Ozonation of methylenecyclopropanes†
Rui
Sang
,
Xiang-Ying
Tang
and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China. E-mail: mshi@mail.sioc.ac.cn
First published on 23rd June 2014
Abstract
Ozonation of methylenecyclopropanes bearing gem-disubstituted electron-withdrawing groups (EWG) gave ring-opened oxidative products in moderate to good yields. For MCPs in which EWGs are two methoxycarbonyl groups, the ozonation gave oxidative cyclization products in methanol at −78 °C in the presence of CuCl; for MCPs in which EWGs are one methoxycarbonyl and one trifluoromethyl group, the ozonation produced α-diketones in ethyl acetate (EA) at −78 °C.
Methylenecyclopropanes (MCPs) as highly strained but easily available small rings have been widely used in organic synthesis as versatile building blocks through a variety of ring-opening processes.1 After careful survey of the reaction patterns of MCPs, we found that the oxidative ring-opening modes of MCPs are limited. The well known examples are the oxidation of MCPs with peracids or SeO2 and H2O2 or other oxidants to give cyclobutanone derivatives.2a–e Another important issue is the oxidative addition of 1,3-dicarbonyl compounds with MCPs via a one electron transfer process mediated by Mn(OAc)3 or CAN2f–j as well as the photo-induced oxidation of MCPs with O2 to give the corresponding 1,2-dioxolane via a charge transfer complex.3 On the other hand, the ozonolysis of alkylidenecyclopropanes to give the corresponding oxidative ring-opened products has also been disclosed by several groups.4 In this paper, we wish to report two different oxidative ring-opening modes of MCPs, in which the cyclopropane has a gem-disubstituted ester group [(CO2Me)2] (MCPs 1) or a gem-disubstituted ester and a trifluoromethyl group [CF3(CO2Me)] (MCPs 2) as electron-withdrawing groups (EWG),5 upon treating with ozone. We will disclose in this paper that the two different ring-opening oxidation products can be exclusively obtained under different conditions.
The initial examinations were carried out upon treating MCP 1a or MCP 2a with ozone in the presence of a variety of reductants and the results are shown in Tables 1 and 2, respectively. We found that the oxidative cyclization product 3a and α-diketone 4a′ were obtained in dichloromethane (DCM) upon treatment of 1a with ozone in a variety of reductants at −78 °C (1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reductant = 1:1.3) (Table 1, entries 1–8). To obtain 3a as the sole product, we carefully examined the solvent effects using CuCl as the reductant and found that in ethanol or methanol, 3a could be obtained exclusively in 87% or 88% yield, respectively (Table 1, entries 9–16). The reductant CuCl is crucial in this reaction to give 3a exclusively and using NiCl2 or FeCl2 as the reductant gave 3a and 4a′ as a product mixture (Table 1, entries 17–19). The examination of the employed amount of CuCl and the reaction temperature revealed that using 2.0 equiv. of CuCl afforded 3a in 89% yield under the standard conditions and the oxidation reaction should be carried out at −78 °C (Table 1, entries 20–23). The structure of 3a has been fully assigned with NMR spectroscopic data (see ESI†).
