
A simple and practical catalytic electron transfer system composed of TiCl4 and metallic Yb: application in carbonyl olefination and insight into the mechanism†
Lili
Zhang
a,
Xiaying
Yu
a,
Lixin
Zhang
a,
Xigeng
Zhou
*ab and
Yanghui
Lin
*a
aDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200433, People's Republic of China. E-mail: xgzhou@fudan.edu.cn; lyanghui@fudan.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai 200032, People's Republic of China
First published on 15th July 2014
Abstract
The McMurry-type olefinations have become a powerful tool in organic synthesis. However, the use of stoichiometric amounts of unmanageable Ti species and the generation of a large amount of salts limit their applications on the industrial scale. This paper outlines a simple and practical catalytic electron transfer (ET) system composed of TiCl4 and Yb, which turned out to be effective for homo- and cross-deoxygenative coupling of aldehydes and ketones. Specifically, the method is based upon the strategy that the low valent Ti species abstracts oxygen from carbonyl and delivers them to highly oxophilic Yb in an unprecedented manner. Remarkably, the present process is operationally simple, minimizes the generation of chemical waste, and makes the only by-product Yb2O3 as a solid easily removed and utilized; furthermore, the yield improved significantly of that obtained in the stoichiometric version in our system. Our mechanistic data not only provide the direct evidence that Ti(IV) can facilitate the part deoxygenation of pinacol at room temperature, but also demonstrate, for the first time, that for bulky ketones the turnover-limiting step is the Ti-mediated carbonyl coupling rather than the subsequent deoxygenation of the resulting pinacolate intermediates, which are different from the previous observations.
Introduction
The selective construction of C–C double bonds continues to be an important goal in organic synthesis. The McMurry reaction is one of the most broadly utilized methods for the carbonyl olefination as it allows direct formation of crucial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and thus reduces the number of steps in synthesis of natural or biologically relevant compounds.1–3 Despite tremendous advances in this area, most well-established methods require the use of excess titanium reagents and reductants even in the presence of activating reagents4 and ligands,5 and involve the stoichiometric production of metal salts (e.g. metal chloride and alkoxide) which are often elusive to remove from the reaction mixture and/or have occasionally adverse effects.6 Pioneering work reported in 1995 by Fürstner and Hupperts showed that the reductive coupling of aromatic oxoamides to indoles can be performed catalytically in low-valent Ti using a chlorosilane as the final oxygen trap (Scheme 1).7 Recently, Barrero and coworkers reported that Cp2TiCl2 could effect the catalytic transformation of benzylic aldehydes to 1,2-diarylethenes in the presence of excess Mn and Me3SiCl.6 Notably, the introduction of chlorosilanes not only leads to the generation of large amounts of metal chlorides, but also probably increases the consumption of reductants due to their potential reaction with chlorosilanes.8 In view of economical benefit and environmental impact, further improvements in this area are desirable. One of the most ideal methods for carbonyl reductive-olefination, in our opinion, would be the use of a single metal as both the reductant and oxygen-trapper in the presence of a suitable catalyst without the requirement of additives; however, no example of such a reaction has been reported so far.
C bonds and thus reduces the number of steps in synthesis of natural or biologically relevant compounds.1–3 Despite tremendous advances in this area, most well-established methods require the use of excess titanium reagents and reductants even in the presence of activating reagents4 and ligands,5 and involve the stoichiometric production of metal salts (e.g. metal chloride and alkoxide) which are often elusive to remove from the reaction mixture and/or have occasionally adverse effects.6 Pioneering work reported in 1995 by Fürstner and Hupperts showed that the reductive coupling of aromatic oxoamides to indoles can be performed catalytically in low-valent Ti using a chlorosilane as the final oxygen trap (Scheme 1).7 Recently, Barrero and coworkers reported that Cp2TiCl2 could effect the catalytic transformation of benzylic aldehydes to 1,2-diarylethenes in the presence of excess Mn and Me3SiCl.6 Notably, the introduction of chlorosilanes not only leads to the generation of large amounts of metal chlorides, but also probably increases the consumption of reductants due to their potential reaction with chlorosilanes.8 In view of economical benefit and environmental impact, further improvements in this area are desirable. One of the most ideal methods for carbonyl reductive-olefination, in our opinion, would be the use of a single metal as both the reductant and oxygen-trapper in the presence of a suitable catalyst without the requirement of additives; however, no example of such a reaction has been reported so far.
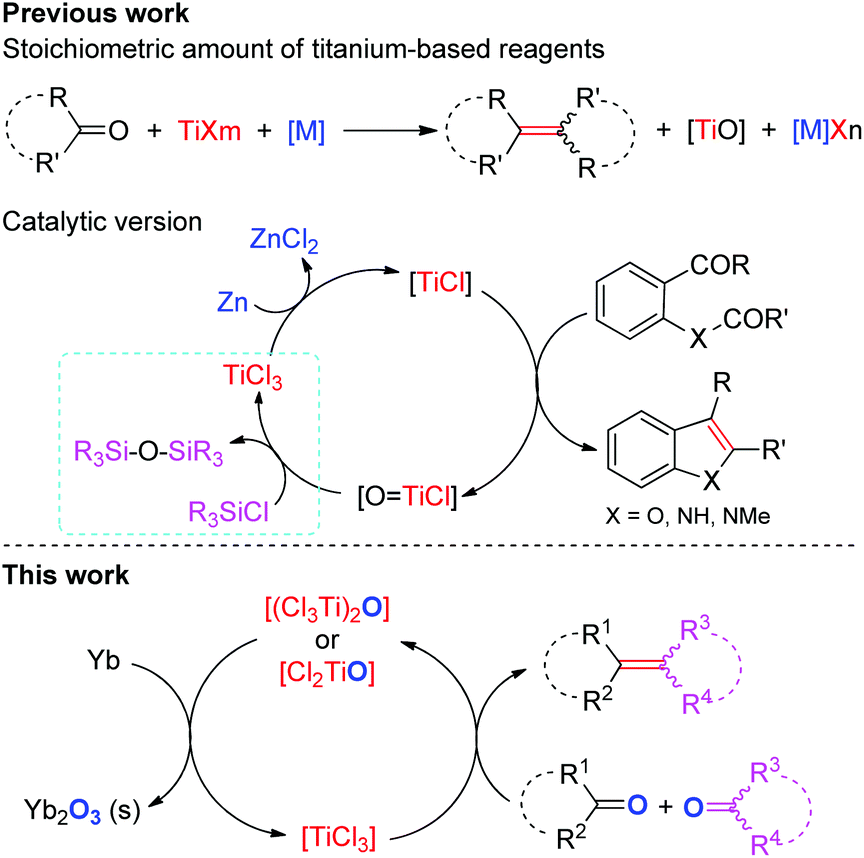 | ||
| Scheme 1 Comparison of the prior work to the current work. | ||
In addition to the synthetic utility and challenges described above, these carbonyl olefinations also posed an interesting mechanistic question. Although it has been widely accepted that species with low valence states of Ti were required in this reductive process, there is little information concerning the structures and reactivities of the intermediates.1 The mechanistic subtleties toward reductants, additives, reaction conditions and ancillary ligands in these systems remain yet to be clarified. In particular, the reactivity of the reducing agents toward the titanium oxo or oxychloride species resulting from the deoxygenation step remains unexplored. Moreover, the factors that control stereochemistry of products are not well understood yet.9 Consequently, detailed mechanistic studies involving the reactivity of related Ti species toward the reducing agents and substrates are of great importance in unraveling factors that promote or deter carbonyl olefination. The development of a new strategy for the extrusion of oxygen from the titanium oxo species would permit one to enable the designing of new reductants and to incorporate the stoichiometric reductive carbonyl coupling reaction in some catalytic versions.
