
Sonogashira cross-coupling under non-basic conditions. Flow chemistry as a new paradigm in reaction control†
Svatava
Voltrova
a and
Jiri
Srogl
*ab
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo nam. 2, 166 10 Prague, Czech Republic. E-mail: jsrogl@uochb.cas.cz
bDepartment of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, 911 Partners Way, P. O. Box 7905, 27695, USA
First published on 26th August 2014
Abstract
The flow regime was implemented as a non-chemical alternative for the reaction control and detrimental acid byproduct sequestration in transition metal catalyzed cross-coupling reactions. The concept was illustrated on the Sonogashira reaction and resulted in a non-basic version of the cross-coupling protocols.
Introduction
Ever since the seminal work of Guldberg and Waage, synthetic chemists have used many ways to manipulate chemical equilibrium and thus improve reaction outcomes. Amongst various tactics for driving reactions to completion, the most common and practical approach has been the continual removal of reaction products.1 In order to attain such a situation, physical (distillation or precipitation of reaction products) and chemical (product sequestration) methods have traditionally been used, leading over the years to their irreplaceable role in organic synthesis. While the conventional control of the reaction equilibrium has been extremely efficient, it is not the only way to affect the balance and tip it in the right direction. Continuous flow processes have recently become a new way to execute traditional chemical reactions.2–5 Securing their position amongst the valuable synthetic tools for their intrinsically excellent mass and energy transport parameters, the continual flow systems could be, in addition, explored as an avenue for reaching a non-equilibrium state with a consequent positive synthetic impact.6Some of the foremost examples of modern synthetic methods are the chemistries that help connect two, often highly functionalized entities, while forming a new bond between two atoms of the respective subunits in the process. In the most common scenario, one of the cross-coupling reaction partners possesses formally electrophilic (organic halides, etc.) properties while the other provides electrons (reactive organometallics, olefins, acetylenes) in the transition metal catalyzed process.7 A prime example of such chemistry is the Sonogashira protocol, which couples organic halides with terminal acetylenes.
Based on the mechanism, a formal acid byproduct is formed, which obviates the obligatory addition of a stoichiometric base (sequestering thus refractory acid). The way the acid affects the reaction equilibrium is depicted at the bottom of Scheme 1.
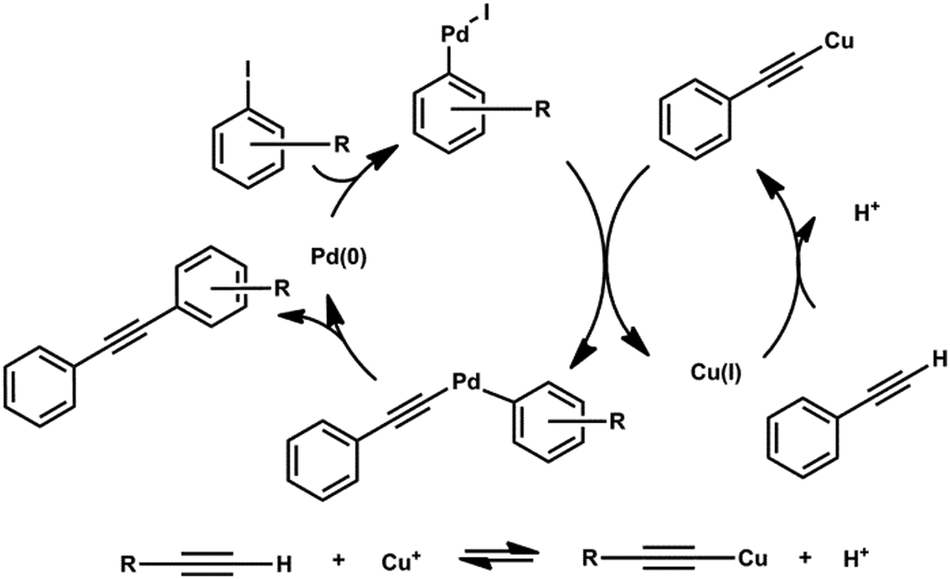 | ||
| Scheme 1 Mechanism of Sonogashira cross-coupling reaction. | ||
Results and discussion