:reductant = 1:1.3) (Table 1, entries 1–8). To obtain 3a as the sole product, we carefully examined the solvent effects using CuCl as the reductant and found that in ethanol or methanol, 3a could be obtained exclusively in 87% or 88% yield, respectively (Table 1, entries 9–16). The reductant CuCl is crucial in this reaction to give 3a exclusively and using NiCl2 or FeCl2 as the reductant gave 3a and 4a′ as a product mixture (Table 1, entries 17–19). The examination of the employed amount of CuCl and the reaction temperature revealed that using 2.0 equiv. of CuCl afforded 3a in 89% yield under the standard conditions and the oxidation reaction should be carried out at −78 °C (Table 1, entries 20–23). The structure of 3a has been fully assigned with NMR spectroscopic data (see ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Reductant |
1a:reductant |
Solvent | T (°C) | Yieldb (%) | |
| 3a | 4a′ | |||||
| a MCP 1a (0.5 mmol, 1 equiv.) was dissolved in 5.0 mL solvent at T °C, and then O3 was bubbled until the solvent became blue. The reductant (x equiv.) was then added at T °C. The reaction mixture naturally returned to room temperature and was further stirred for 5 h. b Isolated yields. c The crude product containing some other complex mixtures. | ||||||
| 1 |

|
1:1.3 |
DCM | −78 | 42 | 41 |
| 2 | Ph3P | 1:1.3 |
DCM | −78 | 67 | 31 |
| 3 | Zn | 1:1.3 |
DCM | −78 | 42 | 29 |
| 4 | S | 1:1.3 |
DCM | −78 | 52 | 30 |
| 5 | Na2NO2 | 1:1.3 |
DCM | −78 | 55 | 37 |
| 6 | Na2SO3 | 1:1.3 |
DCM | −78 | 60 | 22 |
| 7 | Na2S2O3 | 1:1.3 |
DCM | −78 | 62 | 25 |
| 8 | CuCl | 1:1.3 |
DCM | −78 | 80 | 18 |
| 9 | CuCl | 1:1.3 |
EA | −78 | 55 | 31 |
| 10 | CuCl | 1:1.3 |
THF | −78 | — | — |
| 11 | CuCl | 1:1.3 |
Acetone | −78 | 83 | 14 |
| 12 | CuCl | 1:1.3 |
Et2O | −78 | 68 | 30 |
| 13 | CuCl | 1:1.3 |
PE | −78 | 32 | 5 |
| 14 | CuCl | 1:1.3 |
Toluene | −78 | 62 | 22 |
| 15 | CuCl | 1:1.3 |
EtOH | −78 | 87 | — |
| 16 | CuCl | 1:1.3 |
MeOH | −78 | 88 | — |
| 17 | NiCl2 | 1:1.3 |
MeOH | −78 | 59 | 17 |
| 18 | FeCl2 | 1:1.3 |
MeOH | −78 | 67 | 4 |
| 19 | CuCl | 1:0 |
MeOH | −78 | 33 | 41 |
| 20 | CuCl | 1:0.5 |
MeOH | −78 | 52 | 27 |
| 21 | CuCl | 1:2 |
MeOH | −78 | 89 | — |
| 22 | CuCl | 1:3 |
MeOH | −78 | 88 | — |
| 23 | CuCl | 1:2 |
MeOH | 0 | 80c | — |
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Reductant |
2a:reductant |
Solvent | T (°C) | Yieldb (%) | |
| 3a′ | 4a | |||||
| a MCP 2a (0.5 mmol, 1 equiv.) was dissolved in 5 mL solvent at −78 °C, and then O3 was slowly bubbled until the solvent became blue. The reductant was then added at −78 °C. The reaction mixture naturally returned to room temperature with stirring and was further stirred for 5 h. b Isolated yields. | ||||||
| 1 | CuCl | 1:1.3 |
MeOH | −78 | 27 | 37 |
| 2 |

|
1:1.3 |
DCM | −78 | 12 | 62 |
| 3 | Ph3P | 1:1.3 |
DCM | −78 | 8 | 70 |
| 4 | Zn | 1:1.3 |
DCM | −78 | 20 | 47 |
| 5 | S | 1:1.3 |
DCM | −78 | 23 | 42 |
| 6 | NaNO2 | 1:1.3 |
DCM | −78 | 17 | 51 |
| 7 | Na2SO3 | 1:1.3 |
DCM | −78 | 15 | 57 |
| 8 | — | — | DCM | −78 | Trace | 68 |
| 9 | — | — | Acetone | −78 | Trace | 65 |
| 10 | — | — | EA | −78 | Trace | 70 |
| 11 | — | — | Toluene | −78 | Trace | 63 |
| 12 | — | — | Et2O | −78 | 4 | 58 |
| 13 | — | — | MeOH | −78 | Trace | 53 |
On the other hand, using MCP 2a as the substrate, the presence of the reductant gave the corresponding 3a′ and 4a as a product mixture in 64–78% total yields in methanol or DCM at −78 °C (Table 2, entries 1–7). We also identified that 4a could be obtained as a major product in 68% yield along with a trace of 3a′ in the absence of the reductant (Table 2, entry 8). Its structure could be proved by the condensation of ethyl 3,3,3-trifluoro-2-oxopropanoate (0.5 mmol, 1 equiv.) and 1-phenylpropane-1,2-dione in the presence of DABCO (see ESI†). The examination of solvent effects revealed that carrying out the reaction in ethyl acetate (EA) afforded 4a in 70% yield and this served as the best conditions for the formation of 4a (Table 2, entries 8–13). The different oxidation products obtained from 1a and 2a are presumably due to the different EWGs.