Synthetic utility of rare earth metals as the reductants is receiving increased attention because of their unique reactivities and inherent advantages over divalent rare earth complexes, such as stability in air, non-toxic, cheaper and more commercial availability, easy manipulation and store, as well as the higher atom efficiency through three-electronic transfer.8b,10 Disappointingly, the highly kinetic inert and thermodynamic stability of rare earth metals under conventional conditions restrict their applications. Although several methods for activation of rare earth metals have been developed and offer a scaffold for interesting applications in organic synthesis, their application is still relatively limited.10 Thus, the development of simple and practical electron transfer reagents, especially catalytic systems that can improve the electron-releasing ability of rare earth metals and thus are suitable for selective organic transformations, is required. Although several diversified reductions of ketones and aldehydes with rare earth metals were reported, on the other hand, the olefination product has never been obtained selectively.11
Herein, we report a catalytic ET system composed of TiCl4 and Yb and its application in transformation of carbonyls to olefins, in which the Ti(IV) accepts electrons from metallic Yb and delivers them to the substrate and the resulting precipitate Yb2O3 is easily removed. Some unprecedented mechanistic features are observed under our catalytic conditions. The process exhibits broad scope, simple manipulation, good environmental tolerance and a saving on the metal-containing reagents through the multi-electron reductive effect of metallic Yb and avoidance of additives. Furthermore, the present results represent a convenient way for catalytic activation of commercial rare earth metals as an off-the-shelf reagent.
Results and discussion
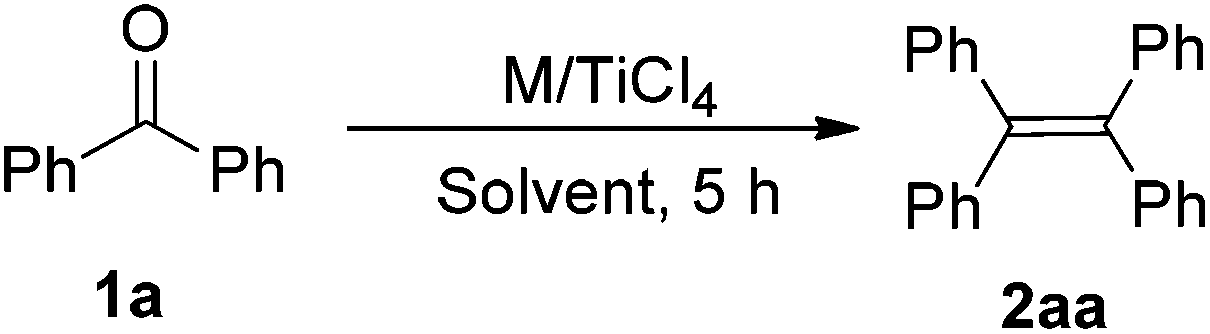
Our research is based on the premise that the reducing agent not only provides more than two electrons but also should participate in the oxygen abstraction. To test this premise, we selected rare earth metals as reducing agents. Table 1 summarizes the results of optimization of an M/TiCl4-mediated reductive coupling of benzophenone (1a) to tetraphenylethene (2aa). In the absence of TiCl4, the reaction did not take place (entry 1). The reaction of 1a with a mixture of Yb and TiCl4 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.1 at 70 °C for 24 h gave the deoxygenated coupling product 2aa in 91% yield (entry 2). It was found that 2aa could be obtained in almost quantitative yield when TiCl4 was adjusted to 0.3 equivalent at 70 °C (entry 4). Notably, the reaction can also proceed smoothly at room temperature (entries 5 and 6), which is extremely rare. The loading of one equivalent of TiCl4 gave 2aa in 95% yield (entry 7), which meant a decrease in the efficiency of transformation when compared to the catalytic version. Unceasingly increasing the amount of Yb would fasten the reaction rate, but did not affect the reaction products (entry 8). Of all solvents tested, THF performed the best (entry 4). No desired product was obtained, when the reaction was carried out in non-coordinated solvents such as toluene (entry 10). Examination of different rare earth metals revealed Yb as the best reductant (entries 4, 11, 12). In addition, the use of Fe or Sn in place of the lanthanide metal did not give the desirable product (entries 13 and 14). Based on the stoichiometric ratio of ketone/Yb/TiCl4, the present deoxygenated coupling reaction not only makes full use of the superiority that rare earth metals can provide three reductive electrons, but also makes rare earth metals as a basic oxygen-trapper. Clearly, the present reducing system makes a significant saving on the molar amount of metal-based reagents compared with other reducing systems.2,4,6,7 To the best of our knowledge, this is the first example that the reductant plays a basic oxygen-trapper in the titanium-based reduction protocol.
:1:0.1 at 70 °C for 24 h gave the deoxygenated coupling product 2aa in 91% yield (entry 2). It was found that 2aa could be obtained in almost quantitative yield when TiCl4 was adjusted to 0.3 equivalent at 70 °C (entry 4). Notably, the reaction can also proceed smoothly at room temperature (entries 5 and 6), which is extremely rare. The loading of one equivalent of TiCl4 gave 2aa in 95% yield (entry 7), which meant a decrease in the efficiency of transformation when compared to the catalytic version. Unceasingly increasing the amount of Yb would fasten the reaction rate, but did not affect the reaction products (entry 8). Of all solvents tested, THF performed the best (entry 4). No desired product was obtained, when the reaction was carried out in non-coordinated solvents such as toluene (entry 10). Examination of different rare earth metals revealed Yb as the best reductant (entries 4, 11, 12). In addition, the use of Fe or Sn in place of the lanthanide metal did not give the desirable product (entries 13 and 14). Based on the stoichiometric ratio of ketone/Yb/TiCl4, the present deoxygenated coupling reaction not only makes full use of the superiority that rare earth metals can provide three reductive electrons, but also makes rare earth metals as a basic oxygen-trapper. Clearly, the present reducing system makes a significant saving on the molar amount of metal-based reagents compared with other reducing systems.2,4,6,7 To the best of our knowledge, this is the first example that the reductant plays a basic oxygen-trapper in the titanium-based reduction protocol.
