in this study are calculated by adding the Gibbs free energy correction calculated at B3LYP/6-31G(d) and the single-point energy calculated at the M06/6-31+G(d,p) method.28
2.2 Model reaction
In the present study, the homoenolate and Stetter reactions of enal react-1 and nitroalkene react-2 in producing the (3S, 4R) configuration product prod-homo and the byproduct prod-set were chosen as the model reactions (Scheme 4).12a Consistent with Rovis's experiment, NHC catalyst 5f, HOEt and NaOAc were chosen as the catalyst, solvent, and base, respectively.12a The mechanisms of the homoenolate and Stetter reactions are named Path-homo and Path-set respectively. In Rovis’ experiment, the ratio of the yield of homoenolate and Stetter product is higher than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the enantioselectivity (ee) and diastereoselectivity (dr) of homoenolate product are 93% and 17:1, respectively.
:1, and the enantioselectivity (ee) and diastereoselectivity (dr) of homoenolate product are 93% and 17:1, respectively.
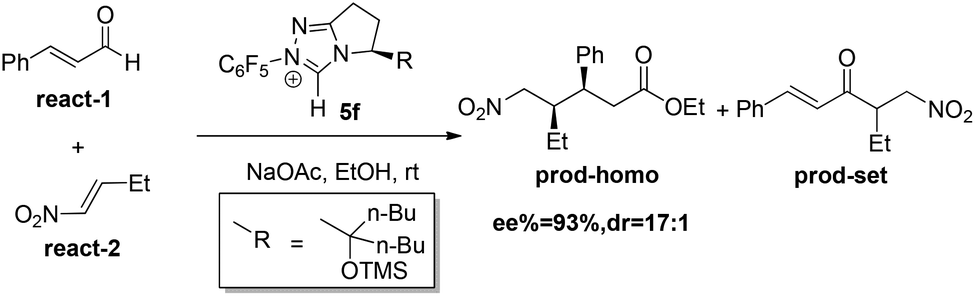 | ||
| Scheme 4 Model reaction used in the calculation. | ||
3. Results and discussion
3.1 Mechanisms for Path-homo and Path-set
Nucleophilic attack of 5f on react-1. As shown in Fig. 1, with the aid of OAc− (from the base NaOAc), deprotonation29 of the original catalyst 5f first occurs to yield the active catalyst cat-5f and HOAc (the conjugate acid of NaOAc). This process is endoergonic by 2.7 kcal mol−1. Then cat-5f attacks the substrate react-1via the C0–C1 forming transition state TS1 to generate the zwitterionic intermediate Int1. The free energy barrier of this step is 18.7 kcal mol−1 (5f + react-1 → TS1).
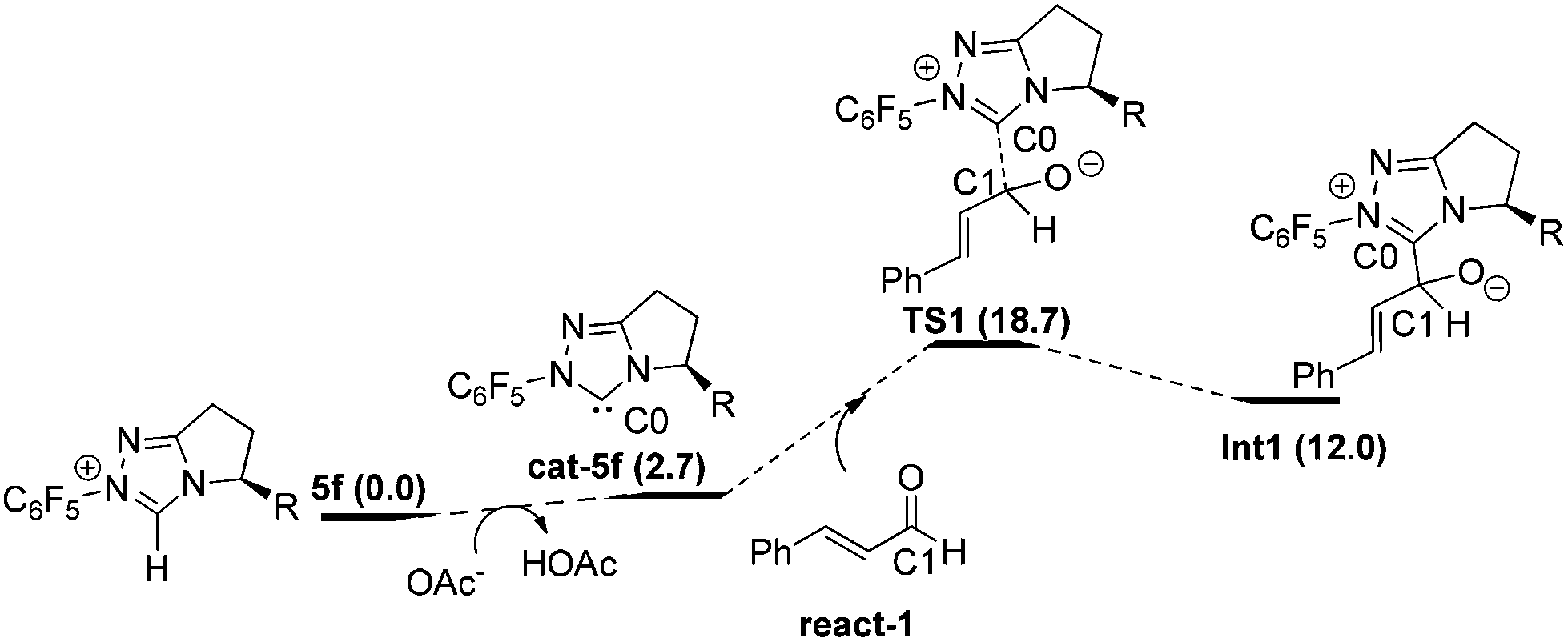 | ||
| Fig. 1 Nucleophilic attack of 5f to react-1. | ||
Proton transfer of Int1. Fig. 2 shows the detailed transformations and the energy demands of Path-a, b, c and d. In Path-a, a direct 1,2-proton transfer step occurs via the three-centered transition state TS2 to generate the Breslow intermediate Int2. The energy barrier of this process is 37.4 kcal mol−1 (Int1 → TS2), and the high energy barrier is in line with the kinetic investigation by White and Leeper.30 In Path-b, all attempts at locating the transition state (TS3) for the intermolecular proton transfer step between two Int1 molecules failed. The reason might be related to the high steric hindrance between the two carbene structures, which makes it difficult for the carbonyl oxygen atom of one Int1 to get close to the C1–H bond of another Int1. In Path-c, the indirect hydride transfer from the C1 atom to the C0 atom of the carbene first occurs via the three-centered transition state TS4-C0 to generate the intermediate Int3. Then with the aid of solvent HOEt, deprotonation of the C0 and protonation of the O atom occurs simultaneously via the five-centered transition state TS5 to obtain the Breslow intermediate Int2. The energy barrier of Path-c is 55.4 kcal mol−1 (Int3 → TS5). In Path-d, the hydride transfer from the C1 atom to the –C6F5 group first occurs via the six-centered transition state TS4-Ar. The free energy of this transition state is 44.3 kcal mol−1.
