
Highly efficient C–C cross-coupling for installing thiophene rings into π-conjugated systems†
Juan Song‡*a,
Fuliang Wei‡a,
Wei Suna,
Xiao Caoa,
Chao Liu *b,
Linghai Xie*a and
Wei Huanga
*b,
Linghai Xie*a and
Wei Huanga
aKey Laboratory for Organic Electronics and Information Display and Institute of Advanced Materials, Nanjing University of Posts and Telecommunications, Nanjing, 210023, P. R. China. E-mail: iamjsong@njupt.edu.cn; iamlhxie@njupt.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, P. R. China. E-mail: chao.liu@whu.edu.cn
First published on 15th July 2014
Abstract
Thiophene, as one of the most common structural units in functional organic materials, was efficiently installed into π-conjugated systems via a simple, ligand-free Suzuki coupling. The reaction proceeded with high efficiency and good functional-group compatibility while requiring 0.02 mol% of palladium catalyst.
Thiophene and its derivatives represent an important class of five-membered heterocyclic compounds in both organic synthesis and materials science.1 In the past few years, thiophene-based π-conjugated compounds have become a highly interdisciplinary field of research. Thousands of papers related to the field of materials science have been published. Various functionalized thiophenes have been extensively used for OPVs, DSSCs, OLEDs, OFETs, etc.2–4 The reason why these compounds are attractive is their characteristic properties such as stable oxidation states, unique electronic, optical, and redox properties, excellent charge transport properties and so on,2a,5 which make them one of the most crucial assets for application in materials science.
Selected examples of molecular material containing thiophene–arene frameworks are listed in Scheme 1. P3HT (I), a commercially available material, was the most commonly used donor for OPVs.6 For FTn (II), it has been reported that the color of the emissions can be controlled by varying the conjugation length of the oligothiophene core.7 C251 (III) was described as one of the most efficient materials for DSSCs which presented a power conversion efficiency of 9.3% at the 100 mW cm−2 and simulated AM 1.5 conditions.8 Besides, the phenyl-capped oligothiophenes a-PnT (IV) have also been widely investigated.9 All these materials shared a common characteristic: they were π-conjugated systems containing thiophene units. Thus, developing highly efficient C–C bonds for installing thiophene rings into π-conjugated systems of molecular materials is appealing and important.
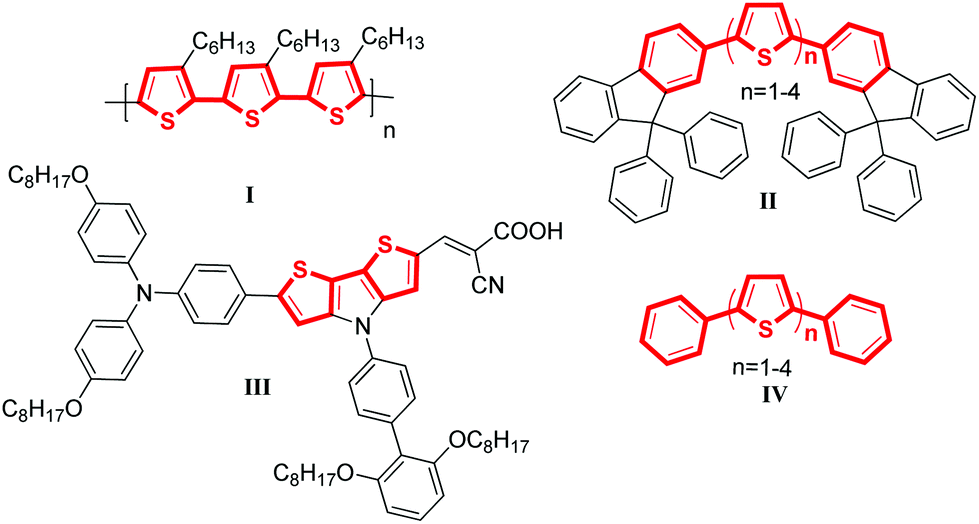 | ||
| Scheme 1 Selected examples of molecular material containing thiophene–arene frameworks. | ||
The most exploited reaction for the preparation of thiophene-based π-conjugated compounds is the coupling of halogenated thienyls with metalated counterparts.10 Among these coupling reactions, the palladium-catalyzed Suzuki reaction is more preferable owing to its generality and ability to tolerate a wide range of important organic functional groups. It offers an abundance of possibilities to transfer aryl groups into π-conjugated systems. Using Suzuki couplings, many thiophene-based materials have been synthesized.11 Herein, we disclose an effective synthetic approach for the preparation of thiophene-based π-conjugated molecules via a simple, ligand-free Suzuki coupling with a low catalyst loading (0.02 mol%).