|
|
|||||
|---|---|---|---|---|---|
| Entry | M (equiv.) | TiCl4 (equiv.) | Solvent | Temp (°C) | Isolated yield (%) |
| a 1a (1.0 mmol), solvent (8 mL) for 5 h under N2. b Reaction time is 24 h. c Reaction time is 72 h. d Reaction time is 2 h. | |||||
| 1 | Yb(1) | 0 | THF | 70 | 0 |
| 2b | Yb(1) | 0.1 | THF | 70 | 91 |
| 3 | Yb(1) | 0.2 | THF | 70 | 94 |
| 4 | Yb(1) | 0.3 | THF | 70 | 99 |
| 5b | Yb(1) | 0.3 | THF | 25 | 74 |
| 6c | Yb(1) | 0.3 | THF | 25 | 90 |
| 7 | Yb(1) | 1.0 | THF | 70 | 95 |
| 8d | Yb(2) | 0.3 | THF | 70 | 99 |
| 9 | Yb(1) | 0.3 | DME | 70 | 97 |
| 10 | Yb(1) | 0.3 | Toluene | 70 | 0 |
| 11 | Sm(1) | 0.3 | THF | 70 | 95 |
| 12 | Dy(1) | 0.3 | THF | 70 | 90 |
| 13 | Fe(1) | 0.3 | THF | 70 | 0 |
| 14 | Sn(1) | 0.3 | THF | 70 | 0 |
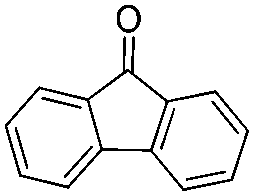
Having established an optimal protocol, we next investigated the generality and the scope of this transformation via variation of ketones and aldehydes. As illustrated in Table 2, the reaction conditions described above are effective for the transformation of a number of ketones and aldehydes, various alkenes can be efficiently synthesized from this reaction in good to excellent yields. In general, the trans-isomer was predominantly obtained when the reaction involved the diastereoselectivity. Aromatic ketones with neutral and electron-donating substituents such as methyl and methoxy groups on the phenyl ring gave the deoxygenated products 2 in nearly quantitative yields (entries 4–6). The presence of electron-withdrawing groups such as chloride in the para-position of benzene rings seems to negatively affect the yields, but the E/Z selectivity remained essentially constant (entries 7–9). Naphthyl methyl ketone (1j) worked well under this condition, yielding 2jj in 88% yield, albeit with low E/Z selectivity (entry 10). Furthermore, 9-fluorenone (1k) was also a suitable substrate, and the desirable product 2kk was generated in 89% yield (entry 11). 1-(Furan-2-yl)ethanone and 1-(thiophen-2-yl)ethanone provided the corresponding products in almost quantitative yields too (entries 12 and 13). Gratifyingly, less reactive aliphatic ketone (1n) worked smoothly under the optimal reaction conditions, affording 2nn in 81% yield (entry 14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Product |
E:Z ratiob |
Isolated yield (%) |
| a TiCl4 (0.3 mmol), Yb (1 mmol), carbonyl compound (1 mmol), THF (8 mL) for 5 h at 70 °C under N2. b The E/Z ratio measured by 1H NMR. c Reaction time is 7 h. | ||||||
| 1 | 1a | C6H5 | C6H5 | 2aa | 99 | |
| 2 | 1b | C6H5 | CH3 | 2bb | 94:6 |
99 |
| 3 | 1c | C6H5 | CH3CH2 | 2cc | 82:18 |
98 |
| 4 | 1d | 4-MeC6H4 | CH3 | 2dd | 79:21 |
99 |
| 5 | 1e | 4-MeOC6H4 | CH3 | 2ee | 78:22 |
99 |
| 6 | 1f | 4-MeOC6H4 | 4-MeOC6H4 | 2ff | 99 | |
| 7 | 1g | 4-ClC6H4 | 4-ClC6H4 | 2gg | 90 | |
| 8 | 1h | 3-ClC6H4 | CH3 | 2hh | 77:23 |
96 |
| 9c | 1i | 3,4-Cl2C6H3 | CH3 | 2ii | 79:21 |
90 |
| 10 | 1j | 1-Naphthyl | CH3 | 2jj | 60:40 |
88 |
| 11 | 1k |
|
2kk | 89 | ||
| 12 | 1l | 2-Furanyl | CH3 | 2ll | 49:51 |
98 |
| 13 | 1m | 2-Thiophenyl | CH3 | 2mm | 83:17 |
97 |
| 14c | 1n | PhCH2 | PhCH2 | 2nn | 81 | |
| 15 | 1o | C6H5 | H | 2oo | 100:0 |
95 |
| 16 | 1p | 4-ClC6H4 | H | 2pp | 100:0 |
95 |
| 17 | 1q | 4-MeOC6H4 | H | 2qq | 100:0 |
94 |
| 18 | 1r | 4-CH3C6H4 | H | 2rr | 100:0 |
98 |
| 19 | 1s | 2-Furanyl | H | 2ss | 100:0 |
90 |
| 20c | 1t | PhCHCH |
H | 2tt | 84:16 |
86 |
Subsequently, utility of various aldehydes was examined. To our delight, various aromatic aldehydes 1o–1s smoothly underwent the McMurry-type olefination, giving 2oo–2ss in excellent yields with complete E selectivity (Table 2, entries 15–19). Cinnamaldehyde is clearly compatible with the reaction (Table 2, entry 20).