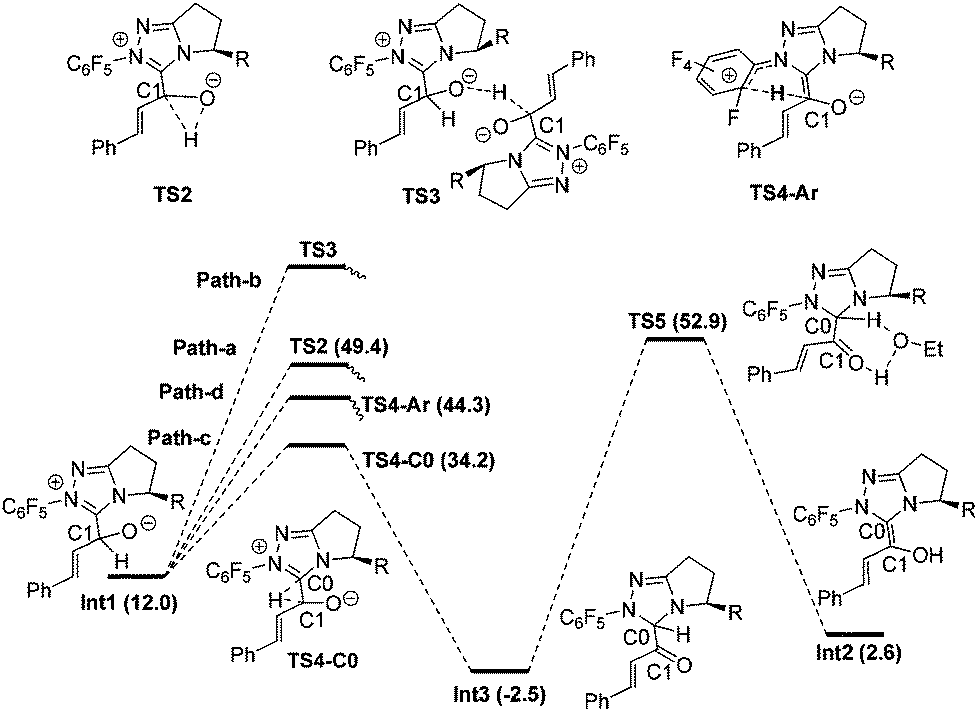 | ||
| Fig. 2 The mechanisms in the absence of HOAc. | ||
From Fig. 2, we found that the Path-a, b, c and d are all unlikely to occur due to the high energy demands and structural limitations.
Fig. 3 shows the detailed transformations and the energy demands of HOAc assisted Path-e and Path-f. In Path-e, the cleavage of C1–H bond and formation of hydroxyl O–H bond are supposed to occur simultaneously in the five-membered ring transition state TS6 or seven-membered2b ring transition state TS6-O. However, the elongated C1–H bond always reformed, and both TS6 and TS6-O turned into Int4 after optimization. In other words, the cleavage of C1–H bond with acid is difficult. The reason might be related to the fact that only a partial negative charge was formed on OAc, thus making it difficult to capture the proton of the C1–H bond. Indeed, a mechanism in which the OAc− anion directly participated in the reaction (Path-f) was found to be much more feasible. As shown in Fig. 3, the O atom of Int1 is first protonated by HOAc to give the intermediate Int4 and OAc−, then Int4 undergoes OAc− assisted C1–H cleavage via transition state TS7 to give the Breslow intermediate Int2. The energy barrier of Path-f is 12.7 kcal mol−1 (Int4 → TS7).31
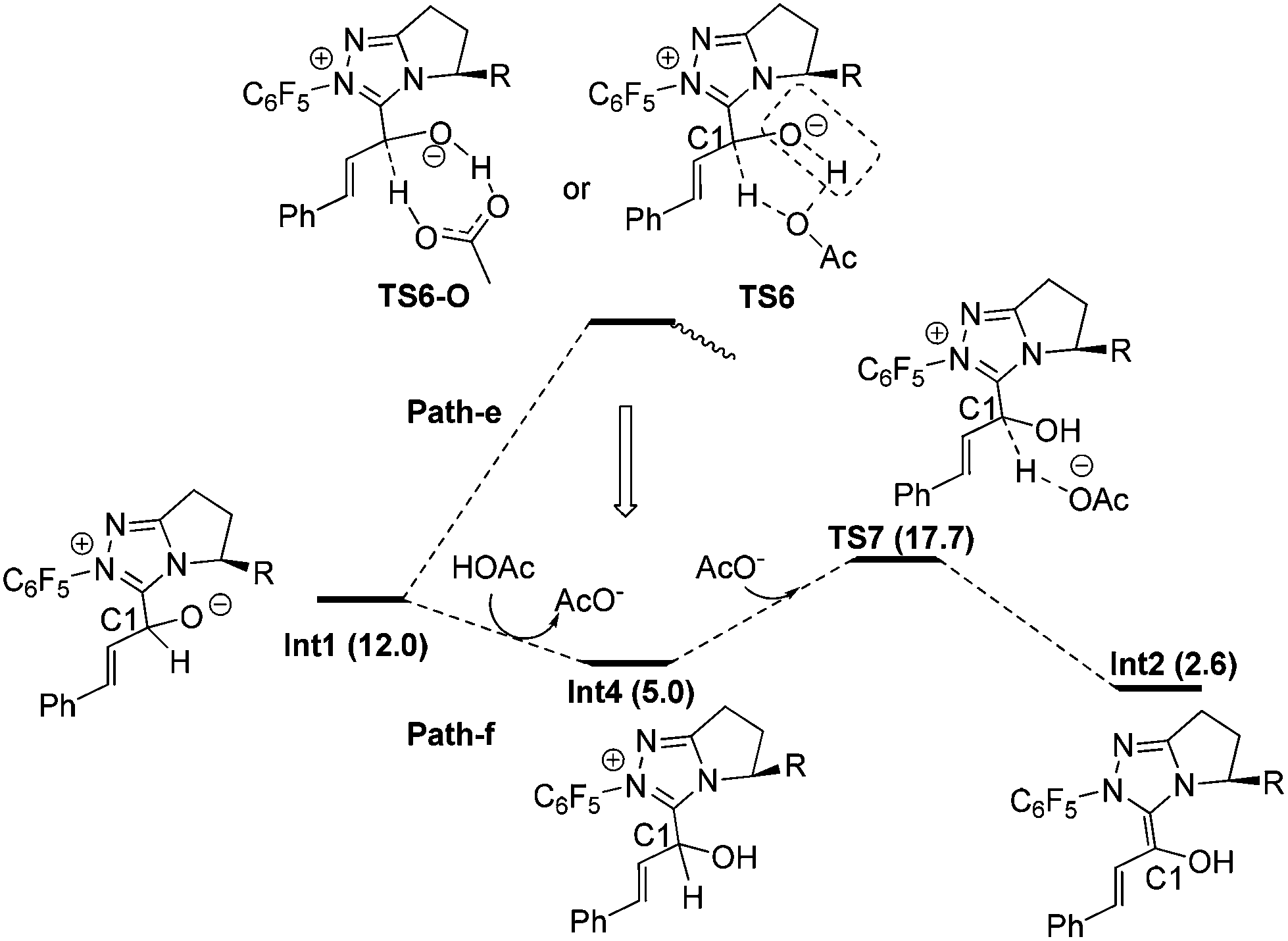 | ||
| Fig. 3 The mechanisms with the aid of HOAc. | ||
From the above calculation results and discussion, it may be concluded that the HOAc mediated stepwise Path-f is energetically more feasible than all the other paths (a–e). In paths a–e, the C1–H cleavage occurs before or simultaneously with O-protonation, whereas in Path-f protonation of the O atom occurs prior to the C1–H cleavage. This intriguing phenomenon suggests that the C1–H bond cleavage step can be greatly facilitated by the O atom protonation. The reason can be understood since in TS7 the negative charge accumulated on C1 during the proton dissociation can be dispersed by the positive charge located on the carbene ring. In contrast, the generated negative charge cannot be dispersed when it is close to the negatively charged O atom, thereby destabilizing the corresponding transition states (Path a–d). To verify this hypothesis, the transition state for the direct deprotonation of C1–H in the intermediate Int1 was located (TS-g in ESI†32). Consistent with our proposal, the relative free energy of TS-g is 28.8 kcal mol−1 higher than that of Int1. Therefore, we conclude that the charge dispersion during C1–H bond cleavage process determines the most favourable mechanism (i.e. Path-f).