Our initial investigation was focused on the condition screening of the reaction between 2-bromothiophene and phenylboronic acid. Detailed information is listed in Table 1. The initial base screening revealed that K3PO4·3H2O, Na2CO3 and KOAc were less effective than K2CO3 in the presence of 2 mol% catalyst in 2 mL DMF at 80 °C for 12 h (Table 1, entries 1–4). Then we tried to decrease the catalyst loading, and surprisingly found that lower catalyst loading also exhibited excellent reactivity in this transformation, among which 0.02 mol% Pd(OAc)2 gave the best result (Table 1, entries 5–8).
|
|
|||
|---|---|---|---|
| Entry | Pd (mol%) | Base | Yieldb (%) |
| a Unless otherwise noted, the reaction was carried out with 1 (0.5 mmol), 2 (0.6 mmol), base (1.0 mmol), solvent (2 mL), 120 °C, 12 h. b The yield was determined by GC analysis with naphthalene as the internal standard. c 80 °C. d 1 mL DMF. e Isolated yield. f No catalyst was added. | |||
| 1c | 2 | K3PO4·3H2O | 68 |
| 2c | 2 | Na2CO3 | 74 |
| 3c | 2 | KOAc | 60 |
| 4c | 2 | K2CO3 | 87 |
| 5 | 0.2 | K2CO3 | 92 |
| 6d | 0.02 | K2CO3 | 99(92 ) |
| 7 | 0.01 | K2CO3 | 89 |
| 8 | 0.005 | K2CO3 | 88 |
| 9f | — | K2CO3 | N.D. |
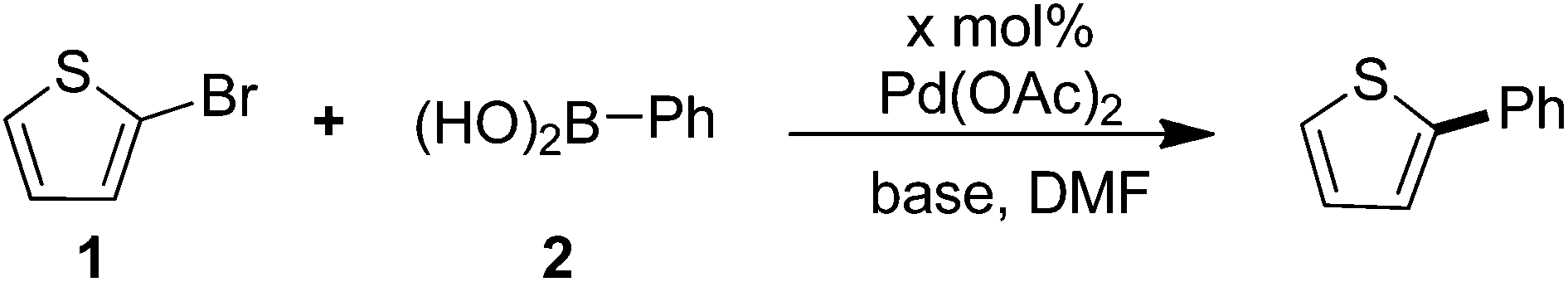
Notably, the catalyst loading could be lowered to 0.005 mol%, along with a good yield of the desired product (Table 1, entry 8). However, no product was detected in the absence of Pd(OAc)2 (Table 1, entry 9). Herein, the combination of reaction conditions including 0.02 mol% of Pd(OAc)2, 1.0 equiv. of 2-bromothiophene, 1.2 equiv. of arylboronic acid, and 2.0 equiv. of K2CO3 in DMF at 120 °C for 12 h was determined to be optimal conditions.