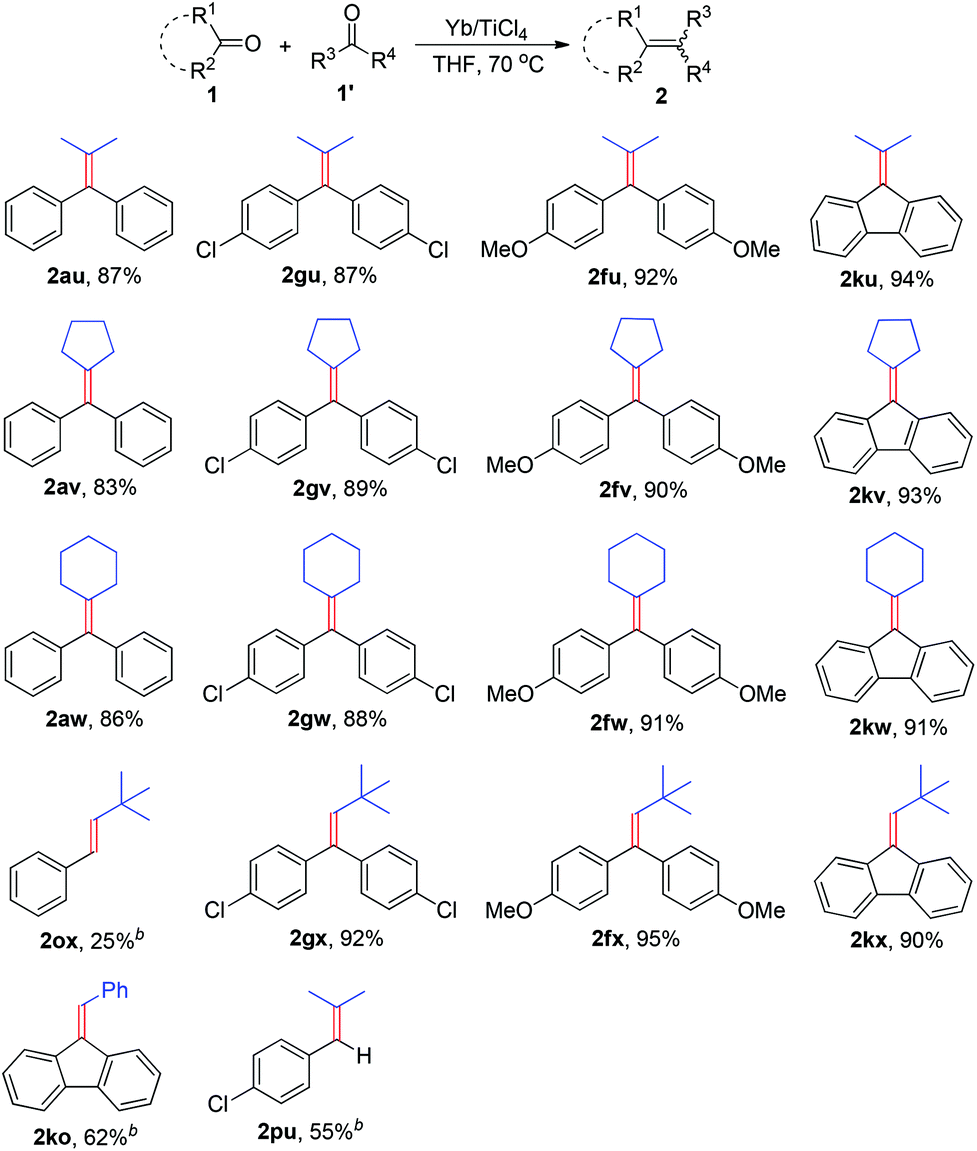
The discovery of reagent systems that allow the selective manipulation of functional groups is crucial for advancements in synthesis. Of particular value are reagent systems that allow functional groups with apparently similar reactivities to be distinguished. Compared to the impressive development of homo-coupling of carbonyl compounds to olefins, the cross-coupling between two different carbonyl compounds has received only scant attention.2k,6 The major reason may be attributed to the difficulties for controlling required selectivity due to the facile homologous coupling pathways available, in most cases this kind of reaction will generate a roughly statistical mixture of the possible coupling products. Therefore, the use of such a reaction in organic synthesis remains a challenge. In seeking to further broaden the scope of this work, we checked the ability of the Yb/TiCl4 reductive system to promote the cross-coupling reaction between different ketones (Table 3). When a variety of diary ketones and one equivalent of aliphatic ketone were subjected to the standard reaction conditions, the desired cross-coupling products were obtained in 83–94% isolated yields. For example, acetone, cyclopentanone and cyclohexanone reacted with one equivalent of benzophenone to give the corresponding cross-coupling products 2au–2aw in 83–87% yields. Aromatic ketones bearing additional functional groups (1f, 1g) led efficiently to the coupling products, which increased the potential of this transformation. The reaction conditions described above are also effective for the cross-coupling of 9-fluorenone (1k) with aliphatic ketones, various fulvalenes can be efficiently synthesized from this reaction in excellent yields (2ku–2kw). Encouraged by the above results, we further envisioned that combination of aldehydes with ketones could be subjected to the transformation in the current experimental conditions. Treatment of a mixture of aromatic ketone and pivalaldehyde with the Yb/TiCl4 reductive system gave the expected cross-coupling products 2fx, 2gx and 2kx in excellent yields. The cross-coupling of aromatic aldehydes with ketones was also conducted under the same conditions, but it gave 2ko and 2pu in moderate yields together with the formation of a small amount of diarylethenes. This protocol proved to be applicable, less efficiently, to the deoxygenative cross-coupling between aromatic aldehyde and aliphatic aldehyde (2ox).
Having demonstrated that the lanthanide metals are able to reduce carbonyls to alkenes in the presence of a catalytic amount of TiCl4, we were interested in gaining insight into the reaction mechanism. Initially, we studied the metal-containing components in the reaction mixture when Ph2CO had been completely consumed. To our delight, the clear solution was concentrated and cooled at −18 °C to give TiCl3(THF)3 (75%), which was confirmed by single crystal X-ray diffraction analysis. The resulting gray precipitate was filtered, washed with hot THF and analyzed by complexometric titration, supporting the selective formation of Yb2O3. In contrast to the observations in reaction of TiCl4 or TiCl3 with other metals, wherein titanium species in the zero- or divalent state, such as [Ti(MgCl2)·nTHF],2b,12 is generally involved as the active reducing species, the reaction of TiCl4 with 1.2 equiv. Yb in THF at 70 °C for 1 h gave TiCl3(THF)3 in 82% yield (Scheme 2). Furthermore, we examined the stoichiometric reaction of TiCl3 with Ph2CO and PhCOMe. Only a trace amount of olefination product was observed in the reaction of TiCl3 with Ph2CO or PhCOMe even with prolonged heating in THF (70 °C), and the starting materials were recovered