With the identification of the best reaction conditions, we next turned our attention to study the scope and limitations of these two oxidative ring-opening reactions and the results are summarized in Tables 3 and 4, respectively. A variety of MCPs 1 and 2 with aryl groups bearing different substituents have been tested and the corresponding oxidative products 3a–3g and 4a–4g were obtained in moderate to good yields without the observation of significant electronic effects (Table 3, entries 1–6 and Table 4, entries 1–6). As for substrates 1h and 2h having a naphthyl substituent, and substrates 1i and 2i in which the aryl groups have two substituents, the reactions also proceeded smoothly, delivering the corresponding products 3h and 4h in 43% and 73% yields and 3i and 4i in 87% and 58% yields, respectively (Table 3, entries 7 and 8 and Table 4, entries 7 and 8). Employing 1k as the substrate gave the desired product 3k in 38% yield (Table 3, entry 10). In the cases of aliphatic MCP 1j, in which R = benzyl group, the corresponding oxidative cyclized product 3j was obtained in 61% yield (Table 3, entry 9). However, in the case of 2j, the ozonation also gave the corresponding oxidative cyclized product 3j′ as a single diastereoisomer on the basis of NMR spectroscopic data rather than the desired α-diketone product 4j (see ESI†) (Table 4, entry 9). At the present stage, we cannot perfectly explain this observation, perhaps because an aromatic group is also required to stabilize the cyclic species B1 or B2 to give the corresponding α-diketone product (Scheme 3).
|
|
||
|---|---|---|
| Entrya | R | 3, yieldb (%) |
| a MCP 1 (0.5 mmol, 1 equiv.) was dissolved in 5 mL MeOH at −78 °C, and then O3 was slowly bubbled until the solvent became blue. CuCl (2 equiv.) was added at −78 °C and the reaction mixture naturally returned to room temperature with stirring and was further stirred for 5 h. b Isolated yields. | ||
| 1 | 4-BrC6H4, 1b | 3b, 57 |
| 2 | 4-ClC6H4, 1c | 3c, 62 |
| 3 | 4-MeC6H4, 1d | 3d, 62 |
| 4 | 3-MeC6H4, 1e | 3e, 70 |
| 5 | 2-MeC6H4, 1f | 3f, 67 |
| 6 | 4-MeOC6H4, 1g | 3g, 67 |
| 7 | 2-Naphthyl, 1h | 3h, 43 |
| 8 | 3,5-Me2C6H3, 1i | 3i, 87 |
| 9 | Bn, 1j | 3j, 61 |
| 10 | 3,5-Br2C6H3, 1k | 3k, 38 |
|
|
||
|---|---|---|
| Entrya | R | 4, yieldb (%) |
| a MCP 2 (1 mmol, 1 equiv.) was dissolved in 5 mL EA at −78 °C, and then O3 was slowly bubbled until the solvent became blue. The reaction mixture naturally returned to r.t. with stirring and was further stirred for 5 h. b Isolated yields. c The structure of 4j′ is the oxidative cyclized product similar to that of 3j. | ||
| 1 | 4-BrC6H4, 2b | 4b, 64 |
| 2 | 4-ClC6H4, 2c | 4c, 61 |
| 3 | 4-MeC6H4, 2d | 4d, 63 |
| 4 | 3-MeC6H4, 2e | 4e, 65 |
| 5 | 2-MeC6H4, 2f | 4f, 67 |
| 6 | 4-MeOC6H4, 2g | 4g, 72 |
| 7 | 2-Naphthyl, 2h | 4h, 73 |
| 8 | 3,5-Me2C6H3, 2i | 4i, 58 |
| 9 | Bn, 2j | 3j ′,c 21 |
For MCP 5a, the reaction gave the corresponding oxidative cyclization product 6a in 33% yield along with a double bond cleaved aldehyde in 23% yield in the presence of a reductant under the standard conditions (Scheme 1).4e In the absence of a reductant, the corresponding oxidative cyclized product 6b was formed in 64% yield (Scheme 1). In the case of disubstituted MCP 5b, 2,2-diphenylcyclobutanone 6b was formed in 28% yield along with benzophenone in 49% yield (Scheme 1).4c In all these cases, none of the corresponding α-diketone could be identified.
