Intramolecular C–H⋯F hydrogen bonding-induced 1,2,3-triazole-based foldamers†
Yan-Hua
Liu
a,
Liang
Zhang
b,
Xiao-Na
Xu
a,
Zhi-Ming
Li
*b,
Dan-Wei
Zhang
b,
Xin
Zhao
*a and
Zhan-Ting
Li
*ab
aShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: xzhao@mail.sioc.ac.cn; Fax: +86 21 64166128; Tel: +86 21 54925122
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: ztli@fudan.edu.cn
First published on 10th April 2014
Abstract
This paper reports the first series of artificial secondary structures that are induced by intermolecular C–H⋯F hydrogen bonding. Oligomers that contain two, four, six, and eight 1,2,3-triazole units have been designed and prepared by connecting the neighbouring 1,2,3-triazole units with 4,6-difluoro-m-phenylene linker(s). Two triphenylmethyl groups are appended at the ends of the backbones to suppress the stacking of the backbones and provide solubility. X-Ray analysis and 1H NMR and two-dimensional 1H–19F heteronuclear Overhauser enhancement spectroscopic (HOESY) experiments support that the 1,2,3-triazole backbones adopt folded or helical conformations due to the formation of continuous three-centred C–H⋯F hydrogen bonding. Quantum chemical calculations reveal that the longest 8-mer foldamer can form a one-turn helical cavity with a diameter of ca. 1.7 nm. Halide anion competition experiments show that the intramolecular C–H⋯F hydrogen bonding is more stable than the well-established intermolecular C–H⋯X− (X = Cl and I) hydrogen bonding.
Introduction
In the last two decades, chemists have shown considerable interest in constructing foldamers, the artificial secondary structures that are stabilized by non-covalent interactions.1–3 Particularly, foldamers of aromatic backbones have found extensive applications as synthetic receptors for various molecules and ions.4 They are also useful preorganized frameworks for assembling advanced supramolecular architectures,5 facilitating the formation of macrocyclic and capsular structures,6 and developing new biologically active molecules.7 Due to its strength and directionality, hydrogen bonding has been most widely used to drive the formation of folded conformations for aromatic oligomers.3 In this context, oligomeric aromatic amides have been