unchanged. However, TiCl3 easily abstracts the oxygen from 2,3-diphenyloxirane, giving stilbene (2oo) in almost quantitative yield (Scheme 3). Significantly, the treatment of a 1:2 mixture of TiCl3 and Ph2CO with 1/3 equiv. of Yb at room temperature gave a mixture of tetraphenylpinacol and tetraphenylethene in 16.2% and 22.8% electron yields (based on the total number of reductive electrons that TiCl3 and Yb provide as much as possible), respectively. The product distribution depended on the reaction temperature. Heating the reaction mixture resulted in an increase of the olefin production (Scheme 4). At this point, addition of a further 1 equiv. of Yb led to the complete consumption of the starting material Ph2CO, tetraphenylethene being the only product formed. Given the fact that Yb is not able to reduce Ti to a valence lower than 3 under mild conditions and that sole Yb does not promote dimerization of Ph2CO under present conditions (Table 1, entry 1), it was reasonable to assume that the ET from Yb to a Ti ketyl is similar to the ET from Ti to another carbonyl in the reductive-coupling process, according to the mechanistic proposal depicted in Scheme 7. The isolation of olefin as the main product from the 3:6:1 reaction of TiCl3, Ph2CO and Yb clearly demonstrates that the coupling step is rate determining and not the subsequent fission of the C–O bonds, which is in contrast to the previous observation.6,9
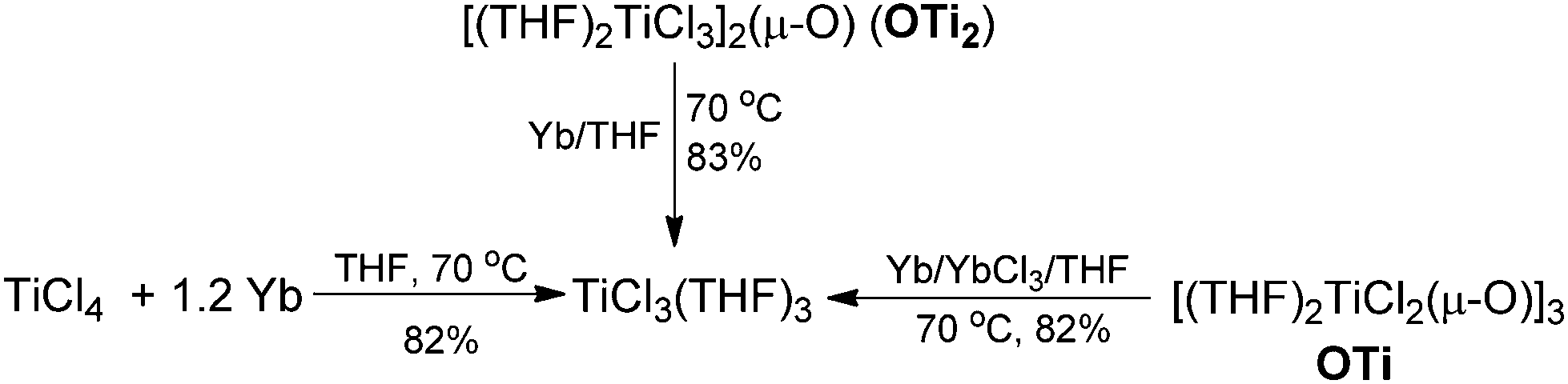 | ||
| Scheme 2 Formation and regeneration of TiCl3. | ||
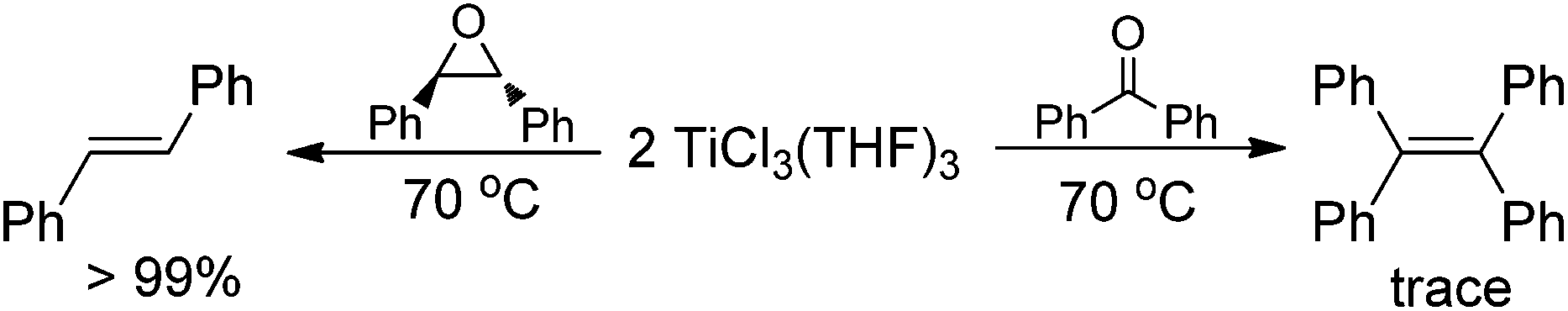 | ||
| Scheme 3 Reaction of TiCl3 with epoxide and ketone. | ||
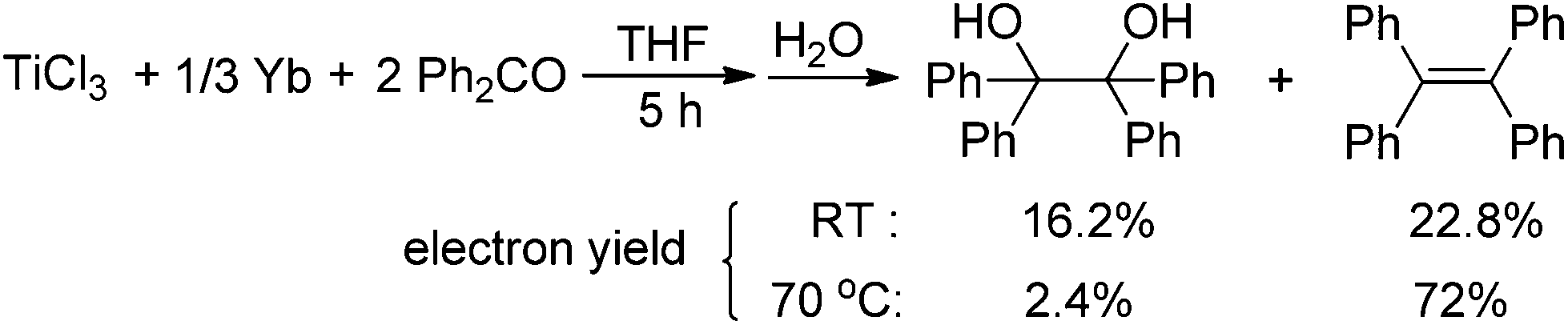 | ||
| Scheme 4 Examination of rate-determining step. | ||
To further assess whether Ti(IV) pinacolates are intermediates in the catalytic olefination reactions, tetraphenylethane-1,2-diol (H2TPL) and 1,2-diphenylethane-1,2-diol (H2DPL) were prepared. Salt-metathesis reactions of TiCl4 with 1 equiv. of Na2TPL at room temperature, generated in situ by stirring H2TPL with 2 equiv. of NaH as described by Woo,13 provided [(THF)2TiCl2(μ-O)]3 (OTi, Fig. 1) in 82% yield, indicating that Ti(IV) tetraphenylpinacolate is unstable. Adding 2/3 equiv. of Yb to the above mixture gave tetraphenylethene in 97%. Encouraged by this result, we subsequently treated Na2TPL with 2 equivalent of TiCl4 under the same conditions. A different Ti oxychloride [(THF)2TiCl3]2(μ-O) (OTi2)14 was obtained with high efficiency (83% yield) after 24 h at room temperature. Treatment of Na2TPL with 2 equiv. of TiCl4 followed by reacting with 2/3 equiv. of Yb gave tetraphenylethene in 90% yield (Scheme 5). Interestingly, switching Na2TPL to Na2DPL resulted in the isolation of the expected pinacolate Ti(IV) complex (ROTi), which can be partly transformed to OTi2 and 2,3-diphenyloxirane when heating at 70 °C. This difference between the reactivities of TPL[TiCl3(THF)2]2 and DPL[TiCl3(THF)2]2 is not surprising, since the epoxide elimination should be more facile for larger sterically crowding pinacolate than for the less sterically crowding one. Consistent with this, it was found that the mononuclear Ti pinacolate seems to undergo the epoxide elimination more easily than the dinuclear congener ROTi under the same conditions. In contrast, no olefination product was observed in the reaction of [(Me3Si)2N]2YbCl with H2TPL and subsequent Yb at 70 °C even for 24 hours. These results with pinacol probes support the intermediacy of a Ti(IV) pinacolate, which liberates alkene through a stepwise O-atom-transfer. The stepwise deoxygenation of Ti(IV) pinacolates indicates that the stereochemistry of the resulting alkenes mainly originates from the pinacol coupling of carbonyls.