A series of experiments was carried out to underscore the key importance of the acid in the reaction mechanism: when the base was omitted from the reaction mixture, no product was detected. When aryl iodide 1a was treated in the separate Pd catalyzed reaction with Cu phenylacetylide 2a, likely the reaction intermediate,8 the reaction proceeded smoothly to completion regardless of the presence of a base. In order to support the concept and demonstrate the detrimental role of acid in the system, the reaction setting was supplemented with HI. The HI aliquots were injected into the reaction mixture and the cross-coupling product formation was monitored by GC and HPLC. For the effects of the acid addition see Fig. 1.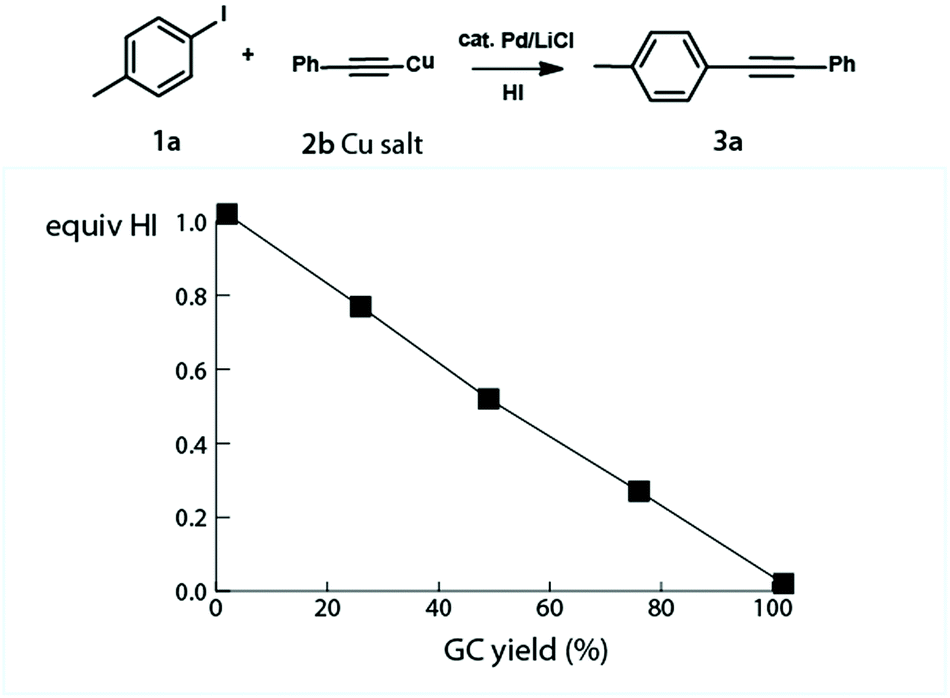 | ||
| Fig. 1 HI, the formal cross-coupling by-product, was added to the reaction mixture to assess its detrimental role in the reaction system. Conversion to 3a is correlated with the amount of HI added. | ||
The experimental results have clearly identified the importance of the Cu acetylide formation and have pointed to the central role which the acid plays in the cross-coupling scenario.
Encouraged by recent developments in commercially accessible continuous flow systems, we wanted to demonstrate the viability of the non-chemical equilibrium manipulation approach by applying flow conditions to non-basic acid sequestration.9 In analogy with the batch experiments, aryl iodides 1, known to undergo an efficient oxidative addition to palladium(0), were used as reaction substrates together with terminal acetylenes. In contrast with the common setting, the dissolved substrates, aryl iodides 1 and terminal alkynes 2, were, in the present study, driven through a flow apparatus instead.
The central component of the reactor – pair of heated short columns – contained both Cu and commercially available Pd catalysts on a solid support (Table 1).10
|
|
||||
|---|---|---|---|---|
| Entry | R | R′ | Yield GC (%) | Yield isol. (%) |
| 3a | 4-CH3 | Phenyl | 74 | 60 |
| 3b | H | Phenyl | 76 | 74 |
| 3c | 4-CH3 | 2-Hydroxypropane-2-yl | 64 | 58 |
| 3d | 2-CH3-4-NO2 | Phenyl | 80 | Dec. |
| 3e | H | Cyclopropyl | 94 | 72 |
| 3f | 4-CH3 | Pyridine-3-yl | 80 | 73 |
| 3g | H | 2-Bromophenyl | 63 | 50 |
| 3h | 4-CH3 | 4-Acetylphenyl | 83 | 61 |
| 3i | H | 4-Fluorophenyl | 91 | 73 |
| 3j | 4-CH3 | 4-Fluorophenyl | 73 | 62 |
| 3k | 4-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
Pyridine-3-yl | 56 | 45 |
| 3l | 4-CH3 | 2-Bromophenyl | 74 | 58 |
| 3m | H | Pyridine-3-yl | 79 | 65 |
| 3n | 4-CH3 | Cyclopropyl | 98 | 85 |
| 3o | H | 2-Hydroxypropane-2-yl | 80 | 63 |
| 3p | 4-CHO |
Phenyl | 92 | 75 |
General procedure for Sonogashira coupling in flow regime
Substituted iodobenzene 1 (0.5 mmol) and aryl acetylene 2 (0.6 mmol) were dissolved in dried THF–DMA 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : 1 (10 mL) and passed through the cartridges packed with Escat™ 1241 (5% Pd on alumina powder):(0.1% Cu2O on alumina powder) = 17:1 at 80 °C in the same solvent composition.