extensively investigated because of the structural diversity and modifiability of the aromatic segments and the high ability of the amide unit to serve as a hydrogen bonding donor.3,8 In most cases, the folding of the backbones is driven by strong intramolecular N–H⋯O or N–H⋯N hydrogen bonding.3 However, N–H⋯F or N–H⋯Cl hydrogen bonding has also been demonstrated to realize this process.8c,9–11
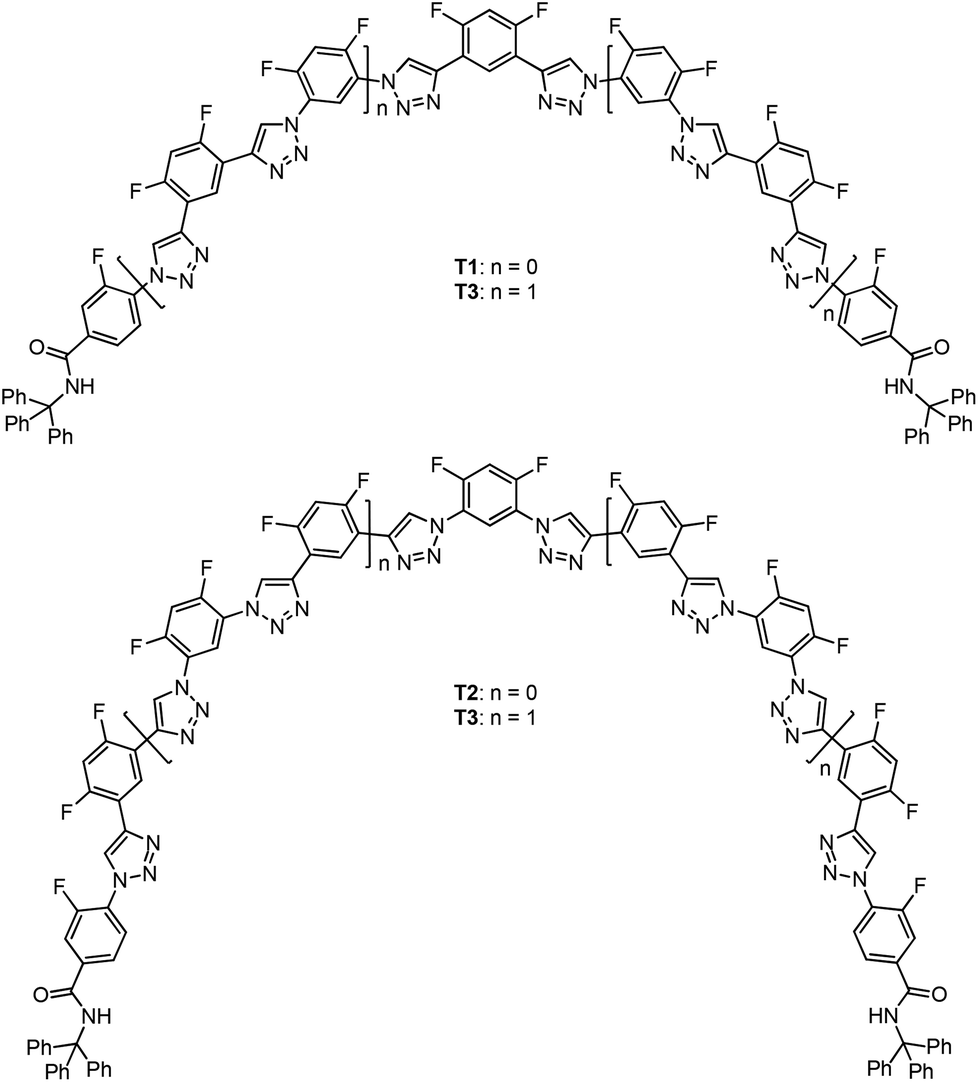
Results and discussion
Because the newly designed 1,2,3-triazole backbones bear no flexible chains, two bulky triphenylmethyl groups were appended at the ends of the backbones to suppress their stacking and provide solubility in organic solvents. The synthetic route for 2-mer T1 is shown in Scheme 1. Thus, acid 1 was first treated with oxalyl chloride to afford acyl chloride 2, which then reacted with amine 3 to produce 4. This nitro derivative underwent Pd-catalyzed hydrogenation to give amine 5, which was treated with sodium nitrite in hydrochloric acid to produce azide 6. Finally, the “click” 1,3-cycloaddition reaction of 6 with diacetylene 7 afforded both T1 and 8.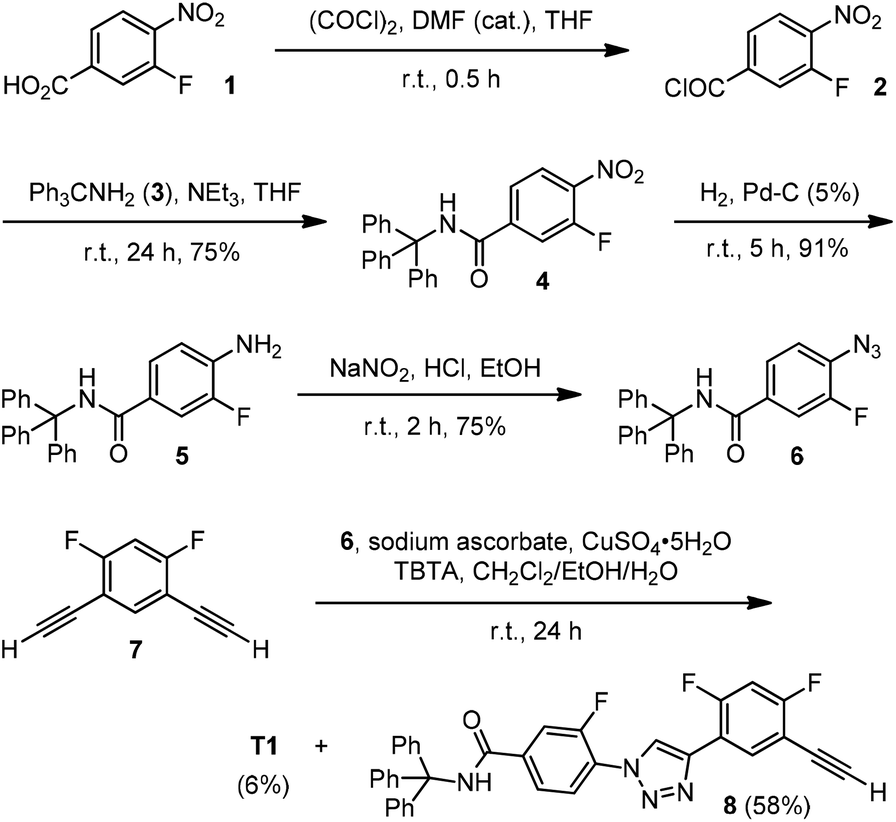 | ||
| Scheme 1 The synthesis of compounds T1 and 8. | ||
For the synthesis of 4-mer T2 (Scheme 2), amine 9 was first converted to azide 10 by treating with sodium nitrite in trifluoroacetic acid (TFA). The azide was then reacted with 7 to afford triazole derivative 11, which was further reduced with iron and ammonium chloride in aqueous ethanol to give amine 12. The amine was then reacted with sodium nitrite in TFA to produce azide 13. The 1,3-cycloaddition of 13 with excess of 7 afforded diacetylene 14. Compound T2 was finally prepared from its reaction with azide 6.
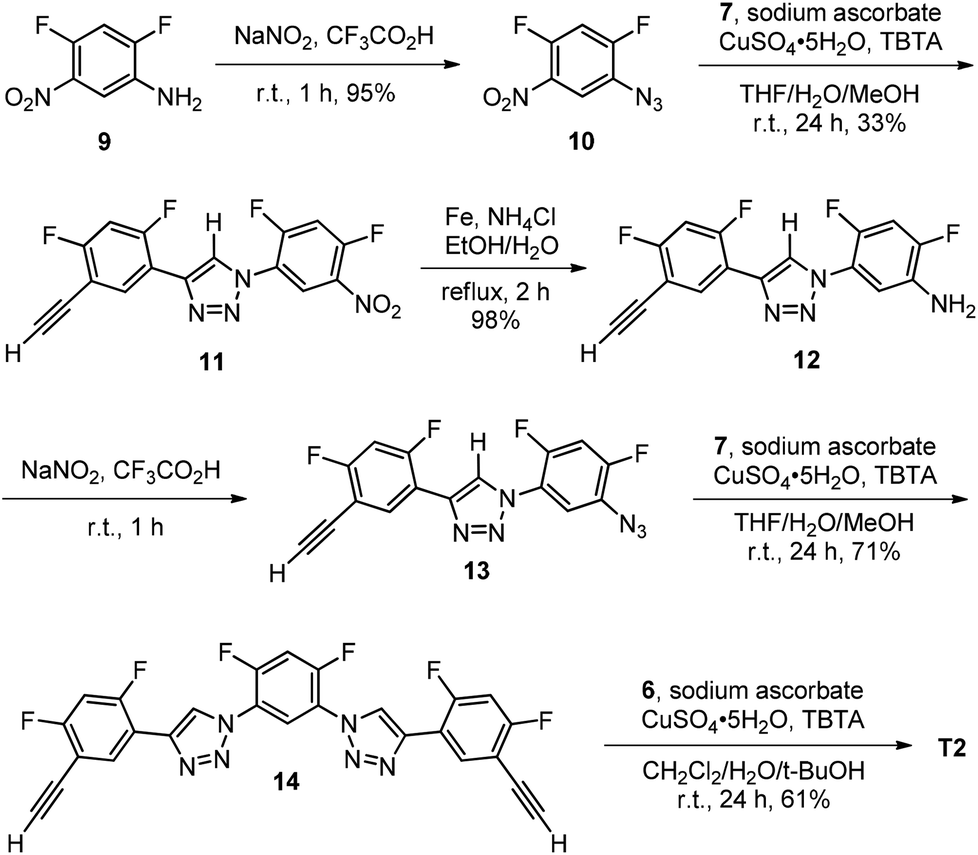 | ||
| Scheme 2 The synthesis of compound T2. | ||
Scheme 3 provides the synthetic route for 6-mer T3. Compound 15 was first prepared from the 1,3-cycloaddition reaction of compounds 7 and 10. The dinitro derivative was then reduced with iron and ammonium chloride by refluxing in aqueous ethanol to afford diamine 16, which was further converted into diazide 16 by treating with sodium nitrite in TFA. Finally, this precursor was reacted with acetylene 8 to afford compound T3.