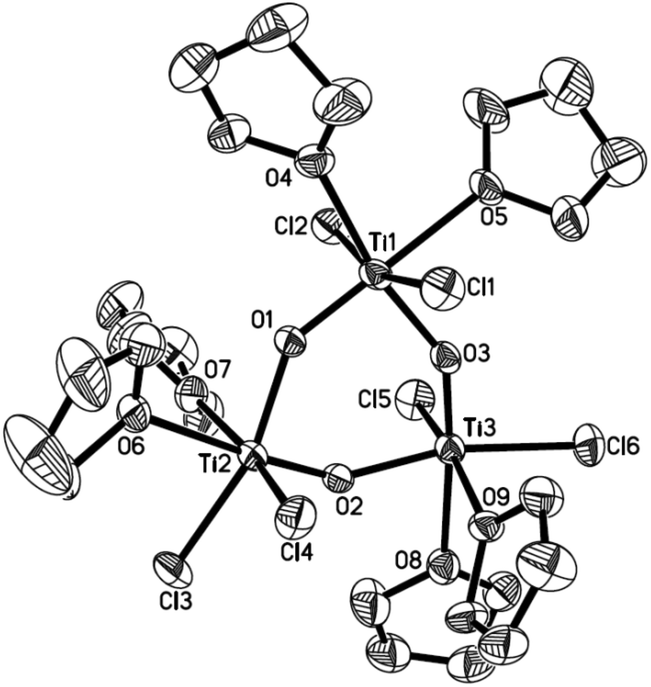 | ||
| Fig. 1 ORTEP structure of OTi. Thermal ellipsoids set at 30% probability. Hydrogen atoms are omitted for clarity. | ||
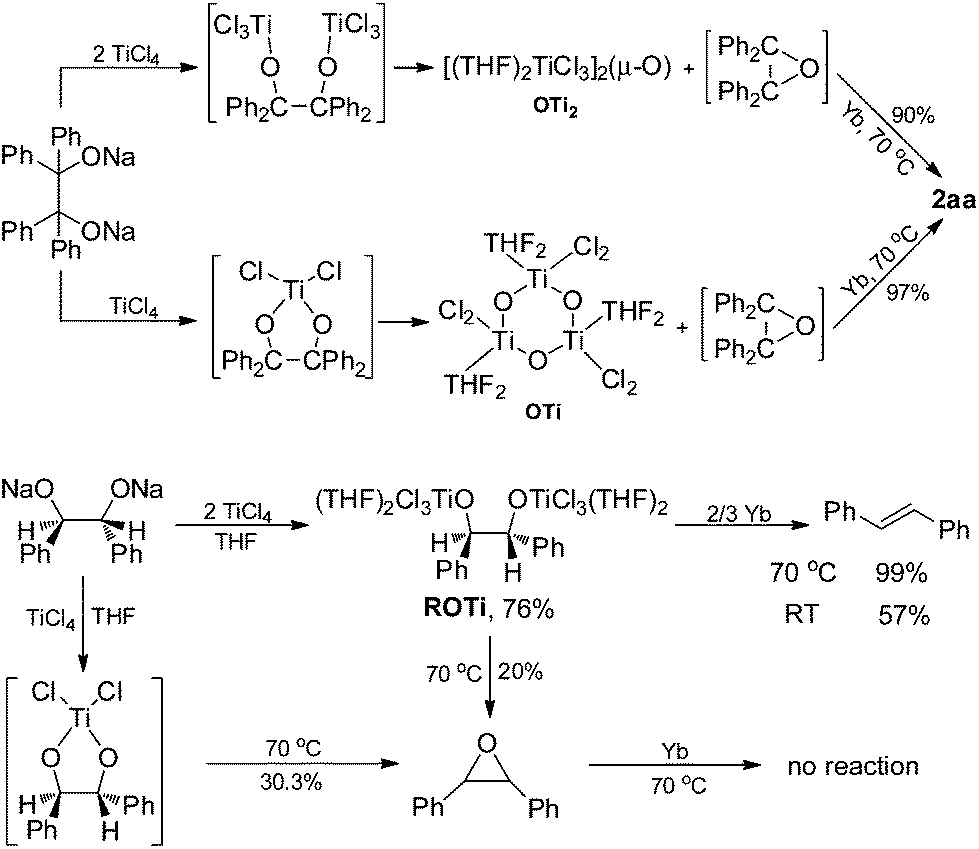 | ||
| Scheme 5 Simulating the deoxygenating processes. | ||
It is well-known that the low-valent titanium-mediated deoxygenation of pinacols is generally carried out via a successively homolytic cleavage of two C–O bonds, the presence of this first radical would facilitate the second C–O cleavage to lead to the corresponding olefin.6 As a result, it is very difficult to isolate the epoxide intermediate and control the stereochemistry of the resulting alkenes in such deoxygenation process. Remarkably, a sequential O-atom-transfer pathway became more pronounced under our catalytic conditions, which should explain excellently complete stereoselective formation of E-1,2-diarylethene products.
Although Ti-promoted pinacolization and deoxygenation coupling of aromatic aldehydes and ketones have been studied extensively, to the best of our knowledge, this is the direct evidence reported to date suggesting that Ti(IV) can facilitate the part deoxygenation of pinacol at room temperature. To gain more information on the mechanism of the catalytic olefination of carbonyls, we conducted catalytic and stoichiometric reactions with isolated OTi and OTi2 complexes. First, we investigated the regeneration of the active titanium species lying on the catalytic reaction (Scheme 2). Reduction of [TiCl3(THF)]2(μ-O) with Yb in THF at 70 °C led smoothly to the regeneration of TiCl3(THF)3. Significantly, reaction of [TiCl2(THF)2(μ-O)]3 with Yb in the presence of YbCl3 in THF at 70 °C for 5 h gave TiCl3(THF)3 too. Second, we performed the catalytic reaction in the presence of OTi2 or OTi (Scheme 6). Indeed, we found that a catalytic amount of OTi2 or OTi (10 mol%) could effect the transformation of Ph2CO to tetraphenylethene in the presence of 1 equiv. of Yb at 70 °C. At this moment, Yb2O3, which can be easily removed, was again proven to be the final acceptor of oxygen. Finally, the present OTi2 or OTi catalyst could perform the deoxygenation of 2,3-diphenyloxirane with Yb to give 2oo in almost quantitative yield.15 However, in the absence of OTi2 or OTi, the deoxygenation did not take place (Scheme 5). This result is consistent with the intermediacy of [TiCl2(THF)2(μ-O)]3 and [TiCl3(THF)2]2(μ-O) in the catalytic reaction.
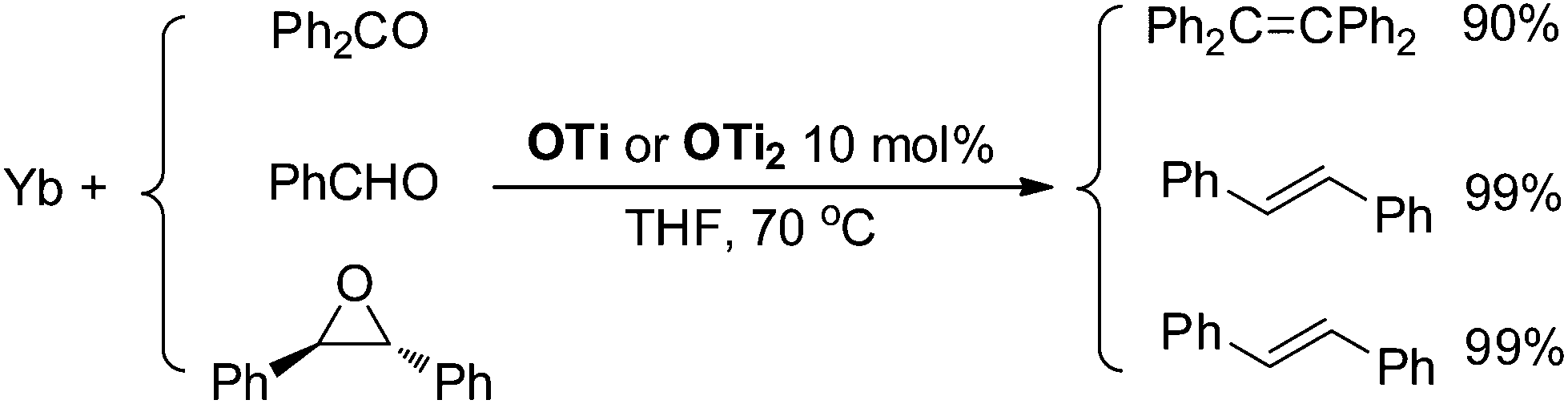 | ||
| Scheme 6 Catalytic deoxygenative coupling of carbonyls and epoxide using OTi and OTi2 as a catalyst. | ||
On the basis of the observations described above and reported previously,6,9 a possible mechanism for the present catalytic olefination of bulky ketones can be proposed as shown in Scheme 7. Reduction of TiCl4 with Yb generates TiCl3, which reacts with ketone to form the Ti ketyl intermediate. The further ET from Yb to the Ti ketyl complex is a prerequisite for coupling of the ketyl with another carbonyl.9 The resulting Ti(IV) pinacolate intermediate undergoes the concerted epoxide elimination to afford OTi. TiCl3 abstracts oxygen from epoxide, giving the olefination product 2. Reduction of the newly formed titanium oxychlorides using Yb leads to the regeneration of TiCl3. An intramolecular oxychloride elimination of Ti(IV) pinacolate followed by an O-atom-transfer from epoxide to Ti(III) species would explain the highly stereoselective formation of the E-alkene product 2. Rare earth metal plays two roles in our system. First, it reduces the Ti(IV) species to generate the active species that promotes the reductive coupling of carbonyls and cleaves the C–O bond of the epoxide intermediate. Second, the rare earth metal accumulates as the final oxygen trap, making the resulting oxide easily removable. Since an aromatic ketone is preferred in the first carbonyl reduction due to the possible formation of a stable ketyl species while for the successive coupling of either the Ti centers sensitive to steric factors or the higher reactivity of aliphatic ketyls compared to aromatic ketyls should prefer less sterically demanding aliphatic aldehydes and ketones over bulky aromatic analogues; the present reducing system completely distinguishes between aromatic and aliphatic substrates, and makes the cross-deoxygenative coupling proceed in good to excellent yield. Therefore, the Yb/TiCl4 promoted homo- and cross-coupling of carbonyl compounds to olefins is an ideal addition to the family of the carbonyl olefinations.