Subsequently, 2-bromothiophene was applied to introduce the thiophene ring into π-conjugated systems with various aryl boronic acids. The results in Scheme 2 demonstrated that this reaction had a high degree of functional group tolerance. A variety of arylboronic acids proceeded efficiently to afford the desired products in excellent yields. Arylboronic acids bearing electron-withdrawing groups including –CHO, –CN, –COOMe were well tolerated and gave excellent yields (3b, 3c, 3h). Electron-donating groups such as methoxyl led to a good yield of the coupling product 3a. Coupling of naphthalene boronic acids also occurred in excellent yields of 95% and 92%, respectively (3e, 3f). Steric hindrance on arylboronic acid or bromothiophene was reasonably tolerated in the reaction. For example, ortho-substituted phenylboronic acid coupled with 2-bromothiophene in a good yield (3k). 3-Methyl-2-bromothiothene also reacted smoothly with 4-methyl-phenylboronic acid under the standard conditions in a high yield (86%, 3i). Notably, fluorene and carbazole borate, which were often selected as building blocks in organic optoelectronic materials,12 showed excellent reactivity and gave the corresponding products in high yields (3d, 3g, 3j).
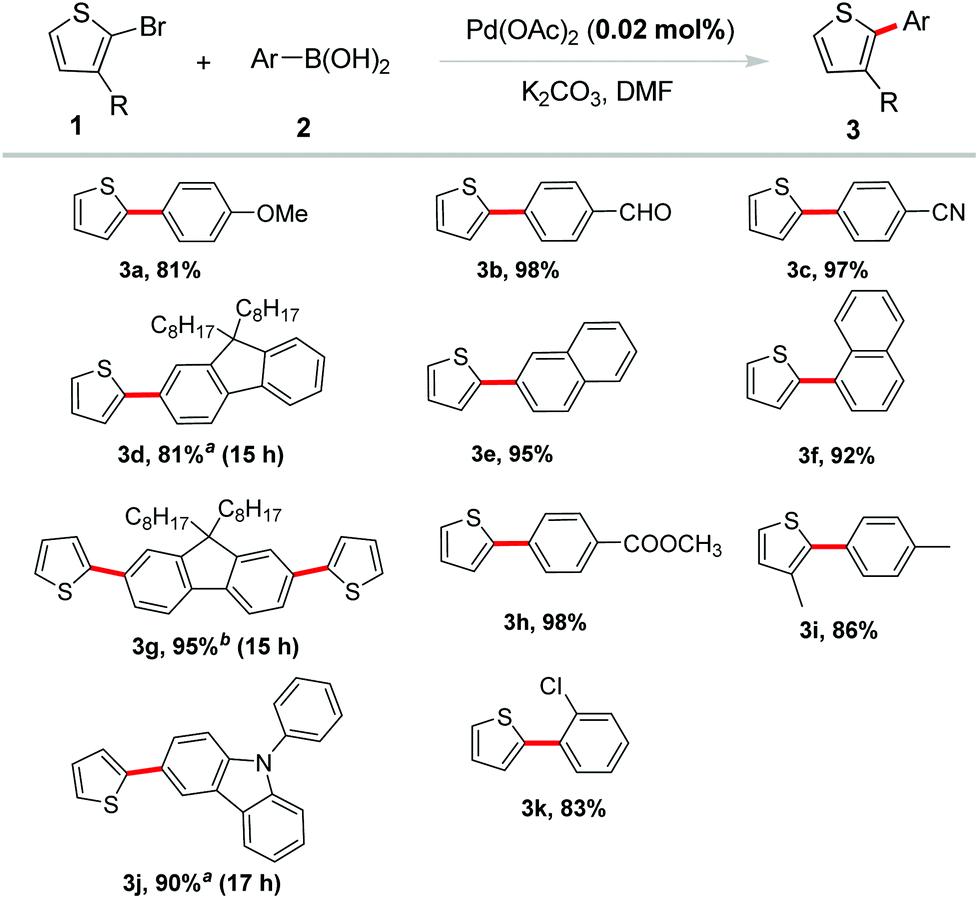 | ||
| Scheme 2 Suzuki coupling of 2-bromothiophenes. Reaction conditions: 0.50 mmol of 1 (1.0 equiv.), 2 (1.2 equiv.), Pd(OAc)2 (0.02 mol%), K2CO3 (2.0 equiv.), DMF (1.0 mL), 12 h, 120 °C. a0.25 mmol of 1 was used. b1 (0.25 mmol), 2 (0.15 mmol). Isolated yield. | ||