Similar to Path-f, an alternative proton transfer process might occur to generate the isomer of Int2 (i.e. another Breslow intermediate Int5). As shown in Fig. 4, the rotation of the C0–C1 single bond occurs first to generate the intermediate Int4′. This intermediate then undergoes OAc− mediated deprotonation viaTS8, and finally results in the formation of Int5. According to the calculation results, the energy barrier of the transformation from Int4 to the Breslow intermediate Int5 (11.2 kcal mol−1) is slightly lower than for Int2 (12.7 kcal mol−1), while the relative Gibbs energy of Int5 (4.0 kcal mol−1) is higher than Int2 (2.6 kcal mol−1).33
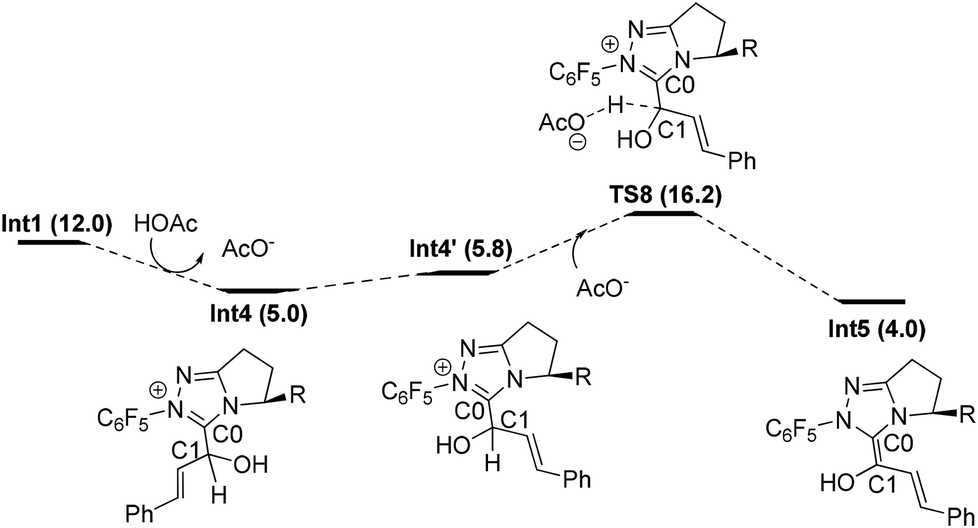 | ||
| Fig. 4 Proton transfer process to generate Int5. | ||
Nucleophilic attack of Int5 and Int2 on react-2. As shown in Scheme 5, nucleophilic attack of the C3 atom (in Int2 and Int5) on the C4 atom (in react-2) might occur via either the Si-face or Re-face, therefore four different modes (3-Si, 4-Si), (3-Si, 4-Re), (3-Re, 4-Si) and (3-Re, 4-Re) are possible in this step. Among these, the (3-Re, 4-Si) attack mode leads to the (3S, 4R) configuration product (prod-homo) in Rovis's experiment.12a As to Int2, the reaction proceeds via the transition state H-TS9-ReSi to directly generate the intermediate H-Int6 (see ESI† for the other three attack modes). Int5 first attacks react-2via the transition state H-TS9′-ReSi to generate the intermediate H-Int6′ (see the other three attack modes in section 3.3). Subsequently, H-Int6′ isomerizes to H-Int6via C0–C1 rotation with an energy release of 0.2 kcal mol−1. In both of the transition states (H-TS9-ReSi and H-TS9′-ReSi), hydrogen bond interactions exist between the nitro group and the hydroxyl (in our study, an open transition state in the absence of the intramolecular hydrogen bond was found to be unfavorable. Please see ESI† for more details). The energy barriers for the nucleophilic attack of Int2 and Int5 are 12.4 (Int2 → H-TS9-ReSi) and 11.3 kcal mol−1 (Int5 → H-TS9′-ReSi). Additionally, the Breslow intermediate Int5 is formed preferentially due to a favorable deprotonation transition state (free energy of TS8 is lower than TS7). Note that the C5 atom of H-Int6 accumulates much negative charge, and therefore it tends to be protonated in the following step.
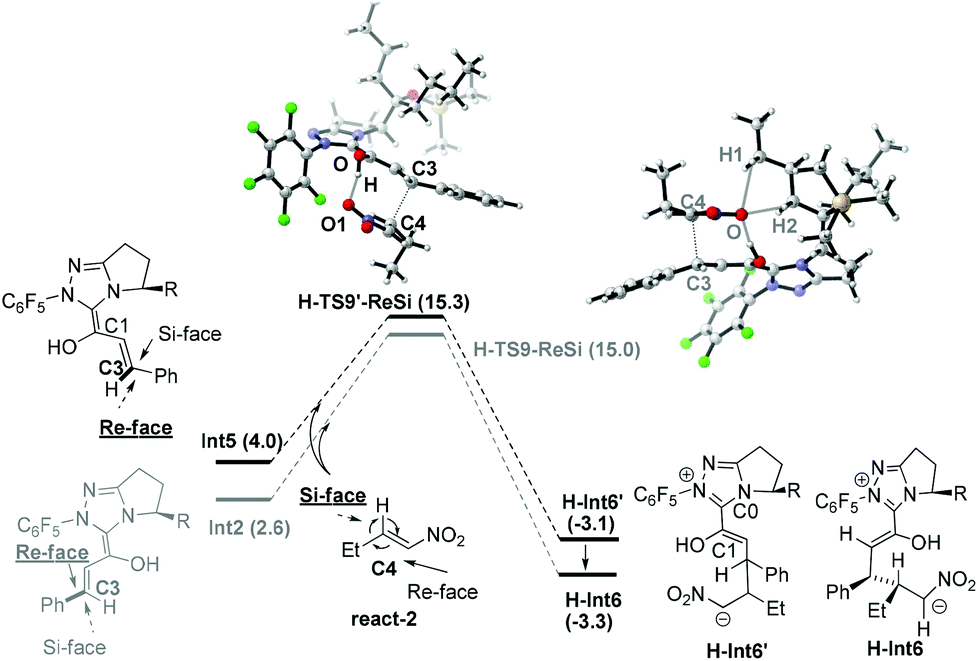 | ||
| Scheme 5 Nucleophilic attack of Int5 and Int2 to react-2. | ||
Protonation of C5 in H-Int6. Fig. 5 shows three possible protonation mechanisms of C5 in H-Int6. Nair et al.9c proposed that the C5 atom of H-Int6 might be directly protonated by the solvent HOEt to give the intermediate H-Int7 (i.e. Path-C5-1). This process is highly endoergonic by 25.6 kcal mol−1 (note: the transition state failed to be located, and the calculations indicate that such process is continuously endoergonic). As mentioned in the introduction, the high energy demand might be attributed to the weak acidity of HOEt, and therefore we investigated the more acidic HOAc as a potential participant in the protonation (i.e. Path-C5-2). From H-Int6, HOAc participated protonation occurs via the transition state H-TS10 to give the intermediate H-Int8 and OAc−. The OAc− anion subsequently captures the proton of hydroxyl in H-Int8 to generate H-Int9. The energy barrier of Path-C5-2 is 14.6 kcal mol−1 (H-Int6 → H-TS10) and it is exoergonic by 7.5 kcal mol−1. Thus, this result seems to suggest that the energy release of hydroxyl deprotonation may compensate the demand of C5 protonation. Thus, we investigated the concerted mechanism (i.e. Path-C5-3). In this mechanism, H-Int6 undergoes intramolecular protonation via the concerted transition state H-TS11 to directly obtain intermediate H-Int9. As expected, the energy barrier of Path-C5-3 is only 7.1 kcal mol−1 (H-Int6 → H-TS11). Therefore, the newly found intramolecular protonation Path-C5-3 is energetically more favorable.