: 1 (10 mL) and passed through the cartridges packed with Escat™ 1241 (5% Pd on alumina powder):(0.1% Cu2O on alumina powder) = 17:1 at 80 °C in the same solvent composition.
A smooth product formation was conveniently followed by UPLC and GC chromatography. The anticipated dramatic decrease of the reaction mixture pH, monitored in real time, complemented the mass balance. No products were observed and the starting material remained unchanged when either of the catalytic components was omitted from the setup.
In an attempt to glean some extra insight into the process, the solid supported catalysts, Cu and Pd, were separated into two different cartridges. Both cartridges were subsequently placed in the flow instrument in a serial fashion (Fig. 2).
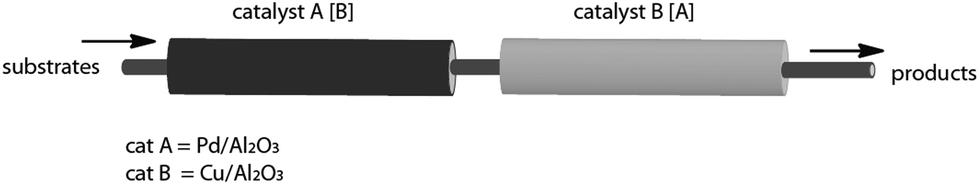 | ||
| Fig. 2 Effect of the catalyst placement. In the first of the two possible situations the cartridge A containing the Pd catalyst was preceding the cartridge B containing Cu. Taking into account the mechanistic scenario of the Sonogashira protocol it was not surprising that there was no product detected.11 In the complementary situation – the cartridge B preceding the cartridge A – no product was observed either. While this observation is somehow unexpected it can be rationalized by a limited motility of Cu acetylide likely formed in the cartridge B.12 | ||
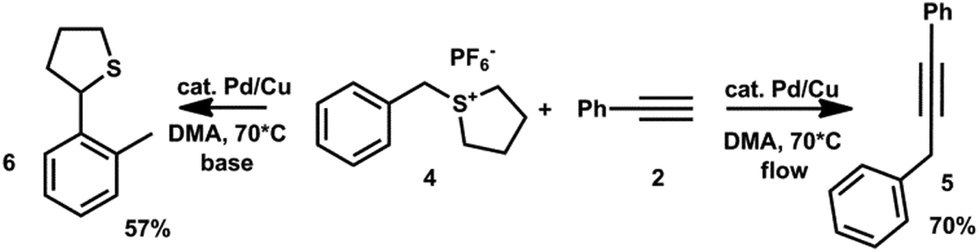
While the primary goal of the study is not a synthetic exploitation of non-basic conditions in a cross-coupling reaction, the practical potential can be illustrated by the Sonogashira cross-coupling of intrinsically base sensitive benzylsulfonium salt 4, which was treated under flow regime conditions.13
Again, the reaction proceeded smoothly leading to the desired coupling product 5 in very good yield. The concurrent control batch experiment only yielded the rearrangement product 6.14
Numerous control experiments were carried out in traditional batch settings with no base present under otherwise identical conditions (catalyst, solvent, temperature). As expected, the batch experiments produced no trace of the desired products, supporting thus the notion of the fundamental differences between flow and batch systems on one side and the importance of acid sequestration on the other.15
Experimental section
General procedure for Sonogashira coupling in flow regime
Continuous-flow reactions were performed on reactor X-Cube™ (ThalesNano Inc., Hungary; two CatCarts™ of 64 mm size, 4 mm i.d. in series connection, residence volume of the system was 6 mL). Substituted iodobenzene 1 (0.5 mmol) and aryl acetylene 2 (0.6 mmol) were dissolved in dried THF–DMA 9:1 (10 mL) and passed through the cartridges packed with a commercially available Pd catalyst on a solid support10 and 0.1% Cu2O (Sigma-Aldrich) on alumina powder (Sigma-Aldrich) in the weight ratio of 17:1 (total amount of the catalyst was 1.9 g, average TON 120–150) at 80 °C in the same solvent composition with the flow rate 0.1–0.2 mL min−1.