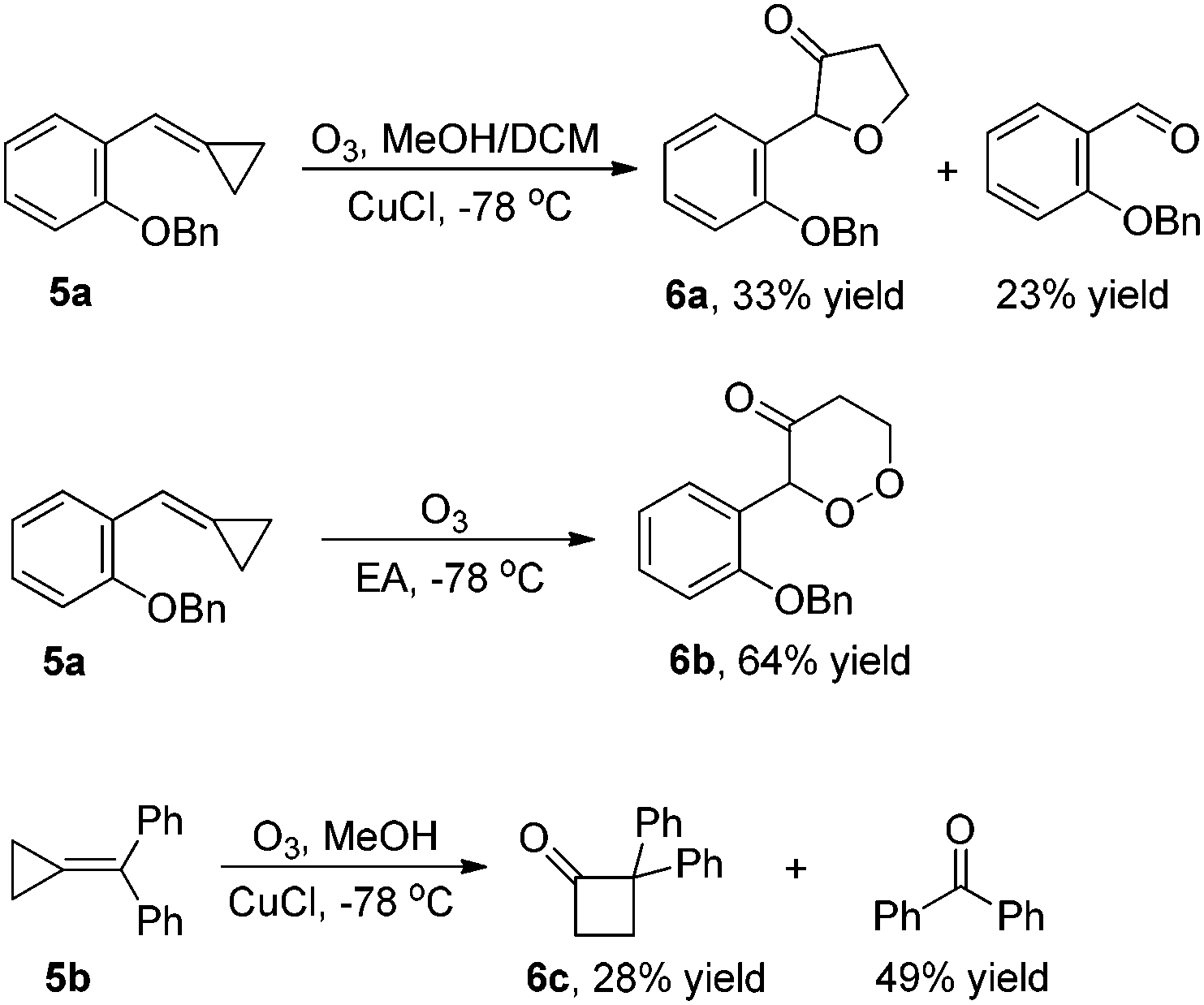 | ||
| Scheme 1 Ozonation of MCPs 5a and 5b. | ||
The control experiment has confirmed that these reactions under the optimized conditions were unaffected by the addition of the radical inhibitor such as TEMPO (1.0 equiv.), rendering unlikely the intervention of a radical pathway (Scheme 2).