As a kind of active components, thiophene-based oligomers and polymers are pervasive in electronic devices and fluorescent materials. In order to elucidate the potential synthetic utility of our methodology in the field of materials science, we extended our studies on the double arylation of 2,5-dibromothiophene under the optimized conditions (Scheme 3). We were delighted to observe that high yields were generally obtained. Phenylboronic acid showed good reactivity and gave the desired product 5a in 90% yield. Reaction of cyano-substituted phenyl boronic acid occurred in an excellent yield of 96% (5b). Naphthalen-2-yl boronic acid and (4-methyl naphthalen-1-yl) boronic acid also coupled with 2,5-dibromothiophene smoothly and gave the corresponding products 5c and 5d in 94% and 82% yields, respectively.
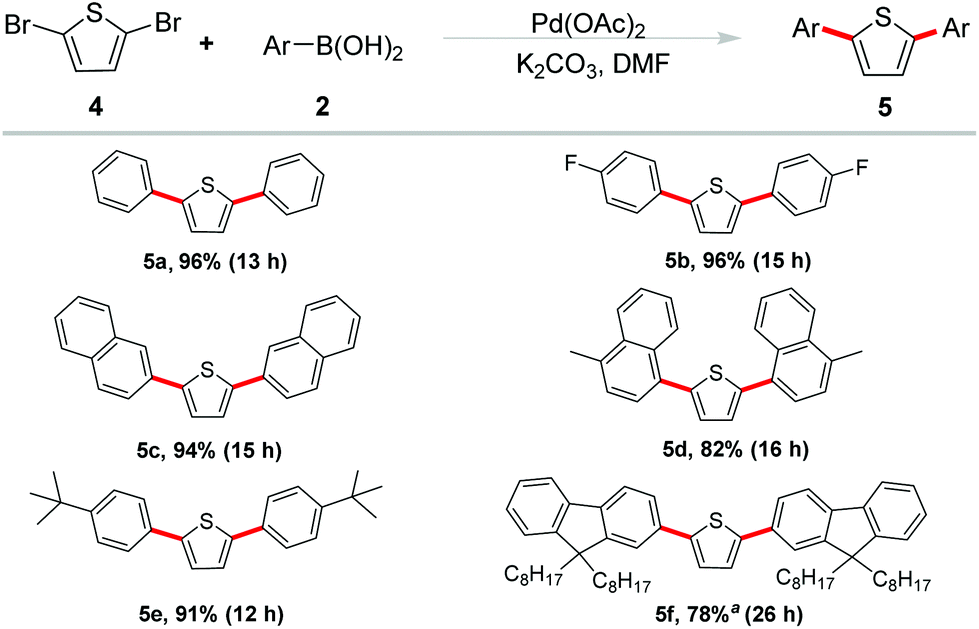 | ||
| Scheme 3 Suzuki coupling of 2,5-dibromothiophene. Reaction conditions: 0.25 mmol of 4 (1.0 equiv.), 2 (2.4 equiv.), Pd(OAc)2 (0.04 mol%), K2CO3 (4.0 equiv.), DMF (1.0 mL), 120 °C, 12 h. a0.125 mmol of 4 was used. Isolated yield. | ||
The tert-butyl functional group has been reported exerting a potent effect on photoelectric properties. It helps to enhance the solubility of materials, increase of steric hindrance to avoid congregating of molecules and so on.13 Therefore, 4-tBu-phenyl boronic acid was selected to couple with 2,5-dibromothiophene and the corresponding coupling product 5e was obtained in 91% yield. Furthermore, another important unit in functional organic materials of 9,9-bioctyl-fluorene was also introduced into the thiophene-based π-conjugated system. The corresponding borate of 9,9-bioctyl-fluorene was employed to react with 2,5-dibromothiophene and gave the product 5f in 78% yield.