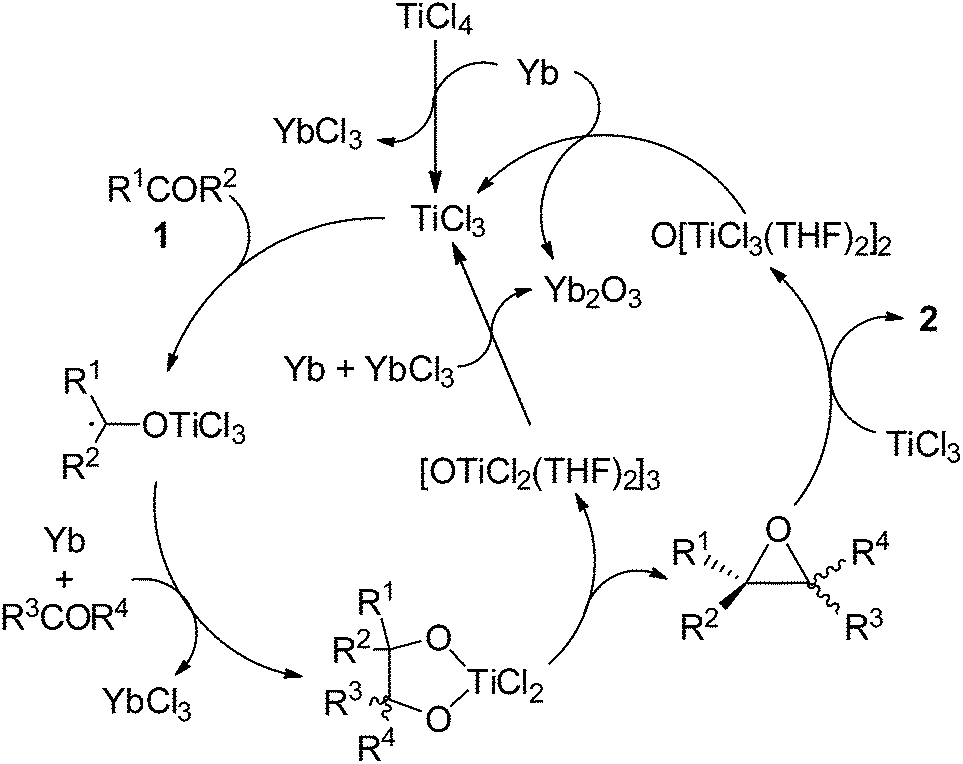 | ||
| Scheme 7 Possible mechanism for the present catalytic olefination of carbonyls. | ||
Conclusions
In summary, we have demonstrated that Yb in combination with a catalytic amount of TiCl4 can serve as an excellent reductive system for deoxygenative coupling of aldehydes and ketones to olefins. Remarkably, the present process is operationally simple and atom-economical, minimizes the generation of chemical waste, and makes Yb2O3 as the only by-product easily removed and utilized; further, the yield improved significantly on that obtained in the stoichiometric version in our system. This result showed that in our reaction conditions aromatic ketone-derived ketyl radicals should prefer to attack an aliphatic ketone to give unsymmetrical olefins.TiCl3, Ti(IV)-pinacolates and oxychlorides were isolated, characterized, and demonstrated to be potential intermediates for reductive olefination of aldehydes and ketones. Our mechanistic data not only provide the direct evidence that Ti(IV) can facilitate the part deoxygenation of pinacols at room temperature, but also prove, for the first time, that for bulky ketones the turnover-limiting step is the Ti-mediated carbonyl coupling rather than the subsequent deoxygenation of the resulting pinacolate intermediates. Such mechanistic insight would bring to light complications involved in the Ti-mediated McMurry olefination and suggest what might be a general role of pinacolates in organic synthesis. Further studies on other applications of this reducing system in organic synthesis are presently under way in our laboratory.
Acknowledgements
We thank the National Natural Science Foundation of China, 973 program (2009CB825300) and the Research Fund for the Doctoral Program of Higher Education of China.Notes and references
- (a) J. E. McMurry and L. R. Krepski, J. Org. Chem., 1976, 41, 3929 CrossRef CAS; (b) J. E. McMurry, Chem. Rev., 1989, 89, 1513 CrossRef CAS; (c) B. Bogdanovic and A. Bolte, J. Organomet. Chem., 1995, 502, 109 CrossRef CAS; (d) T. Wirth, Angew. Chem., Int. Ed. Engl., 1996, 35, 61 CrossRef CAS; (e) A. Fürstner and B. Bogdanovic, Angew. Chem., Int. Ed. Engl., 1996, 35, 2442 CrossRef; (f) M. Ephritikhine, Chem. Commun., 1998, 2549 RSC; (g) A. Gansäuer and H. Bluhm, Chem. Rev., 2000, 100, 2771 CrossRef PubMed.
- (a) M. Sato and K. Oshima, Chem. Lett., 1982, 157 CrossRef CAS; (b) R. Dams, M. Malinowski, I. Westdorp and H. Y. Geise, J. Org. Chem., 1982, 47, 248 CrossRef CAS; (c) S. K. Nayak and A. Banerji, J. Org. Chem., 1991, 56, 1940 CrossRef CAS; (d) J. Szymoniak, J. Besancon and C. Moïse, Tetrahedron, 1992, 48, 3867 CrossRef CAS; (e) T. Eguchi, T. Terachi and K. Kakinuma, Tetrahedron Lett., 1993, 34, 2175 CrossRef CAS; (f) K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannan and E. J. Sorensen, Nature, 1994, 367, 630 CrossRef CAS PubMed; (g) K. C. Nicolaou, J. J. Liu, Z. Yang, H. Ueno, E. J. Sorensen, C. F. Claiborne, R. K. Guy, C. K. Hwang, M. Nakada and P. G. Nantermet, J. Am. Chem. Soc., 1995, 117, 634 CrossRef CAS; (h) A. Fürstner, H. Weintritt and A. Hupperts, J. Org. Chem., 1995, 60, 6637 CrossRef; (i) K. C. Nicolaou, Z. Yang, J. J. Liu, P. G. Nantermet, C. F. Claiborne, J. Renaud, R. K. Guy and K. Shibayama, J. Am. Chem. Soc., 1995, 117, 645 CrossRef CAS; (j) A. Fürstner and G. Seidel, Synthesis, 1995, 63 CrossRef PubMed; (k) D. K. Dutta and D. Konwar, Tetrahedron Lett., 2000, 41, 6227 CrossRef CAS; (l) T. A. Salama, S. S. Elmorsy and A. G. M. Khalil, Tetrahedron Lett., 2007, 48, 4395 CrossRef CAS PubMed; (m) S. V. Bhilare, N. B. Darvatkar, A. R. Deorukhkar, M. S. Rasalkar and M. M. Salunkhe, Synth. Commun., 2007, 37, 3111 CrossRef CAS.