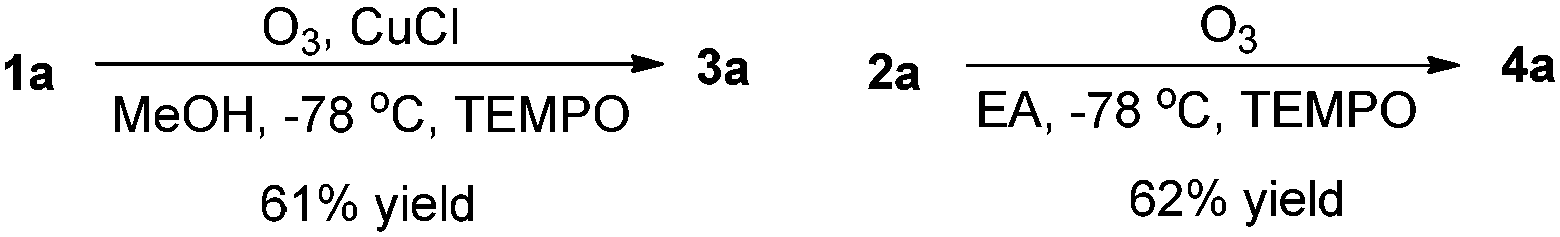 | ||
| Scheme 2 The control experiment in the presence of TEMPO (1.0 equiv.). | ||
The plausible reaction mechanism is depicted below using 1a and 2a as substrate models on the basis of the previous literature and the control experiments (Scheme 3).4e The [3 + 2] cycloaddition of O3 with 1a or 2a gives the intermediate A1 or B1, which undergoes a heterolytic O–O bond cleavage and cyclopropane ring opening to afford the intermediate A2 or B2, respectively. In the case of MCP 1a, the intermediate A2 undergoes rearrangement to afford the intermediate A3, which gives the final product 3avia the Fenton reaction.6 The intermediate A2 can also directly afford 3avia the Fenton reaction. In the case of MCP 2a, the intermediate B2 directly undergoes the O–O bond heterolytic cleavage and proton transfer to give the final product 4a. Presumably due to the strong electron-withdrawing effect of the CF3 substituent, the O–O bond heterolytic cleavage in the intermediate B2 can proceed preferentially under the reaction conditions to give 4a, whereas the intermediate A2 leans to undergo cyclization to give the zwitterionic intermediate A3 or the Fenton reaction. Therefore, the different electronic effects of EWGs in MCPs 1 or 2 cause the different reaction pathways.