4-(Phenylethynyl)toluene (3a). Synthesized from 4-iodotoluene (1a, 0.109 g, 0.5 mmol) and phenylacetylene (2a, 0.062 g, 0.6 mmol), yield 0.0576 g (74%), m.p. 72.5–74.5 °C (ref. 16 72.5–74 °C). 1H NMR spectrum (CDCl3): δ 7.55–7.50 (m, 2H), 7.43 (d, J = 8.1, 2H), 7.37–7.30 (m, 3H), 7.18–7.12 (m, 2H), 2.36 (s, 3H).
1,2-Diphenylethyne (3b). Synthesized from iodobenzene (1b, 0.102 g, 0.5 mmol) and 2a (0.062 g, 0.6 mmol), yield 0.0659 g (60%), m.p. 58.5–60.5 °C (ref. 17 58–60 °C). 1H NMR spectrum (CDCl3): δ 7.55–7.50 (m, 4H), 7.37–7.30 (m, 6H).
2-Methyl-4-p-tolylbut-3-yn-2-ol (3c). Synthesized from 1a (0.109 g, 0.5 mmol) and 2-methylbut-3-yn-2-ol (2b, 0.050 g, 0.6 mmol), yield 0.0505 g (58%), m.p. 48.9–49.1 °C (ref. 18 50–52 °C). 1H NMR spectrum (cf. ref. 19) (CDCl3): δ 7.30 (d, J = 8.1, 2H), 7.11 (d, J = 7.8, 2H), 2.34 (s, 3H), 1.95 (br s, 1H), 1.61 (s, 6H). 13C NMR spectrum ((CD3)2)CO): δ 139.8 (C), 133.1 (2 × CH), 130.9 (2 × CH), 122.3 (C), 96.4 (C), 82.6 (C), 66.1 (C), 33.1 (2 × CH3), 22.3 (CH3).
2-Methyl-4-nitro-1-(phenylethynyl)benzene (3d). Synthesized from 1-iodo-2-methyl-4-nitrobenzene (1c, 0.132 g, 0.5 mmol) and 2a (0.062 g, 0.6 mmol), LRMS (EI): m/z 237 (M+). The product could not be obtained pure after column chromatography.
(Cyclopropylethynyl)benzene (3e). Synthesized from 1b (0.102 g, 0.5 mmol) and ethynylcyclopropane (2c, 0.040 g, 0.6 mmol), yield 0.0512 g (72%), oil. 1H NMR spectrum (cf. ref. 20) ((CD3)2)CO): δ 7.35 (m, 2H), 7.30 (m, 3H), 1.48 (m, 1H), 0.89 (m, 2H), 0.73 (m, 2H). 13C NMR spectrum ((CD3)2)CO): δ 133.2 (2 × CH), 130.1 (2 × CH), 129.4 (CH), 125.9 (C), 95.2 (C), 77.3 (C), 9.8 (2 × CH2), 1.5 (CH).
3-(p-Tolylethynyl)pyridine (3f). Synthesized from 1a (0.109 g, 0.5 mmol) and 3-ethynylpyridine (2d, 0.062 g, 0.6 mmol), yield 0.0705 g (73%), colorless oil. 1H NMR spectrum (cf. ref. 21) (CDCl3): δ 8.75 (d, 1H, J = 1.5 Hz), 8.53 (dd, 1H, J = 5.0, 1.5 Hz), 7.79 (dt, 1H, J = 1.75, 8.0 Hz), 7.44 (d, J = 8.1, 2H), 7.29–7.26 (m, 1H), 7.17 (d, J = 8.1, 2H).