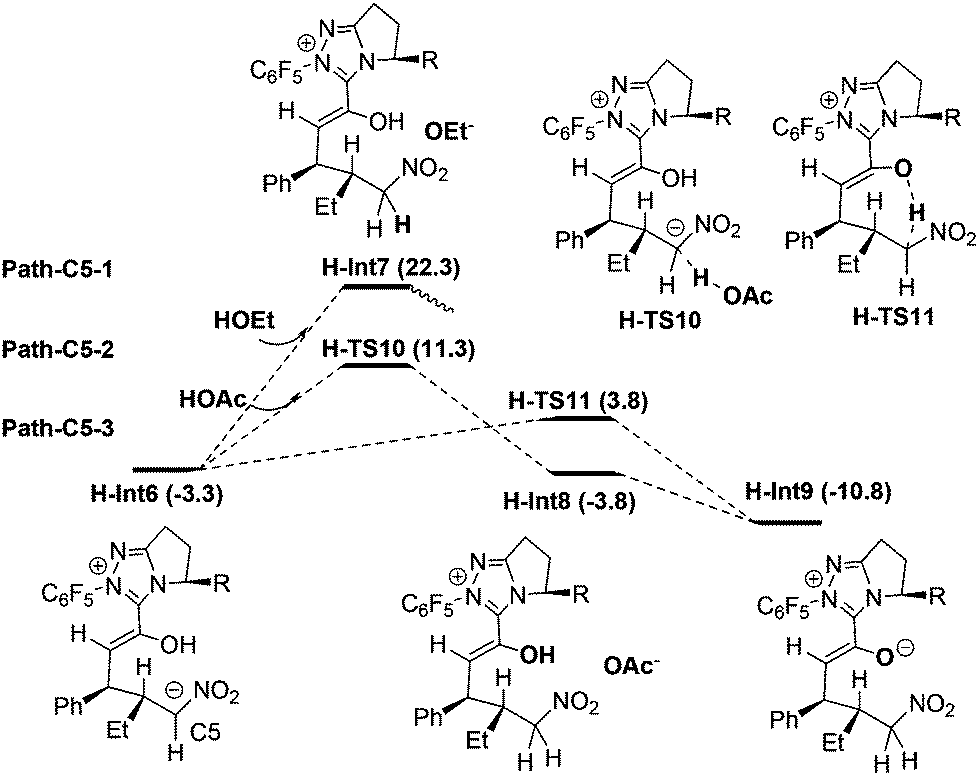 | ||
| Fig. 5 The energy profiles of protonation processes. | ||
Enol–keto tautomerisation and attack of OEt to C1 in H-Int9. As mentioned in the introduction, the obtained intermediate H-Int9 from the above protonation process undergoes enol–keto tautomerisation (i.e. C2 protonation) and attack of HOEt to C1 to obtain prod-homo. The detailed energy profile for this process is given in Fig. 6 (Path-1).
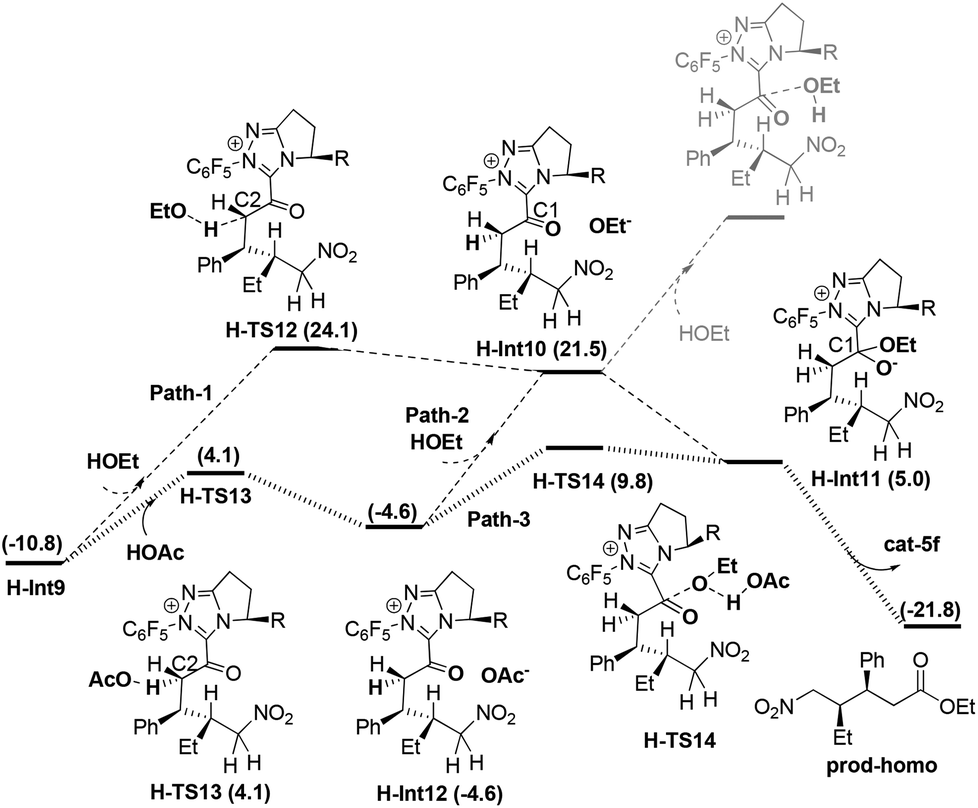 | ||
| Fig. 6 The energy profiles for paths 1–3. | ||
In Path-1, the solvent HOEt first protonates C2 via the transition state H-TS12 which results in an enol–keto tautomerisation. This step generates the intermediate H-Int10 and OEt−, with an energy barrier of 34.9 kcal mol−1 (H-Int9 → H-TS12) and an energy absorption of 32.3 kcal mol−1. Subsequently, HOEt attacks the C1 of H-Int10, resulting in automatic dissociation. Considering that the dissociation of HOEt might be caused by the weak nucleophilicity of HOEt, we allowed the in situ produced OEt− to attack the C1 atom to obtain H-Int11. This process is exoergonic by 16.5 kcal mol−1. Finally the carbene group dissociates from H-Int11 to yield prod-homo. The energy barrier of Path-1 is 34.9 kcal mol−1 (H-Int9 → H-TS12).
As mentioned in Path-C5-1, the weak acidity of HOEt could lead to the high energy demand of the protonation (34.9 kcal mol−1), and we therefore further investigated the more acidic HOAc mediated protonation (Path-2). The intermediate H-Int9 undergoes HOAc-mediated C2 protonation viaH-TS13 to generate H-Int12 and OAc−. As expected, the energy barrier for this step is only 14.9 kcal mol−1. Subsequently, OAc− captures the proton of HOEt, and the produced OEt− attacks the C1 atom. This step is endoergonic by 16.5 kcal mol−1. Thus, the overall energy barrier of Path-2 is 32.3 kcal mol−1 (H-Int9 → H-Int10). Although Path-2 is favored over Path-1, the energy barriers of both processes are too high to be overcome in Rovis's experiment. Thus, we wonder whether there is a more plausible mechanism for the enol–keto tautomerisation and attack of OEt on C1.