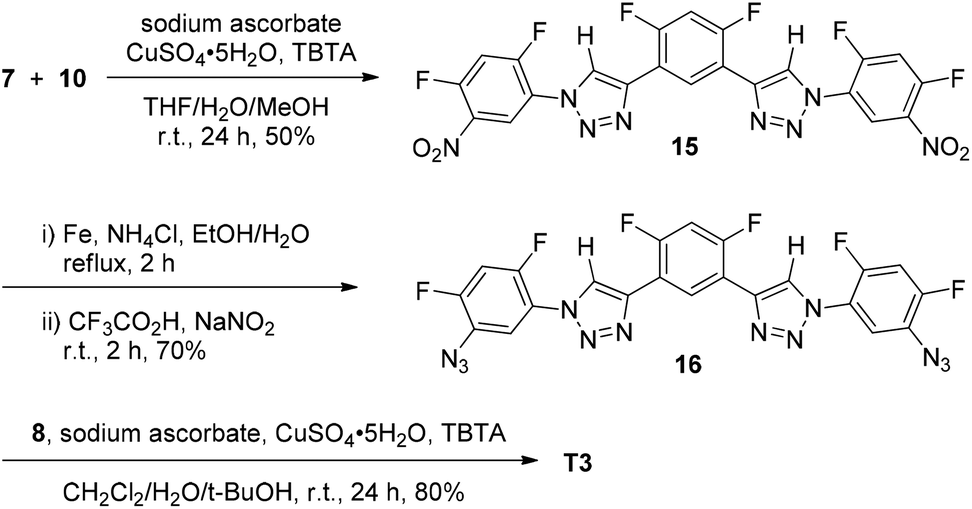 | ||
| Scheme 3 The synthesis of compound T3. | ||
The synthetic route of 8-mer T4 is shown in Scheme 4. Compound 17 was first prepared from the 1,3-cycloaddition reaction of 6 and 11 and then reduced to the corresponding amine by iron and ammonium chloride in aqueous ethanol. The amine intermediate was further treated with sodium nitrite in TFA to afford azide 18. Further 1,3-cycloaddition reaction of 18 with diacetylene 14 gave rise to T4.
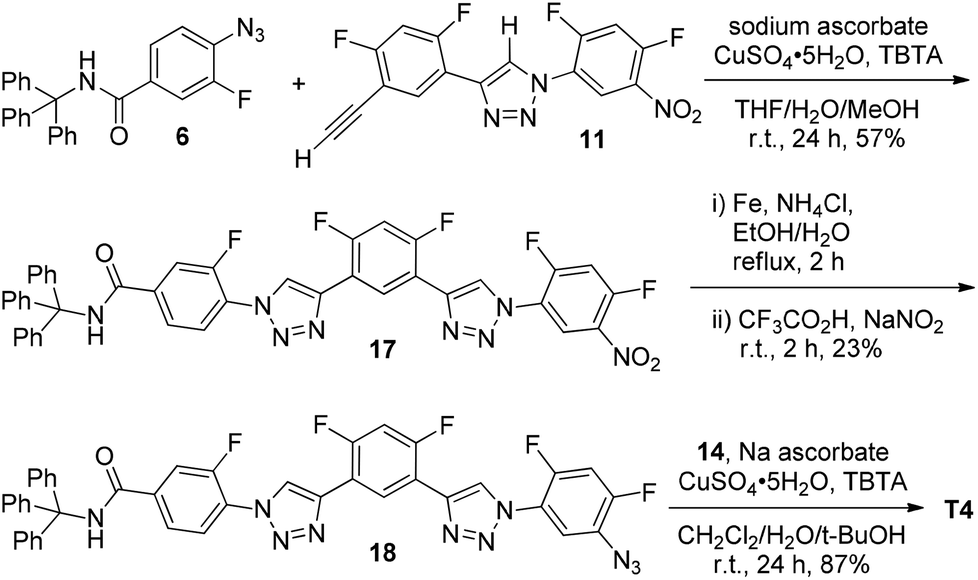 | ||
| Scheme 4 The synthesis of compound T4. | ||
The crystal structures of compounds 8 and 11 were obtained. In the crystal, compound 8 existed in two conformers. For both conformers, the C2–H atom of the 1,2,3-triazole ring was involved in three-centred C–H⋯F hydrogen bonding. The structures of the two conformers are provided in Fig. 1a. For one conformer, the CH⋯F distances were 2.32 and 2.27 Å, respectively. For another conformer, the distances were 2.38 and 2.28 Å, respectively. The crystal structure of compound 11 is shown in Fig. 1b, which shows that this compound also formed a similar three-centred C–H⋯F hydrogen bonding, and the CH⋯F distances were 2.42 and 2.34 Å, respectively. All these values are considerably shorter than the sum (2.67 Å) of the van der Waals radii of the F and H atoms, supporting the stability of this three-centred hydrogen bonding motif.
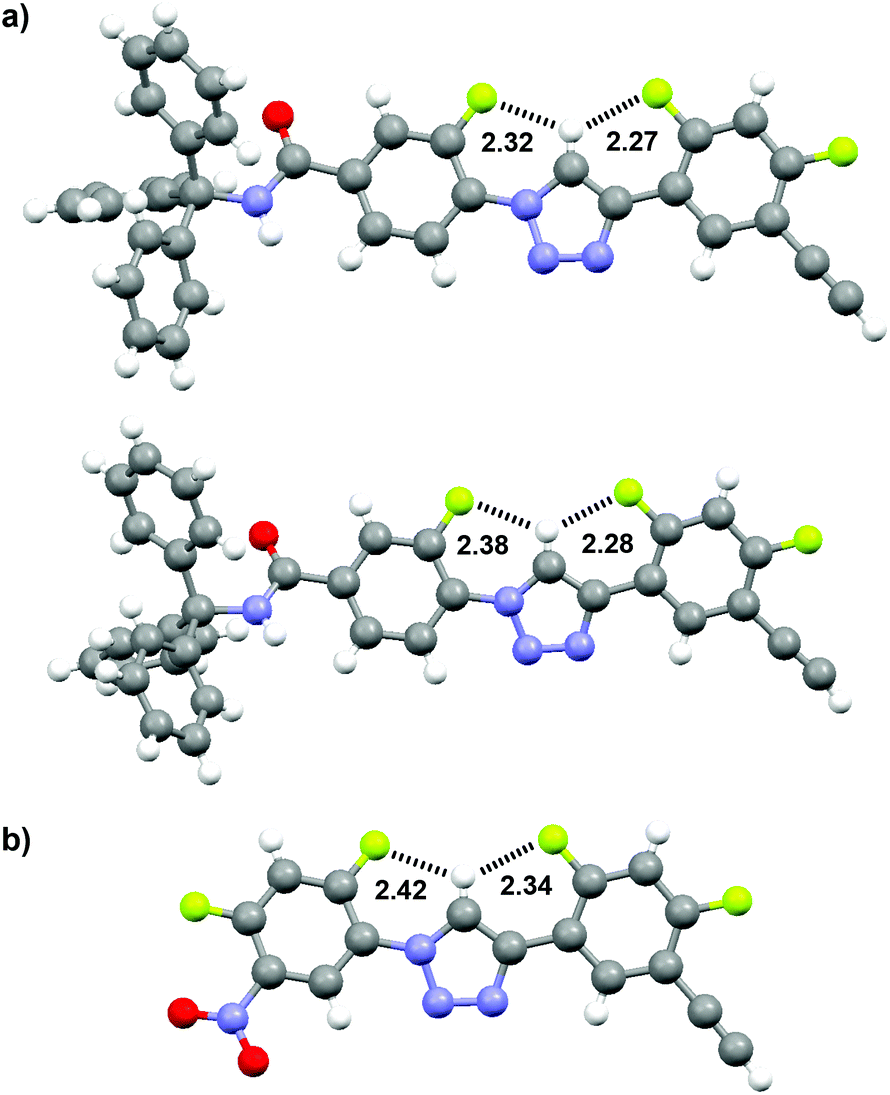 | ||
| Fig. 1 The crystal structures of compounds 8 (two conformers, CCDC 986427) and 11 (CCDC 986426), highlighting the formation of the three-centred C–H⋯F hydrogen bonding. | ||
The 1H NMR spectrum of compounds 8 and 11 in CDCl3 displayed a dd signal for the C2–H atoms. For compound 8, the coupling constant (J) was 3.1 Hz, and for compound 11, the coupling constants were 2.7 and 3.2 Hz, respectively. The splitting should be attributed to the proximity of the two F atoms at the ortho-position of the two attached benzene rings.9a This through-space coupling also supported that the C2–H atoms were engaged in three-centred C–H⋯F hydrogen bonding. The fact that compound 8 gave rise to a triplet implied that the two F atoms approached the C2–H atom to a very comparable extent. Similar triplets were also observed for 17. Adding DMSO-d6 caused the signals to coalesce into singlets, reflecting that the hydrogen bonding was weakened or destroyed in the highly polar solvent. Compounds T1, T2, T3, and 14 also displayed splitting for the signals of their C2–H atoms. However, coupling constants could not be obtained due to signal overlapping or broadening.