 | ||
| Scheme 3 Plausible mechanisms for the formation of 3a and 4a. | ||
MCP 2a can be easily transformed into MCP 8avia hydrolysis under basic conditions and condensation with para-bromobenzylamine in the presence of EDCI and HOBt in DMF. MCP 8a can also undergo ring-opening oxidative cleavage to give 9a in 32% yield via the α-diketone intermediate C under the standard conditions (Scheme 4).7 Its structure has been identified by X-ray diffraction and the CIF data are presented in the ESI†.8
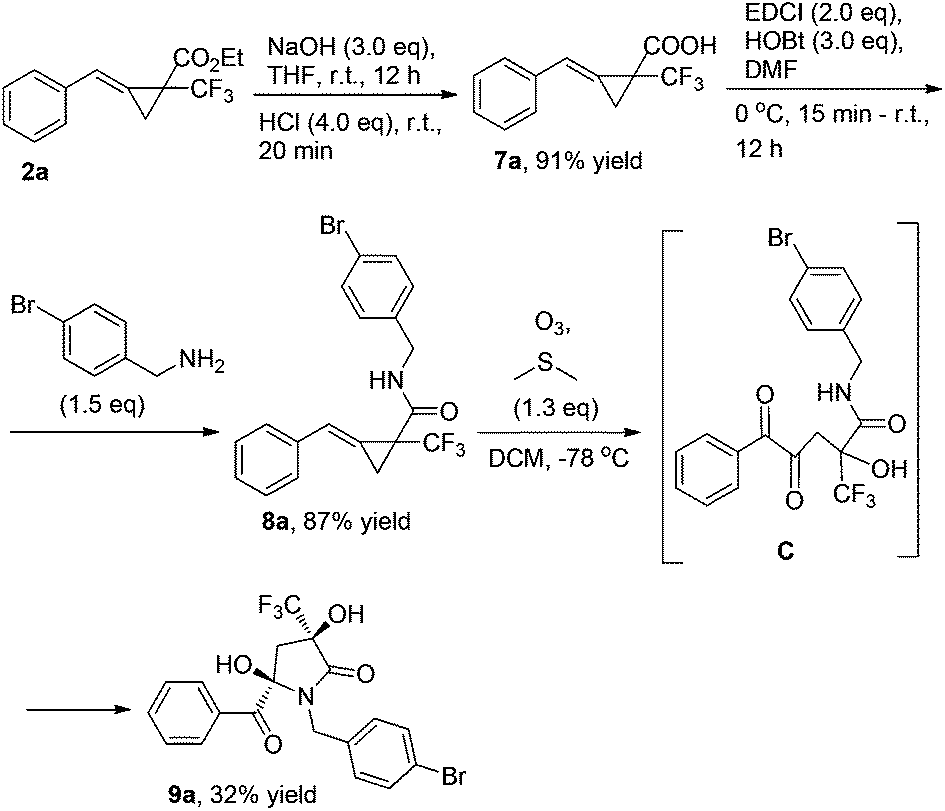 | ||
| Scheme 4 The further transformation of MCP 2a with O3. | ||
In summary, we have developed two easily available ozonation processes for MCPs 1 and 2 bearing gem-disubstituted EWGs. On the control of the oxidative conditions, the ring-opening oxidative cyclization products 3 could be obtained as the sole products for MCPs 1, in which the gem-disubstituted EWGs are two methoxycarbonyl groups, and the ring-opening oxidative α-diketones 4 could be afforded as the major products in most cases for MCPs 2, in which the gem-disubstituted EWGs are one methoxycarbonyl and one trifluoromethyl group, in moderate to good yields. The electronic properties of EWG play a significant role in the reaction outcomes. The related mechanisms have also been proposed. Further investigations to examine the mechanistic details more extensively and the application of this oxidation method are underway in our laboratory.
Acknowledgements
We thank the Shanghai Municipal Committee of Science and Technology (11JC1402600), the National Basic Research Program of China (973)-2010CB833302, the Fundamental Research Funds for the Central Universities, and the National Natural Science Foundation of China for financial support (20472096, 21372241, 20672127, 21361140350, 21102166, 21121062, 21302203 and 20732008).Notes and references
- For the synthesis of MCPs, please see: (a) A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589–636 CrossRef CAS PubMed; (b) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575–608 CrossRef CAS; (c) N. S. Isaacs, Physical Organic Chemistry, John Wiley, New York, 1987, p. 283 Search PubMed; (d) Small Ring Compounds in Organic Synthesis III, ed. A. de Meijere, Springer, Berlin, 1988 Search PubMed; (e) P. Binger and U. Schuchardt, Chem. Ber., 1981, 114, 3313–3324 CrossRef CAS; (f) P. Binger and H. M. Buech, Top. Curr. Chem., 1987, 135, 77–151 CrossRef CAS; (g) W. A. Donaldson, Adv. Met.-Org. Chem., 1991, 2, 269–293 CAS; (h) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed; (i) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111–129 CrossRef CAS; (j) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641–652 CrossRef CAS PubMed; (k) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; (l) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712–9721 CrossRef CAS PubMed; (m) H. Pellissier, Tetrahedron, 2010, 66, 8341–8375 CrossRef CAS PubMed.