These promising results paved the way for the sequential investigations of employing 5,5′-dibromo-2,2′-bithiophene as the substrate. Bithiophene is another active unit in organic semiconductor materials.14 However, in most of the bithiophene-based coupling reactions, without some special ligands or poisonous tin reagents to help facilitate the reaction, it is difficult to obtain satisfactory results.15 Therefore, it is still a challenge to efficiently construct phenyl-capped oligo-thiophenes by Suzuki coupling. Under established optimized reaction conditions, a variety of arylboronic acids were examined to couple with 5,5′-dibromo-2,2′-bithiophene for the feasibility of this methodology as shown in Scheme 4. As expected, phenylboronic acid proceeded well and afforded the coupling product 7a in 90% yield. C–F bonds and –CN, –CHO groups were well tolerated to afford the coupling product in excellent yields (7b–7d). 9,9-Bioctyl-2-fluorene borate and 9-phenyl-3-carbazole boronic acid were also found to be reasonable coupling partners in this reaction (7e–f). Obviously, these transformations provided a straightforward access to the synthesis of biaryl-substituted bithiophene derivatives from readily available and inexpensive dibrominated biothiophene. Among these products, 7a was one of the members of IV (n = 2), its terminal phenyl groups evidently produced additional extension of the π-system.16 It has been reported that the crystal structure of 7b displayed a high close packing. Its HOMO/LUMO levels and film orientation suggested the promising use of this compound as an active layer in FET devices.17 The existence of the formyl functional group in 7d offered the opportunity for further utility in DSSCs.18 Moreover, fluoro-substituted aryl groups usually were beneficial for improving fluorescence quantum efficiency in luminescent materials which allow 7c as a candidate in the material field.19
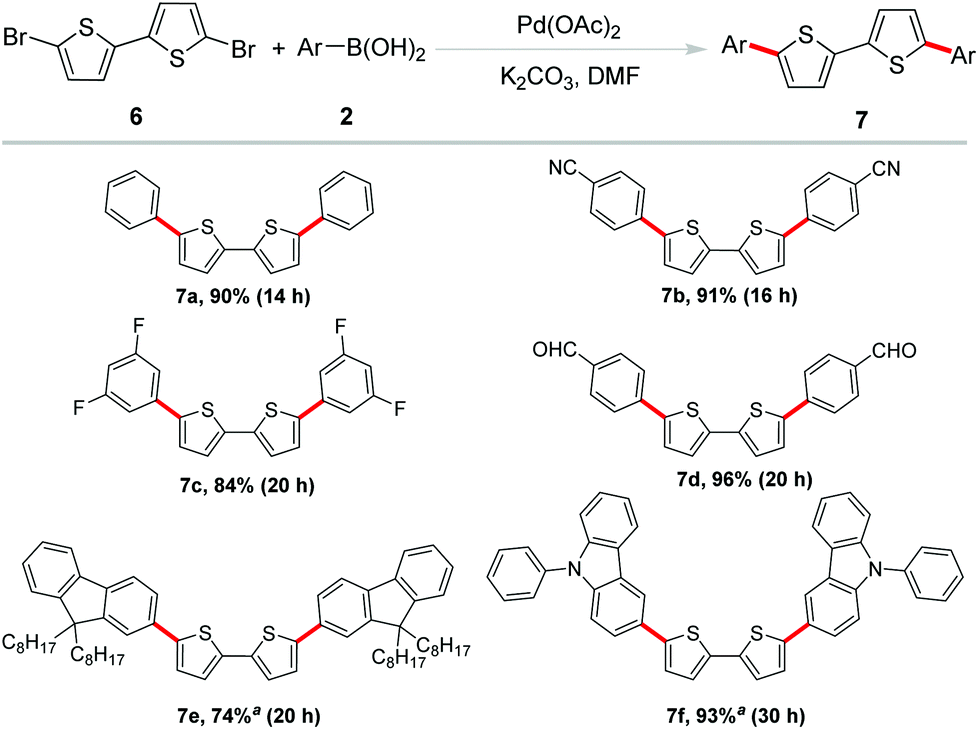 | ||
| Scheme 4 Suzuki coupling of 5,5′-dibromo-2,2′-bithiophene. Reaction conditions: 0.25 mmol of 6 (1.0 equiv.), 2 (2.4 equiv.), Pd(OAc)2 (0.04 mol%), K2CO3 (4.0 equiv.), DMF (1.0 mL), 120 °C, 12 h. a0.125 mmol of 6 was used. Isolated yield. | ||
Conclusions