The signals of the H atoms on the difluorobenzene rings of the triazole oligomers were also split by the neighbouring F atoms. However, these split signals did not coalesce into singlets in DMSO-d6. For T2 and T3, the continuous three-centred hydrogen bonding should force their backbones to form folded conformations (vide infra). Considering the similarity of the backbones, the longest oligomer T4 should also form similar three-centred C–H⋯F hydrogen bonding. The 1H NMR spectrum of T4, however, displayed broad signals of low resolution. This result suggested that a helical conformation should be formed, which might undergo intramolecular stacking and P/M conformational exchange to lead to the decrease of the resolution of the 1H NMR spectrum. The 1H NMR spectrum in DMSO-d6 is of high resolution (ESI†), indicating the formation of a flexible conformation due to the breaking of the C–H⋯F hydrogen bonding in a highly polar medium.
The two-dimensional (2D) 1H–19F heteronuclear Overhauser enhancement spectroscopy (HOESY) is a robust technique which allows for the detection of through-space NOE connectivity between non-bonded H and F nuclei.21 Thus, systematic HOESY experiments were performed in chloroform-d to characterize the folded conformations of the new series of triazole derivatives. The 2D HOESY spectra of compounds 8 and 11 were first recorded and are provided in Fig. 2. For both compounds, intramolecular NOE contacts were observed between the triazole C2–H atom and the neighbouring F-2 and F-3 atoms, supporting the formation of the intramolecular three-centred C–H⋯F hydrogen bonding. In DMSO-d6, similar contacts were not observed, indicating that the hydrogen bonding was weakened or destroyed in this highly polar solvent. In principle, the electrostatic repulsions between the N-2 and N-3 atoms of the 1,2,3-triazole ring and the two adjacent F atoms and the intrinsic co-planarity of the 1,4-diphenyl-1,2,3-triazole backbone should also facilitate the formation of the two NOE contacts. However, the above results indicate that the intramolecular three-centred C–H⋯F hydrogen bonding should play a key role in driving the formation of the NOE contacts, as supported by the DFT calculation for the torsion energies (Fig. 6, vide infra). The 2D NOESY spectrum of compound 8 (40 mM) in CDCl3 was also recorded (ESI†), which did not exhibit any NOE contact between H-2 and H-4 or H-5, which further supports that the hydrogen bonded conformation existed exclusively.
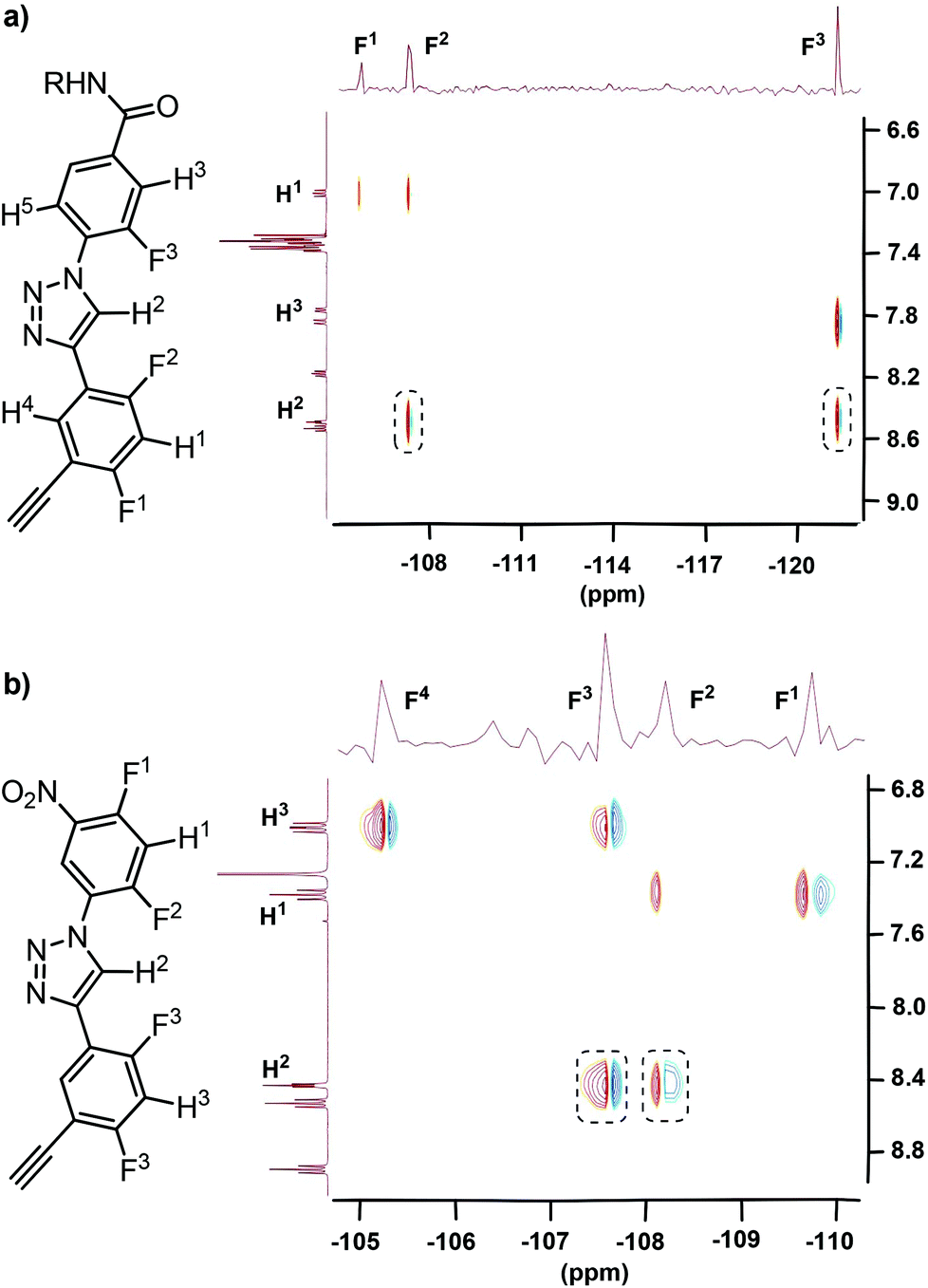 | ||
| Fig. 2 2D 1H–19F HOESY spectra (500 MHz) of compounds 8 (a) and 11 (b) in CDCl3 (5 mM), highlighting the heteronuclear NOE contacts in the dashed boxes. | ||
The 2D 1H–19F HOESY spectra of oligomers T1 and T2 in CDCl3 also exhibited similar NOE contacts between the C2–H atom(s) of the triazole unit(s) and the F atoms on the connected benzene rings (Fig. 3). Similar heteronuclear NOEs were also observed for compound 17 (ESI†), which may be considered as half of T2 by the structures. In highly polar DMSO-d6, no such NOE contacts were observed. In addition, the 1H NOESY spectra of T1 and T2 in CDCl3 did not exhibit NOE contacts between the triazole C2–H atoms and the H atoms at the ortho positions of the connected benzene rings (ESI†). These results indicated that, similar to the short compounds 8 and 11, the two oligomers also formed stable intramolecular three-centred C–H⋯F hydrogen bonding in CDCl3.