One should note that the direct breakage of H–OEt by OAc− is highly endoergonic while the attack of OEt− on the carbonyl is highly exoergonic in Path-2. With this in mind, we tried to investigate Path-3 in which the above two steps occur simultaneously (Fig. 6). From H-Int12, OAc− behaves as a base to capture the H atom of HOEt, meanwhile the OEt group attacks the C1 atom (shown as concerted transition state H-TS14). Gratifyingly, the energy barrier of this step is 14.4 kcal mol−1 (H-Int12 → H-TS14). Finally, the carbene ring dissociates to give prod-homo. Therefore, the overall energy barrier of Path-3 is 20.6 kcal mol−1 (H-Int9 → H-TS14), and this HOAc involved enol–keto tautomerisation and concerted attack of OEt to C1 process is the most favored. (See the ESI† for another three possible mechanisms Paths 4–6.)
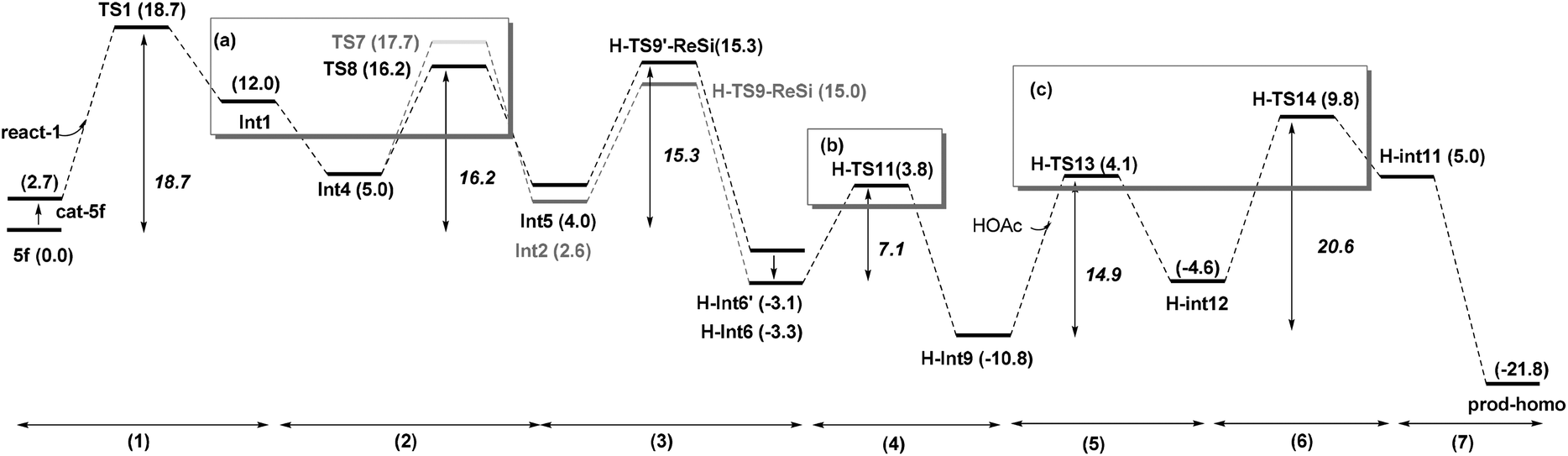 | ||
| Fig. 7 The whole process of Path-homo. | ||
Based on these conclusions, the whole process of Path-homo is shown in Fig. 7. It consists of seven steps: (1) nucleophilic attack of NHC on the enal, (2) HOAc assisted stepwise proton transfer, (3) nucleophilic attack of the Breslow intermediate (C3 atom) on the nitroalkene, (4) intramolecular proton transfer (5) HOAc-mediated protonation of C2, (6) concerted nucleophilic attack of OEt on the carbonyl and (7) carbene dissociation. In the second step, there are two different proton transfer transition states, TS8 and TS7, leading to the Breslow intermediates Int5 and Int2. Since TS8 is more favorable than TS7, the process from Int4 should be Int4 → Int5 → H-TS9′-ReSi → H-Int6′ → H-Int6. The energy barriers of the first six steps are 18.7, 16.2, 15.3, 7.1, 14.9 and 20.6 kcal mol−1 respectively. The sixth step, i.e. the concerted nucleophilic attack of OEt to carbonyl, is the rate determining step with an overall energy barrier of 20.6 kcal mol−1 (H-Int9 → H-TS14).
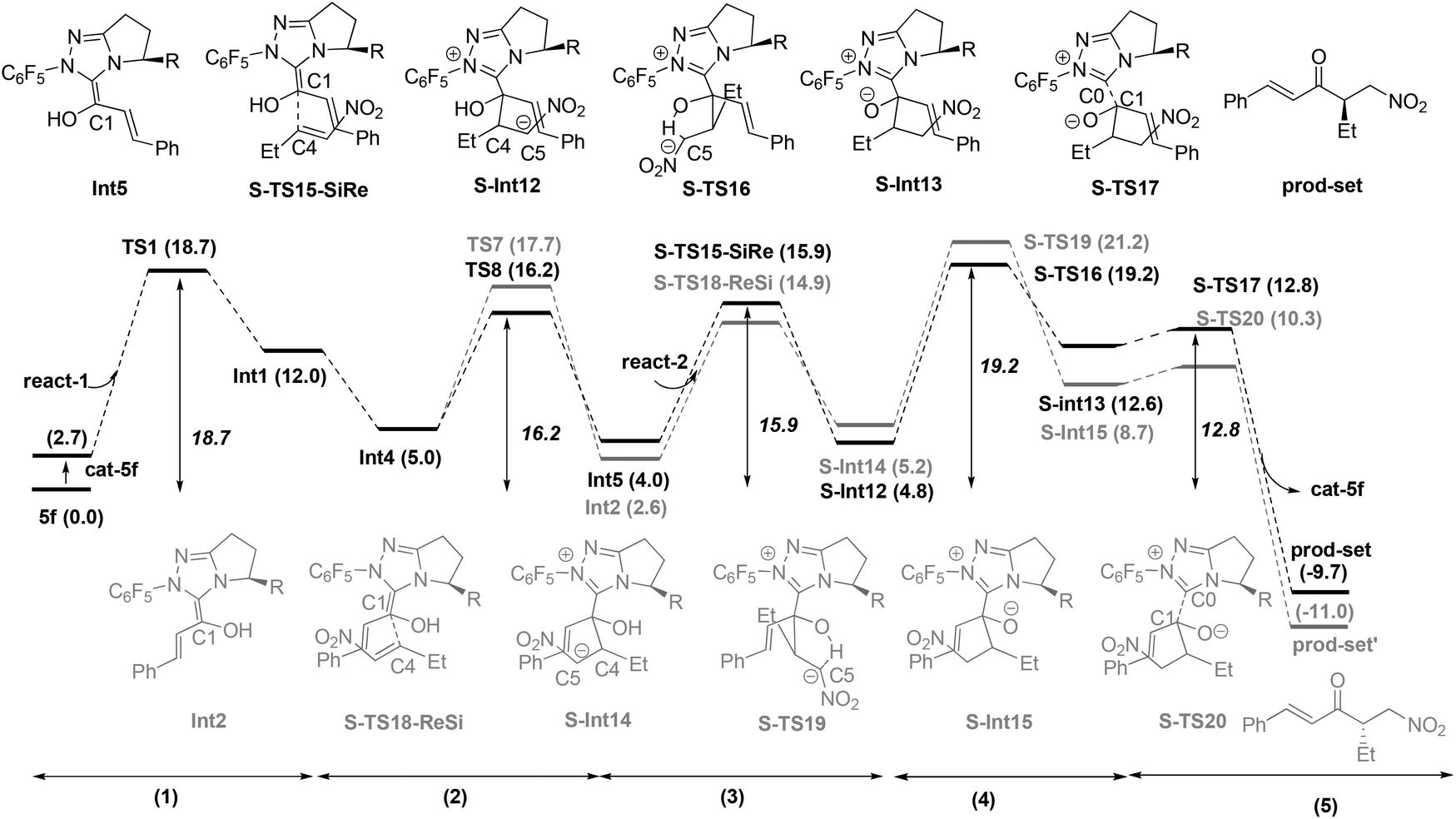 | ||
| Fig. 8 The whole process of Path-set. | ||
The steps following from the Breslow intermediate Int2 resemble those from Int5. First, the Re-face of C1 (in Int2) attacks the Si-face of C4 (in substrate react-2) with an energy barrier of 14.9 kcal mol−1 (5f + react-1 → S-TS18-ReSi).34 Subsequently, intramolecular protonation occurs with an energy barrier of 21.2 kcal mol−1 (5f + react-1 → S-TS19). Finally, product prod-set′ is formed after rapid carbene dissociation. The intramolecular protonation has the highest energy barrier (21.2 kcal mol−1). Thus, the overall energy barriers of the subsequent steps from Int5 and Int2 are 19.2 and 21.2 kcal mol−1, respectively. Therefore the former is more favourable.