1-Bromo-2-(phenylethynyl)benzene (3g). Synthesized from 1b (0.102 g, 0.5 mmol) and 1-bromo-2-ethynylbenzene (2e, 0.108 g, 0.6 mmol), catalyst: (5% Pd on alumina powder)
:(0.1% Cu2O on alumina powder) = 50:1; yield 0.0642 g (50%), isolated as colorless oil. 1H NMR spectrum (cf. ref. 22) ((CD3)2)CO): δ 7.72 (m, 1H), 7.64 (m, 1H), 7.59 (m, 2H), 7.44 (m, 4H), 7.34 (m, 1H). 13C NMR spectrum ((CD3)2)CO): δ 135.2 (CH), 134.4 (CH), 133.3 (2 × CH), 131.9 (2 × CH), 130.8 (CH), 130.5 (CH), 129.5 (CH), 126.9 (C), 126.8 (C), 124.6 (C), 95.6 (C), 89.6 (C).
1-(4-(p-Tolylethynyl)phenyl)ethanone (3h). Synthesized from 1a (0.109 g, 0.5 mmol) and 1-(4-ethynylphenyl)ethanone (2f, 0.087 g, 0.6 mmol), yield 0.0714 g (61%), m.p. 122.7–123.0 °C (ref. 23 126–127 °C). 1H NMR spectrum (cf. ref. 23) ((CD3)2)CO): δ 8.09 (d, J = 8.8, 2H), 7.67 (d, J = 8.6, 2H), 7.47 (d, J = 8.1, 2H), 7.27 (d, J = 7.8, 2H), 2.61 (s, 3H), 2.37 (s, 3H).
1-Fluoro-4-(phenylethynyl)benzene (3i). Synthesized from 1b (0.102 g, 0.5 mmol) and 1-ethynyl- 4-fluorobenzene (2g, 0.072 g, 0.6 mmol), yield 0.0716 g (73%), m.p. 107.8–109.7 °C (ref. 24 108–111 °C). 1H NMR spectrum (cf. ref. 24) (CDCl3): δ 7.53 (m, 4H), 7.35 (m, 3H), 7.05 (m, 2H). 13C NMR spectrum ((CD3)2)CO): δ 165.7 (C), 135.5 (2 × CH), 133.3 (2 × CH), 130.4 (2 × CH), 125.5 (C), 121.4 (C), 117.7 (2 × CH), 117.5 (CH), 90.7 (C), 89.9 (C).
1-Fluoro-4-(p-tolylethynyl)benzene (3j). Synthesized from 1a (0.109 g, 0.5 mmol) and 2g (0.072 g, 0.6 mmol), yield 0.0651 g (62%), m.p. 97.6–98.2 °C (ref. 25 91–92 °C). 1H NMR spectrum (cf. ref. 25) ((CD3)2)CO): δ 7.58 (m, 2H), 7.42 (m, 2H), 7.19 (m, 4H), 2.90 (s, 3H). 13C NMR spectrum ((CD3)2)CO): δ 165.6 (C), 140.6 (C), 135.4 (2 × CH), 133.2 (2 × CH), 131.1 (2 × CH), 121.8 (C), 121.6 (C), 117.6 (2 × CH), 90.9 (C), 89.2 (C), 22.4 (CH3).
4-(Pyridin-3-ylethynyl)benzaldehyde (3k). Synthesized from 4-iodobenzaldehyde (1d, 0.116 g, 0.5 mmol) and 2d (0.062 g, 0.6 mmol), yield 0.0466 g (45%), m.p. 96.5–98.0 °C (ref. 26 98.5–99.3 °C). 1H NMR spectrum (cf. ref. 27) (CDCl3): δ 10.09 (s, 1H), 8.83 (br s, 1H), 8.66 (br s, 1H), 7.99 (d, J = 8.1, 2H), 7.95 (d, J = 6.1, 1H), 7.80 (d, J = 8.1, 2H), 7.49 (br m, 1H).