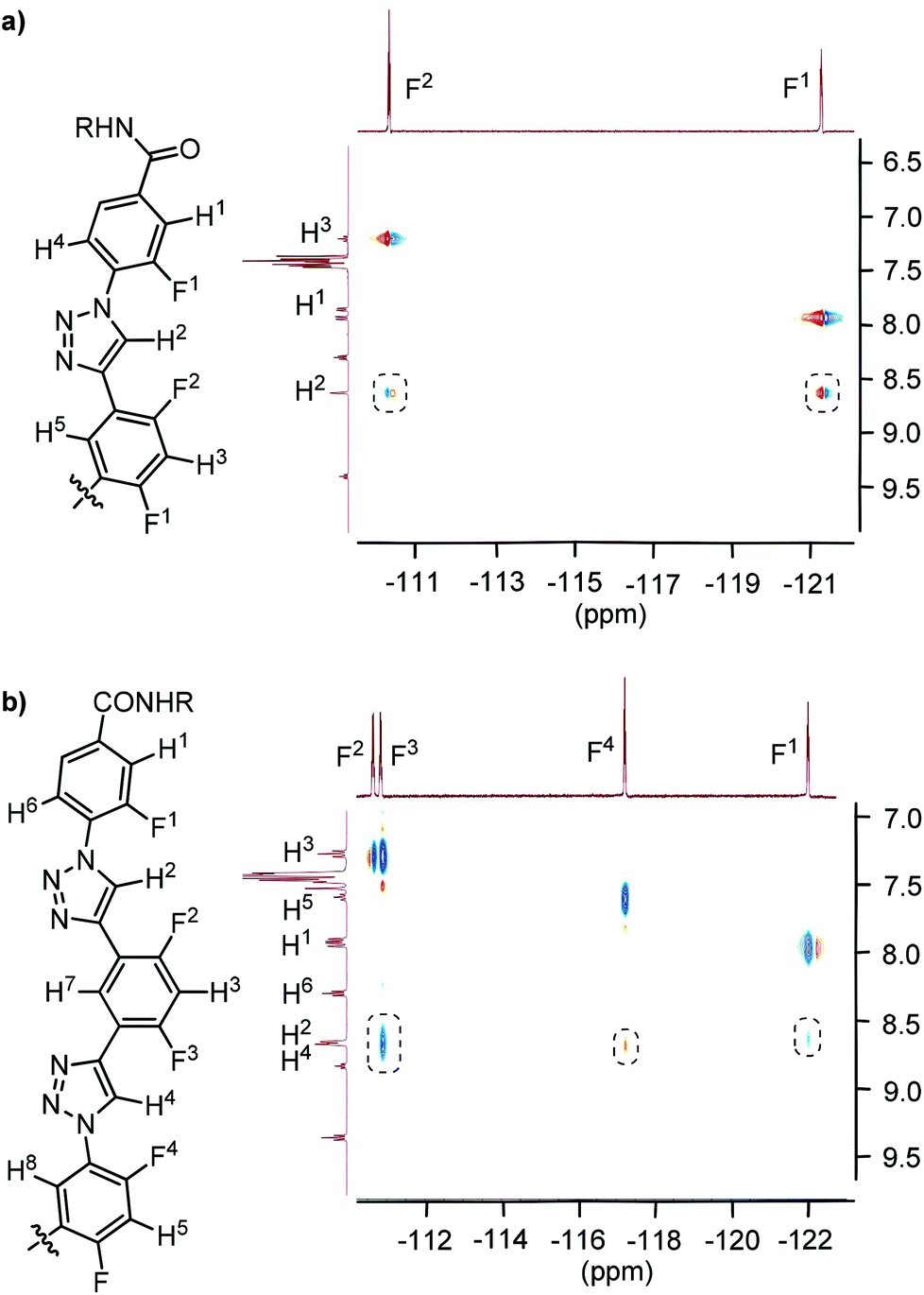 | ||
| Fig. 3 2D 1H–19F HOESY spectra (500 MHz) of compounds T1 (a) and T2 (b) in CDCl3 (5 mM), highlighting the heteronuclear NOE contacts in the dashed boxes. | ||
The HOESY spectrum of 6-mer T3 in CDCl3 is shown in Fig. 4. The 19F signals were identified by comparing the spectrum with those of shorter analogues. Although, due to overlapping, the signals of the F-2, F-3, and F-6 atoms could not be assigned completely, the signals of all the six F atoms exhibited the expected NOE contacting with the C2–H atoms of the triazole rings. The HOESY spectrum of T4 in CDCl3 was not recorded due to the low resolution of its 1H NMR spectrum. However, considering that all the oligomers possess the same backbones, it is reasonable to propose that it also formed a similar continuous intramolecular C–H⋯F hydrogen bonding.