In summary, we have developed an efficient ligandless Suzuki coupling for installing thiophene and bithiophene units into π-conjugated systems. The reaction proceeded with high yields and good functional group compatibility while requiring only 0.02 mol% of palladium catalyst. The simplicity of the reaction procedure coupled with the possibility of obtaining a wide variety of aryl-thiophenes and oligo-thiophenes. This method will certainly attract the attention of material investigators interested in this class of compounds. Studies about further application of this methodology in organic photoelectric materials are currently in progress.Acknowledgements
The project was supported by the 973 Program (2012CB725302), the National Natural Science Foundation of China (21302148, 61274065, 21373114, 51173081, 61136003).Notes and references
- (a) Chemistry of Heterocyclic Compounds, ed. S. Gronowitz, John Wiley & Sons, New York, 1986 Search PubMed; (b) Handbook of Oligo- and Polythiophene, ed. D. Fichou, Wiley-VCH, Weinheim, Germany, 1999 Search PubMed.
- (a) A. Mishra, C. Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed; (b) Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268–18271 CrossRef CAS PubMed.
- (a) J. Gao, L. Li, Q. Meng, R. Li, H. Jiang, H. Li and W. Hu, J. Mater. Chem., 2007, 17, 1421–1426 RSC; (b) Y. Ie, Y. Umemoto, M. Okabe, T. Kusunoki, K.-i. Nakayama, Y.-J. Pu, J. Kido, H. Tada and Y. Aso, Org. Lett., 2008, 10, 833–836 CrossRef CAS PubMed.
- (a) J. Roncali, P. Leriche and A. Cravino, Adv. Mater., 2007, 19, 2045–2060 CrossRef CAS; (b) A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
- P. Frère, J. M. Raimundo, P. Blanchard, J. Delaunay, P. Richomme, J.-L. Sauvajol, J. Orduna, J. Garin and J. Roncali, J. Org. Chem., 2003, 68, 7254–7265 CrossRef PubMed.
- (a) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed; (b) L. M. Chen, Z. Hong, G. Li and Y. Yang, Adv. Mater., 2009, 21, 1434–1449 CrossRef CAS; (c) M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS; (d) A. Iwan and A. Chuchmała, Prog. Polym. Sci., 2012, 37, 1805–1828 CrossRef CAS PubMed.
- K. T. Wong, C. F. Wang, C. H. Chou, Y. O. Su, G. H. Lee and S. M. Peng, Org. Lett., 2002, 4, 4439–4442 CrossRef CAS PubMed.
- N. Cai, J. Zhang, M. Xu, M. Zhang and P. Wang, Adv. Funct. Mater., 2013, 23, 3539–3547 CrossRef CAS.
- (a) C. Moreno Castro, M. C. Ruiz Delgado, V. Hernández, S. Hotta, J. Casado and J. T. López Navarrete, J. Chem. Phys., 2002, 116, 10419–10427 CrossRef CAS PubMed; (b) J. J. Apperloo, L. B. Groenendaal, H. Verheyen, M. Jayakannan, R. A. J. Janssen, A. Dkhissi, D. Beljonne, R. Lazzaroni and J. L. Brédas, Chem. – Eur. J., 2002, 8, 2384–2396 CrossRef CAS; (c) M. A. Ismail, D. W. Boykin and C. E. Stephens, Tetrahedron Lett., 2006, 47, 795–797 CrossRef CAS PubMed.