Based on the above results, the whole process of Path-set consists of five steps: (1) nucleophilic attack of NHC on the enal, (2) HOAc assisted stepwise proton transfer, (3) nucleophilic attack of the Breslow intermediate (C1 atom) on the nitroalkene, (4) intramolecular proton transfer and (5) carbene dissociation. The energy barriers of these five steps are 18.7, 16.2, 15.9, 19.2 and 12.8 kcal mol−1 respectively. The overall energy barrier of Path-set is 19.2 kcal mol−1 (cat-5f + react-1 → S-TS16).
3.2 Discussions on chemoselectivity
Path-homo and Path-set diverge starting from the Breslow intermediate Int5 (Fig. 9). Herein, we use the energetic span model35 to compare Path-homo and Path-set. Since Int5 first undergoes a highly exoergonic nucleophilic attack from the C3 atom and a kinetically facile intramolecular proton transfer to obtain the most stable intermediate H-Int9 in the reaction system, this intermediate is the TDI for Path-homo and Path-set. For Path-homo, the transition state for the attack of OEt on the carbonyl (H-TS14) is the TDTS, and the related δE is 20.6 kcal mol−1. For Path-set, the intramolecular proton transfer transition state S-TS16 is the TDTS, and the related δE is 30.0 kcal mol−1. Therefore, the δE of Path-homo is smaller than for Path-set, and Path-homo has a larger TOF. In addition, the product of Path-homo (prod-homo) is thermodynamically more stable than the product of Path-set (prod-set). Therefore, Path-homo is kinetically and thermodynamically more favored than Path-set, and this conclusion is consistent with Rovis's experiment12a where the homoenolate product was obtained predominantly. Furthermore, the highly exoergonic nucleophilic attack (i.e. the thermodynamic stability of H-Int6) and the facile subsequent steps (intramolecular proton transfer and attack of OEt on the carbonyl) in Path-homo result in the feasibility of the homoenolate reaction.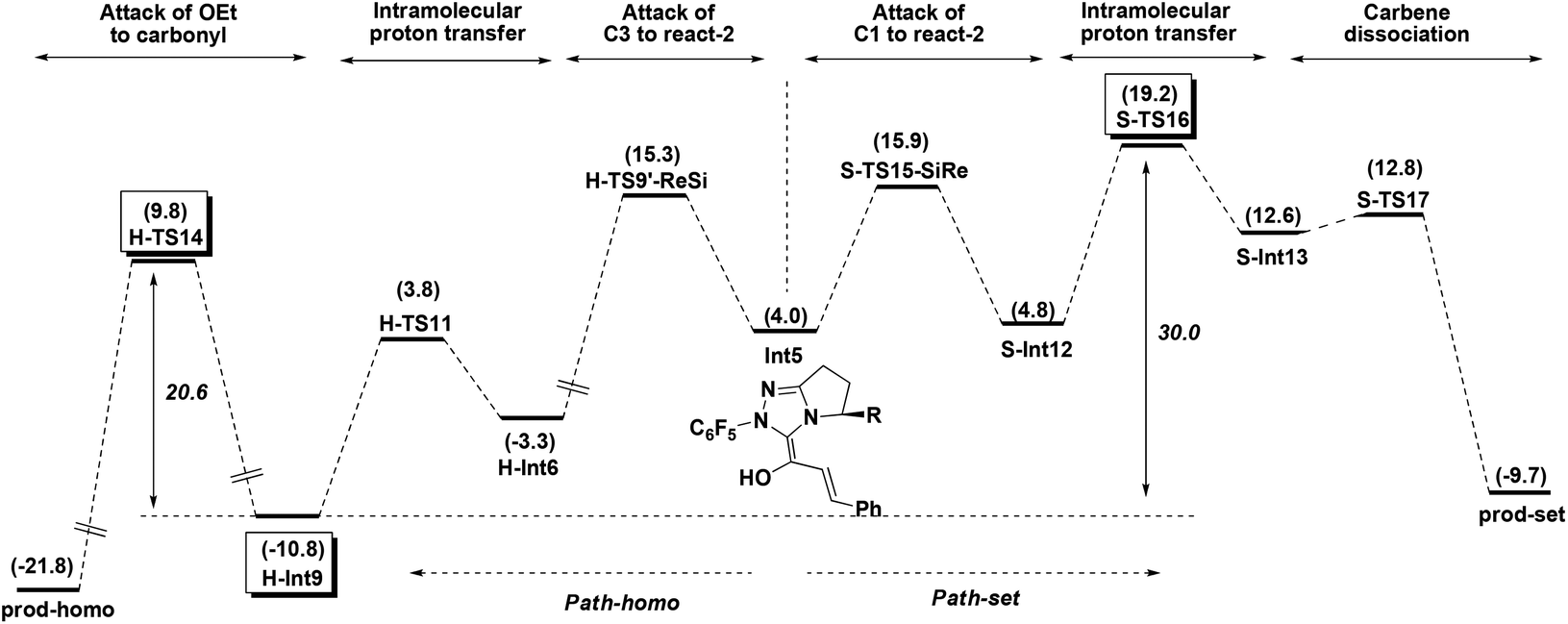 | ||
| Fig. 9 Comparison between Path-homo and Path-set. | ||
3.3 Discussions on the stereoselectivity of Path-homo
The nucleophilic attack of the Breslow intermediate Int5 on the nitroalkene react-2 determines the stereoselectivity of Path-homo. As shown in Fig. 10, four different attack modes (3-Re, 4-Si), (3-Re, 4-Re), (3-Si, 4-Re) and (3-Si, 4-Si) are possible in this step. Their corresponding transition states are H-TS9′-ReSi, H-TS9′-ReRe, H-TS9′-SiRe and H-TS9′-SiSi, and they finally lead to (3S, 4R) (3S, 4S) (3R, 4S) and (3R, 4R) configuration products. The Gibbs free energies of these four transition states are 15.3, 17.8, 18.9 and 23.1 kcal mol−1, respectively. Therefore, the (3-Re, 4-Si) attack mode leading to the syn-configuration product (prod-homo) is the most favourable mechanism. The second most favourable attack mode (3-Re, 4-Re) leads to the anti-configuration product. The calculation results are consistent with the unexpected syn configuration product obtained in Rovis's experiment.12a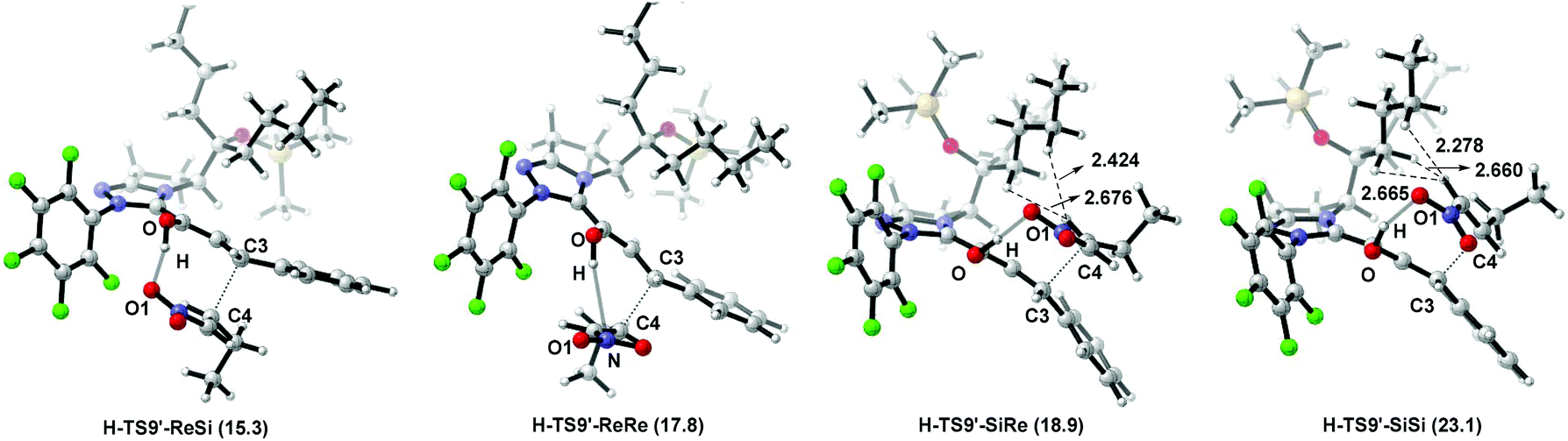 | ||
| Fig. 10 Transition states for nucleophilic attack of Int5 to react-2. | ||
Structural observations on the former two transition states (H-TS9′-ReSi and H-TS9′-ReRe) show that the C4 plane is below the C3 plane (Fig. 10). Therefore, the Et group of substrate react-2 is far away from the alkyl substituent (i.e. R) on the carbene ring, and little steric effect exists between them. In addition, the former transition state is more favored than the latter, because of a larger hydrogen bonding interaction between O–H⋯O1 in the former case (the bond distances are 1.656 Å and 3.099 Å in H-TS9′-ReSi and H-TS9′-ReRe). In contrast, in the latter two transition states (H-TS9′-SiRe and H-TS9′-SiSi), large steric effects exist between –Et and –nBu reflected by H⋯H interactions (bond length of H1⋯H2 and H1⋯H3 in H-TS9′-SiRe are 2.424 Å and 2.676 Å, while they are 2.278 Å and 2.665 Å in H-TS9′-SiSi).36
Based on the above discussion, we propose that the less steric hindrance between Et and nBu and stronger hydrogen bonding interaction between O–H⋯O1 in H-TS9′-ReSi result in the observed syn stereoselectivity.
4. Conclusion