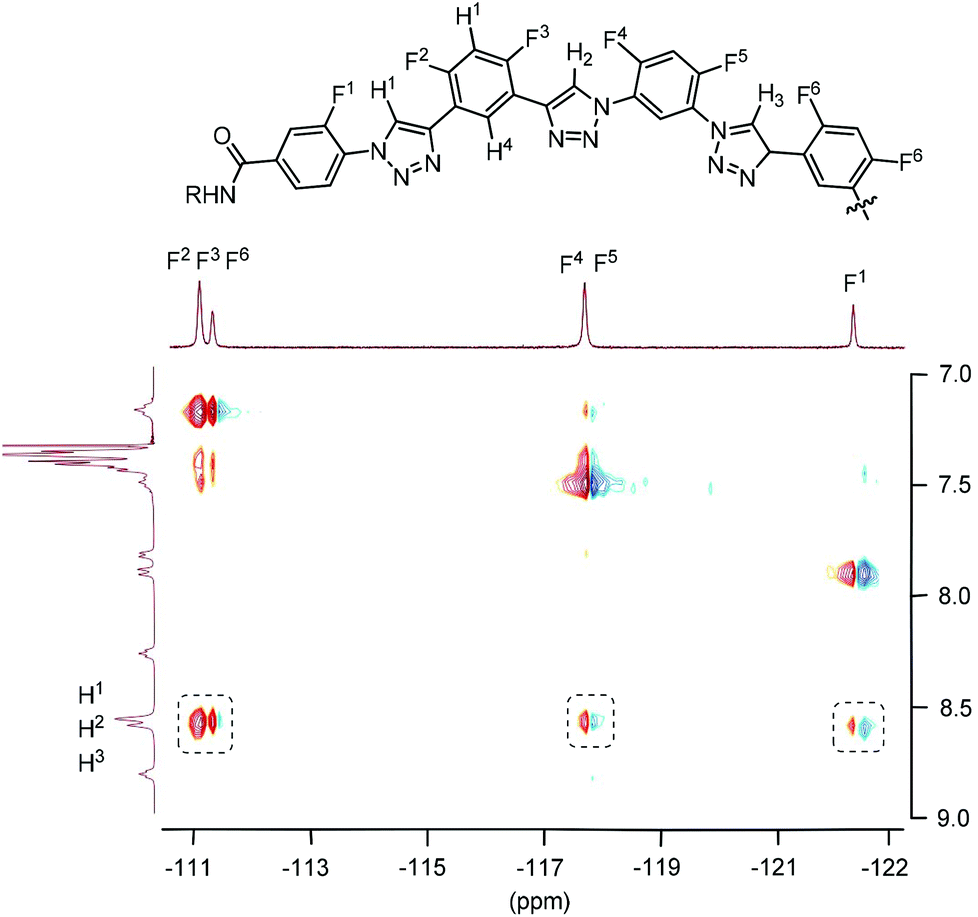 | ||
| Fig. 4 2D 1H–19F HOESY spectra (500 MHz) of compound T3 in CDCl3 (4 mM), highlighting the heteronuclear NOE contacts in the dashed boxes. | ||
The lowest-energy conformations of the 1,2,3-triazole oligomers were also optimized with the M062X/6-31G(d,p) calculation method,22 using the GAUSSIAN09 program. The obtained lowest-energy conformations of T2, T3 and T4 are provided in Fig. 5. As expected, T2 and T3 formed a crescent cavity, while the longest T4 could form one turn with the diameter of the cavity being about 1.7 nm, although the two appended bulky triphenylmethyl groups forced the 1,2,3-triazole backbone to twist considerably.
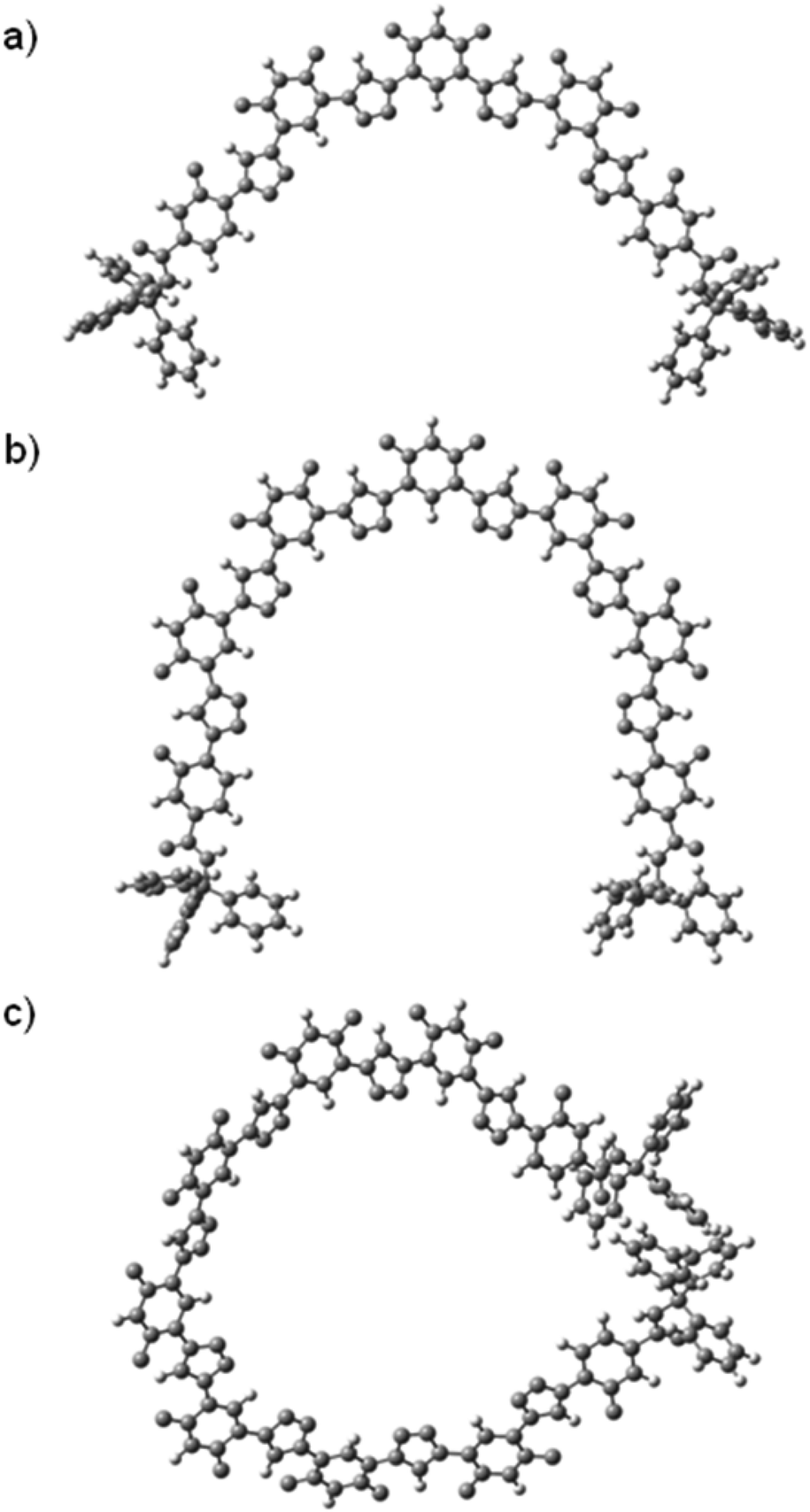 | ||
| Fig. 5 The optimized conformers of oligomers (a) T2, (b) T3, and (c) T4, calculated in chloroform with the Gaussian 09 package using the 6-31G(d,p) basis set. | ||
DFT calculations were further performed for compound T1 to obtain the energy difference between different conformers.20b The conformers were produced by changing the torsions between one of the two triazole rings and the two connected benzene rings, while constraining another triazole ring to be co-planar with the connected benzene rings (Fig. 6). It can be found that, for both cases, the conformer at the torsion angle of 180° was of high energy, which may be attributed to the electrostatic repulsion between the fluorine atoms and the nitrogen atoms of the triazole. For both cases, the minimum-energy conformer was that with the connected benzene rings closely co-planar with the triazole ring and the fluorine atoms pointing to the C2–H of triazole, even though at the torsion angles of about 130° for CNCC and 140° for CCCC, there existed other low-energy conformers. These results should be used as evidence that it was the three-centred C–H⋯F hydrogen bonding that made the main contribution in stabilizing the minimum-energy conformation.