1-Bromo-2-(p-tolylethynyl)benzene (3l). Synthesized from 1a (0.109 g, 0.5 mmol) and 2e (0.108 g, 0.6 mmol), catalyst: (5% Pd on alumina powder)
:(0.1% Cu2O on alumina powder) = 50:1; yield 0.0786 g (58%), isolated as colorless oil. 1H NMR spectrum (cf. ref. 28) ((CD3)2)CO): δ 7.70 (d, J = 8.1, 1H), 7.62 (d, J = 7.6, 1H), 7.48 (d, J = 8.1, 2H), 7.41 (m, 1H), 7.31 (m, 1H), 7.25 (d, J = 7.8, 2H), 2.36 (s, 3H). 13C NMR spectrum ((CD3)2)CO): δ 141.1 (C), 135.1 (CH), 134.4 (CH), 133.3 (2 × CH), 131.7 (2 × CH), 131.2 (CH), 129.4 (CH), 127.2 (C), 126.8 (C), 121.6 (C), 95.9 (C), 89.0 (C), 22.5 (CH3).
3-(Phenylethynyl)pyridine (3m). Synthesized from 1b (0.102 g, 0.5 mmol) and 2d (0.062 g, 0.6 mmol), yield 0.0582 g (65%), m.p. 50.5–52 °C (ref. 29 50–51 °C). 1H NMR spectrum (CDCl3): δ 8.77 (d, 1H, J = 1.5 Hz), 8.55 (dd, 1H, J = 5.0, 1.5 Hz), 7.81 (dt, 1H, J = 1.75, 8.0 Hz), 7.56–7.53 (m, 2H), 7.38–7.36 (m, 3H), 7.30–7.27 (m, 1H).
4-(Cyclopropylethynyl)toluene (3n). Synthesized from 1a (0.109 g, 0.5 mmol) and 2c (0.040 g, 0.6 mmol), yield 0.0663 g (85%), oil. 1H NMR spectrum (cf. ref. 20) ((CD3)2)CO): δ 7.23 (d, J = 8.1, 2H), 7.11 (d, J = 7.8, 2H), 2.30 (s, 3H), 1.46 (m, 1H), 0.87 (m, 2H), 0.70 (m, 2H). 13C NMR spectrum ((CD3)2)CO): δ 139.2 (C), 133.2 (2 × CH), 130.8 (2 × CH), 122.9 (C), 94.3 (C), 77.4 (C), 22.3 (CH3), 9.8 (2 × CH2), 1.6 (CH).
2-Methyl-4-phenylbut-3-yn-2-ol (3o). Synthesized from 1b (0.102 g, 0.5 mmol) and 2b (0.050 g, 0.6 mmol), yield 0.0504 g (63%), m.p. 53.5–53.8 °C (ref. 30 52–54 °C). 1H NMR spectrum (cf. ref. 32) ((CD3)2)CO): δ 7.41 (m, 2H), 2.03 (br s, 1H), 1.62 (s, 6H). 13C NMR spectrum ((CD3)2)CO): δ 133.2 (2 × CH), 130.2 (2 × CH), 129.9 (CH), 125.3 (C), 97.1 (C), 82.6 (C), 66.1 (C), 33.0 (2 × CH3).
4-(Phenylethynyl)benzaldehyde (3p). Synthesized from 1d (0.123 g, 0.5 mmol) and 2a (0.062 g, 0.6 mmol), yield 0.0773 g (75%), m.p. 95.5–98 °C (ref. 31 96.5–98 °C). 1H NMR spectrum (CDCl3): δ 10.02 (s, 1H), 7.87 (ddd, J = 0.4, 1.6, 8.1, 2H), 7.68 (ddd, J = 0.4, 1.6, 8.1, 2H), 7.58–7.54 (m, 2H), 7.36–7.40 (m, 3H).
:(0.1% Cu2O on alumina powder) = 17:1; 0.040 g was added and the reaction mixture heated to 80 °C with stirring under an Ar atmosphere. After 16 h, the UPLC analysis showed complete conversion of the starting sulfonium salt. The reaction mixture was poured into 5% HCl (30 mL) and shaken with ether (5 × 30 mL). The collected ether portion was washed with brine and dried by MgSO4. After evaporation, the residue was purified by column chromatography on silica gel (hexane–ether–acetone (30:1:2). The obtained colorless oil (0.040 g) was identified as 2-o-tolyltetrahydrothiophene (6, Scheme 2), which is the usual product of the Sommelet–Hauser rearrangement of the starting sulfonium salt in the presence of bases.13 Yield 57%, m/z (EI) 178, 1H NMR spectrum (CDCl3) (cf. ref. 13): δ 7.61 (d, J = 7.0, 1H), 7.12–7.24 (m, 3H), 4.76 (dd, J = 8.0, 5.9, 1H), 3.12–3.18 (m, 1H), 3.00–3.08 (m, 1H), 2.40 (s, 3H), 2.25–2.39 (m, 2H), 1.93–2.09 (m, 2H).