- (a) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (b) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed; (c) C. Y. Kuo, Y. C. Huang, C. Y. Hsiow, Y.-W. Yang, C. I. Huang, S. P. Rwei, H. L. Wang and L. Wang, Macromolecules, 2013, 46, 5985–5997 CrossRef CAS.
- (a) Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro. Lett., 2011, 1, 67–70 CrossRef; (b) D. T. Tung, D. T. Tuan, N. Rasool, A. Villinger, H. Reinke, C. Fischer and P. Langer, Adv. Synth. Catal., 2009, 351, 1595–1609 CrossRef CAS; (c) S.-M. T. Toguem, A. Villinger and P. Langer, Synlett, 2010, 909–912 CAS; (d) S. Varello and S. T. Handy, Synthesis, 2009, 138–142 CAS; (e) E. Bey, S. Marchais-Oberwinkler, M. Negri, P. Kruchten, A. Oster, T. Klein, A. Spadaro, R. Werth, M. Frotscher, B. Birk and R. W. Hartmann, J. Med. Chem., 2009, 52, 6724–6743 CrossRef CAS PubMed; (f) A. Oster, S. Hinsberger, R. Werth, S. Marchais-Oberwinkler, M. Frotscher and R. W. Hartmann, J. Med. Chem., 2010, 53, 8176–8186 CrossRef CAS PubMed; (g) Y. Gu and X. Wu, Lett. Org. Chem., 2012, 9, 396–400 CrossRef; (h) S. Shi and Y. Zhang, Green Chem., 2008, 10, 868–872 RSC.
- (a) L. H. Xie, F. Liu, C. Tang, X. Y. Hou, Y. R. Hua, Q. L. Fan and W. Huang, Org. Lett., 2006, 8, 2787–2790 CrossRef CAS PubMed; (b) S. i. Kato, S. Shimizu, H. Taguchi, A. Kobayashi, S. Tobita and Y. Nakamura, J. Org. Chem., 2012, 77, 3222–3232 CrossRef CAS PubMed.
- (a) S. Curran, S. Roth, A. P. Davey, A. Drury and W. Blau, Synth. Met., 1996, 83, 239–243 CrossRef CAS; (b) M. Holzinger, O. Vostrowsky, A. Hirsch, F. Hennrich, M. Kappes, R. Weiss and F. Jellen, Angew. Chem., Int. Ed., 2001, 40, 4002–4005 CrossRef CAS; (c) F. Wang, Y.-H. Lai and M.-Y. Han, Macromolecules, 2004, 37, 3222–3230 CrossRef CAS.
- (a) H. E. Katz, Z. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359–369 CrossRef CAS PubMed; (b) H. Usta, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 9034–9035 CrossRef CAS PubMed; (c) A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- V. P. Reddy, R. Qiu, T. Iwasaki and N. Kambe, Org. Lett., 2013, 15, 1290–1293 CrossRef CAS PubMed.
- S. A. Lee, Y. Yoshida, M. Fukuyama and S. Hotta, Synth. Met., 1999, 106, 39–43 CrossRef CAS.
- W. Porzio, S. Destri, M. Pasini, A. Rapallo, U. Giovanella, B. Vercelli and M. Campione, Cryst. Growth Des., 2006, 6, 1497–1503 CAS.
- (a) H.-Y. Yang, Y.-S. Yen, Y.-C. Hsu, H.-H. Chou and J. T. Lin, Org. Lett., 2009, 12, 16–19 CrossRef PubMed; (b) W. Wu, J. Yang, J. Hua, J. Tang, L. Zhang, Y. Long and H. Tian, J. Mater. Chem., 2010, 20, 1772–1779 RSC.
- M. H. Yoon, A. Facchetti, C. E. Stern and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 5792–5801 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00167b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2014 |