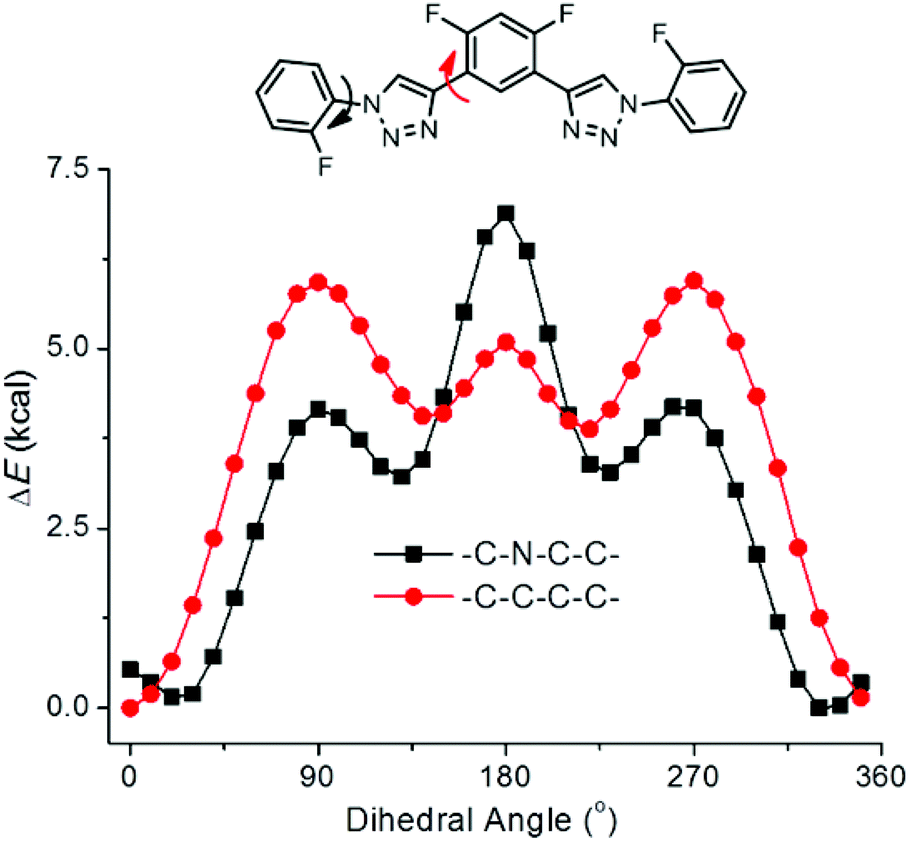 | ||
| Fig. 6 The torsion energies of compound T1 for turning the two connected benzene rings out of the plane of one of the triazole units by incremental steps of 10°. | ||
To further evaluate the stability of the intramolecular C–H⋯F hydrogen bonding, the 1H and 19F NMR spectra of T2 were recorded in CDCl3 in the presence of an incremental amount of tetra(n-butyl)ammonium chloride or iodide (ESI†). It was reported that the meta-phenylene-connected 1,2,3-triazole tetramer can fold to complex chloride or iodide anions by forming four intermolecular C2–H⋯Cl− or C2–H⋯I− hydrogen bonds.13 It was found that, upon adding an excess of tetra(n-butyl)ammonium chloride (up to 140 equiv.) or iodide (up to 110 equiv.), the 1H NMR signals of the C2–H atoms of T2 did not exhibit discernible shifting. In the presence of more amounts of the salts, T2 began to precipitate. These observations indicate that the folded conformation of T2, as shown in Fig. 5a, was quite stable and chloride or iodide anions could not induce the backbone to overturn to form another folded state of smaller cavity. Although the electrostatic repulsion between the F atoms and the N-2 and N-3 atoms may play a role in stabilizing the intramolecular C–H⋯F hydrogen bonding, the fact that the chemical shifting of the C2–H atoms did not undergo any change in the presence of an excess of chloride or iodide anions clearly supports the intrinsic stability of this intramolecular hydrogen bonding and the resulting folded secondary structure of this kind of 1,2,3-triazole oligomers.
Conclusions
In summary, we have demonstrated that intramolecular C–H⋯F hydrogen bonding can be utilized to induce conjugated 1,2,3-triazole oligomers to form folded or helical secondary structures. The stability of the C–H⋯F hydrogen bonding is higher than the well-established intermolecular C–H⋯F hydrogen bonding. As a result, the new folded secondary structures do not undergo a conformational overturn to produce another kind of folded structures of smaller cavity. Different from C–H⋯OR hydrogen bonding-driven folded systems, the new folded frameworks bear no side chains. Thus, both the F atoms and the N-2 and N-3 atoms of the triazole ring may serve as acceptors to further form intermolecular hydrogen bonding for the assembly of new interlocked architectures.We thank the National Natural Science Foundation (91227108 and 21228203), the Ministry of Science and Technology (2013CB834501) of China, and the Science and Technology Commission of Shanghai Municipality (13NM1400200) for financial support.
Notes and references
- Foldamers: Structure, Properties and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007, pp. 431 Search PubMed.
- (a) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS; (b) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed; (c) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed; (d) X. Li and D. Yang, Chem. Commun., 2006, 3367 RSC; (e) B.-B. Ni, Q. Yan, Y. Ma and D. Zhao, Coord. Chem. Rev., 2010, 254, 954 CrossRef CAS PubMed; (f) H.-Y. Hu and C.-F. Chen, Methods Mol. Biol., 2013, 932, 219 CrossRef CAS; (g) Y. Zhao, H. Cho, L. Widanapathirana and S. Zhang, Acc. Chem. Res., 2013, 46, 2763 CrossRef CAS PubMed.