 | ||
| Scheme 2 Cross-coupling of the base sensitive substrate. | ||
:(0.1% Cu2O on alumina powder) = 17:1 at 80 °C at a flow of 0.3 mL min−1. To the solution was then added water (30 mL) and the mixture shaken with hexane (3 × 30 mL). The collected hexane portion was washed with brine and dried using MgSO4. After evaporation, the residue was purified by column chromatography on silica gel (hexane–ether–acetone 30:1:2). The obtained colorless oil (0.052 g) was identified as the Sonogashira product, 1,3-diphenylpropyne (5). Yield 70%, m/z (EI) 192, 1H NMR spectrum (CDCl3) (cf. ref. 33): δ 7.39–7.44 (m, 4H), 7.18–7.36 (m, 6H), 3.81 (s, 2H).
Sonogashira protocol. (a) Representative example: With catalytic amounts (4.4%) of Pd: 4-iodotoluene (1a, 0.109 g, 0.5 mmol) and phenylacetylene (2a, 0.062 g, 0.6 mmol) were dissolved in degassed dried THF–DMA 9
:1 (10 mL). The catalyst (5% Pd on alumina powder):(0.1% Cu2O on alumina powder) = 17:1; 0.050 g was added and the reaction mixture heated to 75 °C while stirring under an Ar atmosphere. After 72 h, less than 2% of the Sonogashira product 3a was formed in the reaction mixture (detection: UPLC-MS, GC-MS). When under the same conditions K2CO3 (0.276 g, 2 mmol) was added, the reaction proceeded smoothly with a GC yield of 86%.
(b) With more than a stoichiometric amount of Pd (equivalent amount of catalyst present in the cartridge): 4-iodotoluene (1a, 0.109 g, 0.5 mmol) and phenylacetylene (2a, 0.062 g, 0.6 mmol) were dissolved in THF–DMA 9:1 (10 mL). The catalyst (5% Pd on alumina powder):(0.1% Cu2O on alumina powder) = 17:1; 1.90 g was added and the reaction mixture heated to 80 °C while stirring under an Ar atmosphere. After 72 h, less than 2% of the Sonogashira product 3a was formed in the reaction mixture (detection: UPLC-MS, GC-MS). The same conditions were used for reactions of iodobenzene (1b, 0.102 g, 0.5 mmol) and phenylacetylene (2a, 0.062 g, 0.6 mmol), with the results giving less than 2–3% of the Sonogashira product 3b (GC-MS).
Conclusions
Overall, the present study describes the role of a flow regime in the development of traditional cross-coupling reactions under non-basic conditions. Even though a certain synthetic advantage of the protocol was illustrated by the transformation of the sensitive substrate, the main value of the present study should not be seen solely in the synthetic exploitation of the reaction conditions for the construction of base sensitive molecules. Rather, its major forte may be found in exposing a less traditional facet of a flow regime as an alternative strategy for reaction mixture composition control.Acknowledgements
This work was supported by the Ministry of Education of the Czech Republic (LH12013).Notes and references
- K. K. Baird, J. Chem. Educ., 1999, 76, 1146 CrossRef.
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384 CrossRef CAS PubMed.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849 RSC.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519 CrossRef CAS PubMed.
- Similar concept in the esterification protocol has been reported in: T. Razzaq, T. N. Glasnov and C. O. Kappe, Eur. J. Org. Chem., 2009, 1321 CrossRef CAS PubMed.
- J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed.
- The Cu phenylacetylide was prepared independently. A. Henke and J. Srogl, Chem. Commun., 2010, 46, 6819 RSC.
- Metal catalyzed cross-coupling reactions present an excellent model for such a study; native to their reaction mechanism are several reversible steps which could be affected by the equilibrium shift. Interesting accounts of cross-coupling reactions in a flow regimen have been published recently underscoring a great potential of flow chemistry. Those accounts however have not dealt with the reaction equilibrium modulation as a tool for acid sequestration. Thus, in the above reaction protocols the requisite base has always been present. For Sonogashira protocol under flow regime, see: (a) Y. Zhang, T. F. Jamison, S. Patel and N. Mainolfi, Org. Lett., 2011, 280 CrossRef PubMed; (b) R. Javaid, H. Kawanami, M. Chatterjee, T. Izhizaka, A. Suzuki and T. M. Suzuki, Chem. Eng. J., 2011, 167, 431 CrossRef CAS PubMed.