Recently, the Rovis group reported a homoenolate reaction between alkyl substituted nitroalkenes and enals, obtaining a syn configuration product accompanied by trace amounts of Stetter byproduct. In the present study, we carried out DFT calculations to investigate the detailed mechanism of the nitroalkene participated homoenolate reaction, and we explain the origin of the homoenolate chemoselectivity and syn stereoselectivity. Our calculations indicate that: Path-homo and Path-set first share the same formation process of Breslow intermediates. This proceeds via HOAc mediated Path-f (rather than Path-a to e), due to the good charge dispersion during the C1–H bond cleavage process. Subsequently, Path-homo consists of five steps: nucleophilic attack of the Breslow intermediate (C3 atom) on the nitroalkene; intramolecular proton transfer (rather than HOEt or HOAc mediated stepwise mechanism, because the energy release of hydroxyl deprotonation can compensate for the demand of the C5 protonation); HOAc involved protonation of C2; concerted nucleophilic attack of OEt on the carbonyl (rather than HOEt or HOAc involved stepwise mechanism, since the attack of OEt− on the carbonyl can provide the energy required in the breakage of H–OEt by OAc−); and final carbene dissociation. Path-set, on the other hand, subsequently consists of three steps: nucleophilic attack of the Breslow intermediate (C1 atom) on the nitroalkene, intramolecular proton transfer and carbene dissociation. Path-homo is more feasible than Path-set. The thermodynamic stability of H-Int6 and the facile intramolecular proton transfer and attack of OEt on the carbonyl in Path-homo result in its feasibility. Furthermore, the stereoselectivity determining step of Path-homo is the attack of the Breslow intermediate on the nitroalkene, in which the steric hindrance and hydrogen bonding determine the syn stereoselectivity.Acknowledgements
We thank the NSFC (21325208, 21172209, 21361140372, 21202006), SRFDP (20123402110051), FRFCU (WK2060190025), CAS (KJCX2-EW-J02), Fok Ying Tung Education Foundation, ChinaGrid project funded by MOE of China, and the super-computer center of Shanghai and USTC.Notes and references
- (a) D. Enders, O. Niemeier and A. Hensler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (c) J. Read de Alaniz and T. Rovis, Synlett, 2009, 1189 CAS; (d) H. U. Vora and T. Rovis, Aldrichimica Acta, 2011, 44, 3 CAS; (e) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (f) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS PubMed; (g) E. M. Phillips, A. Chan and K. A. Scheidt, Aldrichimica Acta, 2009, 42, 55 CAS; (h) J. L. Moore and T. Rovis, Top Curr. Chem., 2010, 291, 77 CrossRef CAS; (i) P. C. Chiang and J. W. Bode, TCI Mail, 2011, 149, 2 Search PubMed; (j) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182 CrossRef CAS PubMed.
- (a) H. Stetter and H. Kuhlmann, in Org. React., ed. L. A. Paquette, Wiley, New York, 1991, vol. 40, pp. 407–496 Search PubMed; (b) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed; (c) D. A. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872 CrossRef CAS PubMed; (d) M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298 CrossRef CAS PubMed; (e) Q. Liu, S. Perreault and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14066 CrossRef CAS PubMed.
- (a) D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J. P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021 CrossRef CAS; (b) D. Enders, K. Breuer and J. H. Teles, Helv. Chim. Acta, 1996, 79, 1217 CrossRef CAS; (c) D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743 CrossRef CAS; (d) R. L. Knight and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 1998, 1891 RSC.
- Z. Q. Rong, W. Zhang, G. Q. Yang and S. L. You, Curr. Org. Chem., 2011, 15, 3077 CrossRef CAS.
- (a) S. P. Lathrop and T. Rovis, J. Am. Chem. Soc., 2009, 131, 13628 CrossRef CAS PubMed; (b) C. M. Filloux, S. P. Lathrop and T. Rovis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20666 CrossRef CAS PubMed; (c) G. L. Zhao and A. Córdova, Tetrahedron Lett., 2007, 48, 5976 CrossRef CAS PubMed; (d) H. Jiang, B. Gschwend, L. Albrecht and K. A. Jørgensen, Org. Lett., 2010, 12, 5052 CrossRef CAS PubMed.
- V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC.
- (a) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS PubMed; (b) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131 CrossRef CAS PubMed; (c) P. C. Chiang, J. Kaeobamrung and J. W. Bode, J. Am. Chem. Soc., 2007, 129, 3520 CrossRef CAS PubMed; (d) M. Rommel, T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 17266 CrossRef CAS PubMed.
- C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS PubMed.