- (a) B. Gong, Chem. – Eur. J., 2001, 7, 4337 CrossRef; (b) I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS; (c) Q. Gan, Y. Wang and H. Jiang, Curr. Org. Chem., 2011, 15, 1293 CrossRef CAS; (d) A. Roy, P. Prabhakaran, P. K. Baruah and G. J. Sanjayan, Chem. Commun., 2011, 47, 11593 RSC; (e) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMedZ. Shi, Y. Song, F. Lu, T. Zhou, X. Zhao, W. Zhang and Z. Li, Huaxue Xuebao, 2013, 71, 51 CrossRef CAS.
- (a) Z.-T. Li, J.-L. Hou, C. Li and H.-P. Yi, Chem. – Asian J., 2006, 1, 766 CrossRef CAS PubMed; (b) H. Juwarker, J.-M. Suk and J. Jeong, Chem. Soc. Rev., 2009, 38, 3316 RSC; (c) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC.
- (a) X. Zhao and Z.-T. Li, Chem. Commun., 2010, 46, 1601 RSC; (b) Q. Gan, Y. Wang and H. Jiang, Curr. Org. Chem., 2011, 15, 1293 CrossRef CAS; (c) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (d) P. Prabhakaran, G. Priya and G. J. Sanjayan, Angew. Chem., Int. Ed., 2012, 51, 4006 CrossRef CAS PubMed.
- (a) B. Gong, Acc. Chem. Res., 2008, 41, 1376 CrossRef CAS PubMed; (b) H. Fu, Y. Liu and H. Zeng, Chem. Commun., 2013, 49, 4127 RSC; (c) W. Q. Ong and H. Zeng, J. Inclusion Phenom. Macrocyclic Chem., 2013, 76, 1 CrossRef CAS PubMed; (d) Y. Yang, W. Feng, J. Hu, S. Zou, R. Gao, K. Yamato, M. Kline, Z. Cai, Y. Gao, Y. Wang, Y. Li, Y. Yang, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590 CrossRef CAS PubMed.
- (a) C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS PubMed; (b) C. G. Cummings and A. D. Hamilton, Curr. Opin. Chem. Biol., 2010, 14, 341 CrossRef CAS PubMed; (c) V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161 CrossRef CAS PubMed.
- (a) H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448 CrossRef CAS PubMed; (b) L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528 CrossRef CAS PubMed; (c) Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715 CrossRef CAS PubMed; (d) Y. Yan, B. Qin, C. Ren, X. Chen, Y. K. Yip, R. Ye, D. Zhang, H. Su and H. Zeng, J. Am. Chem. Soc., 2010, 132, 5869 CrossRef CAS PubMed; (e) V. V. E. Ramesh, G. Priya, A. S. Kotmale, R. G. Gonnade, P. R. Rajamohanan and G. J. Sanjayan, Chem. Commun., 2012, 48, 11205 RSC; (f) S. Zou, L. He, J. Zhang, Y. He, L. Yuan, L. Wu, J. Luo, Y. Wang and W. Feng, Org. Lett., 2012, 14, 3584 CrossRef CAS PubMed; (g) C. Li, X. Zhao, X. Gao, Q. Wang and Z. Li, Chin. J. Chem., 2013, 31, 582 CrossRef CAS.
- (a) C. Li, S.-F. Ren, J.-L. Hou, H.-P. Yi, S.-Z. Zhu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725 CrossRef CAS PubMed; (b) C. Li, Y.-Y. Zhu, H.-P. Yi, C.-Z. Li, X.-K. Jiang and Z.-T. Li, Chem. – Eur. J., 2007, 13, 9990 CrossRef CAS PubMed.
- Q. Gan, F. Li, G. Li, B. Kauffmann, J. Xiang, I. Huc and H. Jiang, Chem. Commun., 2010, 46, 297 RSC.
- C. Ren, S. Xu, J. Xu, H. Chen and H. Zeng, Org. Lett., 2011, 13, 3840 CrossRef CAS PubMed.
- (a) J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325 RSC; (b) X. Li, Chem. – Asian J., 2011, 6, 2606 CrossRef CAS.
- (a) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC; (b) K. P. McDonald, Y. Hua and A. H. Flood, Top. Heterocycl. Chem., 2012, 28, 85 Search PubMed.
- R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926 CrossRef CAS PubMed.
- (a) H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740 CrossRef CAS PubMed; (b) H. Juwarker, J. M. Lenhardt, J. C. Castillo, E. Zhao, S. Krishnamurthy, R. M. Jamiolkowski, K.-H. Kim and S. L. Craig, J. Org. Chem., 2009, 74, 8924 CrossRef CAS PubMed.
- (a) Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630 CrossRef CAS PubMed; (b) Y. Wang, J. Xiang and H. Jiang, Chem. – Eur. J., 2011, 17, 613 CrossRef CAS PubMed.
- (a) Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838 CrossRef CAS PubMed; (b) K. P. McDonald, R. O. Ramabhadran, S. Lee, K. Raghavachari and A. H. Flood, Org. Lett., 2011, 13, 6260 CrossRef CAS PubMed.
- (a) Y. Wang, F. Li, Y. Han, F. Wang and H. Jiang, Chem. – Eur. J., 2009, 15, 9424 CrossRef CAS PubMed; (b) C.-F. Wu, Z.-M. Li, X.-N. Xu, Z.-X. Zhao, X. Zhao, R.-X. Wang and Z.-T. Li, Chem. – Eur. J., 2014, 20, 1418 CrossRef CAS PubMed.
- L.-Y. You, S.-G. Chen, X. Zhao, Y. Liu, W.-X. Lan, Y. Zhang, H.-J. Lu, C.-Y. Cao and Z.-T. Li, Angew. Chem., Int. Ed., 2012, 51, 1657 CrossRef CAS PubMed.
- (a) D. Zornik, R. M. Meudtner, T. E. Malah, C. M. Thiele and S. Hecht, Chem. – Eur. J., 2011, 17, 1473 CrossRef CAS PubMed; (b) B.-Y. Lu, Y.-Y. Zhu, Z.-M. Li, X. Zhao and Z.-T. Li, Tetrahedron, 2012, 68, 8857 CrossRef CAS PubMed.
- (a) C. Yu and G. C. Levy, J. Am. Chem. Soc., 1984, 106, 6533 CrossRef CAS; (b) M. A. Hyland, M. D. Morton and C. Brückner, J. Org. Chem., 2012, 77, 3038 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, calculation details, crystal data, 1H, 19F and 13C NMR spectra, and additional 2D 1H NOESY and 1H–19F HOESY NMR spectra. CCDC 986426–986427. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00047a |
| This journal is © the Partner Organisations 2014 |