- Escat™ 1241 (5% Pd on alumina powder), particle size 50–70 μm, surface area 110 m2 g−1.
- Due to the reversible interaction (oxidative addition) of the substrate and solid supported catalyst and the absence of Cu acetylide the reaction could not proceed in the cartridge A. In the cartridge B the Pd catalyst was absent and, consequently, aryliodide could not react even though Cu acetylide was presumably formed.
- Polymeric nature of Cu acetylide is the origin of acetylide limited solubility. In the stoichiometric experiment with preformed Cu acetylide 2a catalytic LiCl has to be added for solubilization and consequent effective cross-coupling.
- V. K. Aggarwal, H. W. Smith, G. R. Hynd, V. H. Jones, R. Fieldhouse and S. E. Spey, J. Chem. Soc., Perkin Trans. 1, 2000, 3267 RSC.
- For a successful Pd/Cu catalyzed Stille cross-coupling of sulfonium moieties, see: J. Srogl, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1997, 119, 12376 CrossRef CAS.
- The dramatic differences between batch and flow system can be rationalized by relative concentration changes of the par participating molecules at the immobilized metallic reaction centers.
- D. Seyferth and R. Damrauer, J. Org. Chem., 1966, 31, 1965 CrossRef.
- R. S. Glass, ARKIVOC, 2005, 185 CrossRef CAS.
- A. Shchukin, Russ. J. Org. Chem., 2007, 43, 784 CrossRef CAS.
- J. Jiang Cheng, Y. Sun, F. Wang, M. Guo, J.-H. Jian-Hua Xu, Y. Pan and Z. Zhang, J. Org. Chem., 2004, 69, 5428 CrossRef PubMed.
- C.-W. Chia-Wen Li, K. Pati, G.-Y. Lin, S. Md. Abu Sohel, H.-H. Hung and R.-S. Liu, Angew. Chem., Int. Ed., 2010, 49, 9891 CrossRef PubMed.
- K. Park, G. Bae, J. J. MoonChoe, K. H. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS PubMed.
- H.-J. Chen, Z.-Y. Lin, M.-Y. Li, R.-J. Lian, Q.-W. Xue, J.-L. Chung, S.-C. Chen and Y.-J. Chen, Tetrahedron, 2010, 66, 7755 CrossRef CAS PubMed.
- A. R. Gholap, K. Venkatesan, R. Pasricha, T. Daniel, R. J. Lahoti and K. V. Srinivasan, J. Org. Chem., 2005, 70, 4869 CrossRef CAS PubMed.
- M. Bandini, R. Luque, V. Budarina and D. J. Macquarrie, Tetrahedron, 2005, 61, 9860 CrossRef CAS PubMed.
- D. Yang, B. Li, H. Yang, H. Fu and L. Hub, Synlett, 2011, 702 CAS.
- U. S. Sorensen and E. Pombo-Villar, Tetrahedron, 2005, 61, 2697 CrossRef CAS PubMed.
- C. Richardson and C. A. Reed, J. Org. Chem., 2007, 72, 4750 CrossRef CAS PubMed.
- M. Kuhn, F. C. Falk and J. Paradies, Org. Lett., 2011, 13, 4100 CrossRef CAS PubMed.
- Z. Novak, P. Nemes and A. Kotschy, Org. Lett., 2004, 6, 4917 CrossRef CAS PubMed.
- M. Pal, V. K. Subramanian and K. R. Parasuraman Yeleswarapu, Tetrahedron, 2003, 59, 9563 CrossRef CAS PubMed.
- J. Tolosa, C. Kub and U. H. F. Bunz, Angew. Chem., Int. Ed., 2009, 48, 4610 CrossRef CAS PubMed.
- T. Endo and H. Uno, J. Polym. Sci., Polym. Lett. Ed., 1985, 23, 359 CrossRef CAS PubMed.
- G. Rosini and R. Ranza, J. Org. Chem., 1971, 36, 1915 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H and 13C NMR spectra of prepared compounds. See DOI: 10.1039/c4qo00198b |
| This journal is © the Partner Organisations 2014 |