- (a) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS PubMed; (b) V. Nair, M. Poonoth, S. Vellalath, E. Suresh and R. Thirumalai, J. Org. Chem., 2006, 71, 8964 CrossRef CAS PubMed; (c) V. Nair, C. R. Sinu, B. P. Babu, V. Varghese, A. Jose and E. Suresh, Org. Lett., 2009, 11, 5570 CrossRef CAS PubMed; (d) V. Nair, B. P. Babu, S. Vellalatha and E. Suresh, Chem. Commun., 2008, 747 RSC.
- (a) E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 2416 CrossRef CAS PubMed; (b) B. C. David, D. E. A. Raup and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 5345 CrossRef PubMed; (c) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed; (d) D. T. Cohen, B. C. David and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 1678 CrossRef CAS PubMed; (e) M. Wadamoto, E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 10098 CrossRef CAS PubMed.
- B. Maji, L. Ji, S. Wang, S. Vedachalam, R. Ganguly and X. W. Liu, Angew. Chem., Int. Ed., 2012, 51, 8276 CrossRef CAS PubMed.
- (a) N. A. White, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2013, 135, 8504 CrossRef CAS PubMed; (b) X. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466 CrossRef CAS PubMed.
- Mechanism studies of Stetter reaction: (a) J. L. Moore, A. P. Silvestri, J. Read de Alaniz, D. A. DiRocco and T. Rovis, Org. Lett., 2011, 13, 1742 CrossRef CAS PubMed; (b) J. M. Um, D. A. DiRocco, E. L. Noey, T. Rovis and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 11249 CrossRef CAS PubMed; (c) D. A. DiRocco, E. L. Noey, K. N. Houk and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 2391 CrossRef CAS PubMed.
- (a) J. Martí, F. Lopez-Calahorra and J. M. Bofill, THEOCHEM, 1995, 339, 179 CrossRef; (b) F. Lopez-Calahorra, J. Castells, L. Doming, J. Martí and J. M. Bofill, Heterocycles, 1994, 37, 1579 CrossRef.
- K. J. Hawkes and B. F. Yates, Eur. J. Org. Chem., 2008, 5563 CrossRef CAS.
- P. Verma, P. A. Patni and R. B. Sunoj, J. Org. Chem., 2011, 76, 5606 CrossRef CAS PubMed.
- L. R. Domingo, M. J. Aurell and M. Arnó, Tetrahedron, 2009, 65, 3432 CrossRef CAS PubMed.
- Y. Q. He and Y. Xue, J. Phys. Chem. A, 2011, 115, 1408 CrossRef CAS PubMed.
- C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514 RSC.
- J.-S. Zheng, H.-N. Chang, F.-L. Wang and L. Liu, J. Am. Chem. Soc., 2011, 133, 11080 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- The DFT method B3LYP has been used in many theoretical studies for organic reaction systems: (a) Y. Feng, L. Liu, J.-T. Wang, S.-W. Zhao and Q.-X. Guo, J. Org. Chem., 2004, 69, 3129 CrossRef CAS PubMed; (b) Y. Q. He and Y. Xue, J. Phys. Chem. A, 2010, 114, 9222 CrossRef CAS PubMed; (c) R. Shang, Q. Xu, Y.-Y. Jiang, Y. Wang and L. Liu, Org. Lett., 2010, 12, 1000 CrossRef CAS PubMed; (d) Q. Zhang, H. Z. Yu and J. Shi, Acta Phys.-Chim. Sin., 2013, 29, 2321 CAS; (e) C. Wang and L. Liu, Chin. J. Chem., 2012, 30, 1974 CrossRef CAS; (f) M. N. Paddon-Row, C. D. Anderson and K. N. Houk, J. Org. Chem., 2009, 74, 861 CrossRef CAS PubMed; (g) A. E. Hayden and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 4084 CrossRef CAS PubMed.
- A. W. Ehlers, M. Bohme, S. Dapprich, A. Gobbi, A. Hollwarth, V. Jonas, K. F. Kohler, R. Stegmman, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS.
- (a) H. Z. Yu, D. J. Liu, Z. M. Dang, D. R. Wang and Y. Fu, Chin. J. Chem., 2013, 31, 200 CrossRef CAS; (b) M. S. T. Morin, D. J. St-Cyr, B. A. Arndtsen, E. H. Krenske and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17349 CrossRef CAS PubMed; (c) Q. L. Xu, H. Gao, M. Yousufuddin, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2013, 135, 14048 CrossRef CAS PubMed; (d) R. Shang, Z.-W. Yang, Y. Wang, S.-L. Zhang and L. Liu, J. Am. Chem. Soc., 2010, 132, 14391 CrossRef CAS PubMed; (e) A. Adhikary, A. Kumar, B. J. Palmer, A. D. Todd and M. D. Sevilla, J. Am. Chem. Soc., 2013, 135, 12827 CrossRef CAS PubMed.
- A. Hollwarth, M. Bohme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Kohler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- The M06 method has been used in many metal free systems for single-point energy calculations: (a) J. M. Lee, P. Helquist and O. Wiest, J. Am. Chem. Soc., 2012, 134, 14973 CrossRef CAS PubMed; (b) O. L. Griffith, J. E. Anthony, A. G. Jones, Y. Shu and D. L. Lichtenberger, J. Am. Chem. Soc., 2012, 134, 14185 CrossRef CAS PubMed; (c) Y. Chen and M. T. Rodgers, J. Am. Chem. Soc., 2012, 134, 5863 CrossRef CAS PubMed; (d) Y. Chen and M. T. Rodgers, J. Am. Chem. Soc., 2012, 134, 2313 CrossRef CAS PubMed.
- (a) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS; (b) A. Lapworth, J. Chem. Soc., 1903, 83, 995 RSC; (c) H. Z. Yu, Y. Fu, L. Liu and Q.-X. Guo, Chin. J. Org. Chem., 2007, 27, 545 CAS.
- M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124 CrossRef CAS PubMed.
- We took the proton transfer step (Path-f) as an example to investigate the influence of hydrogen bonding, and it turned out that hydrogen bonding induced by solvent is unlikely to occur. Please see ESI† for details.
- Mechanism Path-g was also examined in which C1–H bond of Int1 break first via TS-g. See the ESI† for more details.
- We have investigated the formation of the diastereomer of Int1 and the transition states for conversion of both diastereomers into Int2 and Int5. Detailed results are given in the ESI.†.
- Other three attack mode (1-Si, 4-Si), (1-Re, 4-Re), (1-Re, 4-Si) of Int5, and another three attack mode (1-Si, 4-Si), (1-Re, 4-Re), (1-Si, 4-Re) of Int2 in Stetter reaction are placed in ESI.†.
- (a) S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101 CrossRef CAS PubMed; (b) Z. Li, C. Wang, Y. Fu, Q.-X. Guo and L. Liu, J. Org. Chem., 2008, 73, 6127 CrossRef CAS PubMed; (c) C. Wang, Y. Fu, Q.-X. Guo and L. Liu, Chem. – Eur. J., 2010, 16, 2586 CrossRef CAS PubMed; (d) S.-L. Zhang, Y. Fu, R. Shang, Q.-X. Guo and L. Liu, J. Am. Chem. Soc., 2010, 132, 638 CrossRef CAS PubMed; (e) Y. Y. Jiang, H. Z. Yu and Y. Fu, Organometallics, 2013, 32, 926 CrossRef CAS.
- (a) X. Hong, P. Liu and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 1456 CrossRef CAS PubMed; (b) A. Patel, G. A. Barcan, O. Kwon and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 4878 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full text for ref. 31–34, Cartesian coordinates, free energies and thermal corrections. See DOI: 10.1039/c4qo00036f |
| This journal is © the Partner Organisations 2014 |