- For the direct oxidation of methylenecyclopropanes, see: (a) J. K. Crandall and W. W. Conover, J. Org. Chem., 1978, 43, 3533–3535 CrossRef CAS; (b) J. Salaun, B. Garnier and J. M. Conia, Tetrahedron, 1974, 30, 1423–1426 CrossRef CAS; (c) D. H. Aue, M. J. Meshishnek and D. F. Shellhamer, Tetrahedron Lett., 1973, 48, 4799–4802 CrossRef; (d) J. R. Salaun and J. M. Conia, J. Chem. Soc. D, 1971, 1579–1580 RSC; (e) V. Nair, T. D. Suja and K. Mohanan, Synthesis, 2006, 2531–2534 CrossRef CAS PubMed. For the oxidation of methylenecyclopropanes on the basis of one electron transfer, see: (f) J.-W. Huang and M. Shi, J. Org. Chem., 2005, 70, 3859–3863 CrossRef CAS PubMed; (g) W.-J. Fu and X. Huang, J. Organomet. Chem., 2007, 692, 740–745 CrossRef CAS PubMed; (h) V. Nair, T. D. Suja and K. Mohanan, Synthesis, 2006, 2335–2338 CrossRef CAS PubMed; (i) H. Ikeda, H. Namai, N. Kato and T. Ikeda, Tetrahedron Lett., 2006, 47, 1857–1860 CrossRef CAS PubMed; (j) W. Chen, X. Huang, H. Zhou and L. Ren, Synthesis, 2006, 609–612 CAS.
- (a) Y. Takahashi, T. Miyashi and T. Mukai, J. Am. Chem. Soc., 1983, 105, 6511–6512 CrossRef CAS; (b) T. Miyashi, M. Kamata and T. Mukai, J. Am. Chem. Soc., 1986, 108, 2755–2756 CrossRef CAS.
- (a) F. R. Goss, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., 1923, 123, 327–361 RSC; (b) J. T. Gragson, K. W. Greenlee, J. M. Derfer and C. E. Boord, J. Am. Chem. Soc., 1953, 75, 3344–3347 CrossRef CAS; (c) R. F. Langler, R. K. Raheja, K. Schank and H. Beck, Helv. Chim. Acta, 2001, 84, 1943–1951 CrossRef CAS; (d) C. J. M. Van den Heuvel, A. Hofland, J. C. van Velzen, H. Steinberg and Th. J. de Boer, Recl. Trav. Chim. Pays-Bas, 1984, 103, 233–240 CrossRef CAS; (e) A. de Meijere, I. Erden, W. Weber and D. Kaufmann, J. Org. Chem., 1988, 53, 152–161 CrossRef CAS; (f) G. Buechi and H. Wuest, J. Am. Chem. Soc., 1978, 100, 294–295 CrossRef CAS; (g) K. Igawa, Y. Kawasaki and K. Tomooka, Chem. Lett., 2011, 40, 233–235 CrossRef CAS.
- (a) R. Sang, H.-B. Yang and M. Shi, Tetrahedron Lett., 2013, 54, 3591–3594 CrossRef CAS PubMed; (b) S. Ma and L. Lu, J. Org. Chem., 2005, 70, 7629–7633 CrossRef CAS PubMed; (c) T. Q. Tran, V. V. Diev and A. P. Molchanov, Tetrahedron, 2011, 67, 2391–2395 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–130 CrossRef CAS.
- (a) P.-Q. Huang, S.-L. Wang, Y.-P. Ruan and J.-X. Gao, Nat. Prod. Lett., 1998, 11, 101–106 CrossRef CAS; (b) X. Z. Li, K. Y. Lai, K. M. Wu, D. F. Huang and L. Huang, Eur. J. Med. Chem., 2014, 74, 736–741 CrossRef CAS PubMed.
- The X-ray crystal data of 9a have been deposited in CCDC with number 947607.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 947607. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00160e |
| This journal is © the Partner Organisations 2014 |