- For applications of McMurry reaction, see: (a) D. R. Moreno, G. Giorgi, C. O. Salas and R. A. Tapia, Molecules, 2013, 18, 14797 CrossRef CAS PubMed; (b) Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. M. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210 RSC; (c) Y. L. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660 CrossRef CAS PubMed; (d) Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622 CrossRef CAS PubMed; (e) J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675 RSC; (f) H. Huang, Z. Chen, R. P. Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y. Y. Noh, K. J. Baeg, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 10966 CrossRef CAS PubMed; (g) S. Lahore, U. Narkhede, L. Merlini and S. Dallavalle, J. Org. Chem., 2013, 78, 10860 CrossRef CAS PubMed.
- (a) A. Fürstner, Angew. Chem., Int. Ed. Engl., 1993, 32, 164 CrossRef; (b) S. Talukdar, S. K. Nayak and A. Banerji, J. Org. Chem., 1998, 63, 4925 CrossRef CAS.
- N. Balu, S. K. Nayak and A. Banerji, J. Am. Chem. Soc., 1996, 118, 5932 CrossRef CAS.
- H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. M. Herrador, J. F. Q. Moral and A. F. Barrero, J. Am. Chem. Soc., 2010, 132, 254 CrossRef PubMed.
- A. Fürstner and A. Hupperts, J. Am. Chem. Soc., 1995, 117, 4468 CrossRef.
- (a) Z. Zhu, C. Wang, X. Xiang, C. Pi and X. Zhou, Chem. Commun., 2006, 2066 RSC; (b) Z. Zhu, J. Wang, Z. Zhang, X. Xiang and X. Zhou, Organometallics, 2007, 26, 2499 CrossRef CAS; (c) M. Kira, T. Obata, I. Kon, H. Hashimoto, M. Ichinohe, H. Sakurai, S. Kyushin and H. Matsumoto, Chem. Lett., 1998, 11, 1097 CrossRef.
- M. Paradas, A. G. Campaña, R. E. Estévez, L. Á. de Cienfuegos, T. Jiménez, R. Robles, J. M. Cuerva and J. E. Oltra, J. Org. Chem., 2009, 74, 3616 CrossRef CAS PubMed.
- (a) T. Imamoto, T. Kusumoto, Y. Hatanaka and M. Yokoyama, Tetrahedron Lett., 1982, 23, 1353 CrossRef CAS; (b) L. Wang and Y. Zhang, Tetrahedron, 1998, 54, 11129 CrossRef CAS; (c) R. Yanada, N. Negoro, M. Okaniwa and T. Ibuka, Tetrahedron, 1999, 55, 13947 CrossRef CAS; (d) A. Ghatak, F. F. Becker and B. K. Banik, Tetrahedron Lett., 2000, 41, 3793 CrossRef CAS; (e) S. Talukdar and J. M. Fang, J. Org. Chem., 2001, 66, 330 CrossRef CAS; (f) W. S. Jin, Y. Makioka, T. Kitamura and Y. Fujiwara, J. Org. Chem., 2001, 66, 514 CrossRef CAS; (g) T. Nishino, Y. Nishiyama and N. Sonada, J. Org. Chem., 2002, 67, 966 CrossRef CAS PubMed; (h) T. Nishino, M. Okada, T. Kuroki, T. Watanabe, Y. Nishiyama and N. Sonada, J. Org. Chem., 2002, 67, 8696 CrossRef CAS PubMed; (i) H. B. Kagan, Tetrahedron, 2003, 59, 10351 CrossRef CAS PubMed; (j) X. Xiang, Q. Shen, J. Wang, Z. Zhu, W. Huang and X. Zhou, Organometallics, 2008, 27, 1959 CrossRef CAS; (k) W. Chen, K. Li, Z. Hu, L. Wang, G. Lai and Z. Li, Organometallics, 2011, 30, 2026 CrossRef CAS.
- (a) Z. Hou, K. Takamine, Y. Fujiwara and H. Taniguchi, Chem. Lett., 1987, 2061 CrossRef CAS; (b) Z. Hou, K. Takamine, O. Aoki, H. Shiraishi, Y. Fujiwara and H. Taniguchi, J. Org. Chem., 1988, 53, 6077 CrossRef CAS; (c) Z. Hou, A. Fujita, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1996, 118, 7843 CrossRef CAS; (d) Z. Hou, A. Fujita, Y. Zhang, T. Miyano, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 1998, 120, 754 CrossRef CAS; (e) O. Akiya, T. Hiroki and H. Toshikazu, Tetrahedron Lett., 1999, 40, 7113 CrossRef; (f) S. Fukuzawa, N. Nakano and T. Saitoh, Eur. J. Org. Chem., 2004, 2863 CrossRef CAS.
- (a) L. E. Aleandri, B. Bogdanovíc, A. Gaidies, D. J. Jones, S. Liao, A. Michalowicz, J. Rozière and A. Schott, J. Organomet. Chem., 1993, 459, 87 CrossRef CAS; (b) L. E. Aleandri, S. Becke, B. Bogdanovíc, D. J. Jones and J. Rozière, J. Organomet. Chem., 1994, 472, 97 CrossRef CAS; (c) B. Bogdanovíc and A. Bolte, J. Organomet. Chem., 1995, 502, 109 CrossRef.
- G. Du and L. K. Woo, Organometallics, 2003, 22, 450 CrossRef CAS.
- R. Mahrwald, B. Ziemer and S. Troyanov, Tetrahedron Lett., 2001, 42, 6843 CrossRef CAS.
- For deoxygenation of epoxide, see: (a) J. E. McMurry, M. G. Silvestri, M. P. Fleming, T. Hoz and M. W. Grayston, J. Org. Chem., 1978, 43, 3249 CrossRef CAS; (b) L. M. Atagi, D. E. Over, D. R. McAlister and J. M. Mayer, J. Am. Chem. Soc., 1991, 113, 870 CrossRef CAS; (c) K. G. Moloy, Inorg. Chem., 1988, 27, 677 CrossRef CAS; (d) T. V. RajanBabu, W. A. Nugent and M. S. Beattie, J. Am. Chem. Soc., 1990, 112, 6408 CrossRef CAS; (e) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986 CrossRef CAS; (f) A. F. Mateos, P. H. Teijón and R. R. González, Tetrahedron, 2013, 69, 1611 CrossRef PubMed; (g) Y. Li, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2013, 52, 12636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1000407 and 1000408. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00140k |
| This journal is © the Partner Organisations 2014 |