
Room temperature, open-flask C–H arylation of electron-deficient heteroarenes with triazenes: rapid synthesis of heterobiaryls†
Rui
Wang
* and
John R.
Falck
Division of Chemistry, Department of Biochemistry, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, Texas, 75390-9038 USA. E-mail: rwang9@albany.edu
First published on 7th August 2014
Abstract
Aryl triazenes, a source of aryl radicals, were coupled with heteroarenes via C–H functionalization to produce heterobiaryls in moderate to good yields. Couplings proceeded under an open atmosphere at ambient temperature for 3–24 h. Best results were obtained with electron-deficient heteroarenes, while both electron donating and withdrawing substituents in the triazene moieties were tolerated.
Introduction
Heteroarenes are well represented in both natural products and pharmaceuticals. Nitrogen heterobiaryls are especially important. For example, Crestor, Lamictal, Viagra, Glivec, some of the commercially most successful drugs in the last decade,1 and the natural compound Micrococcin P1 contain the heterobiaryl motif (Fig. 1).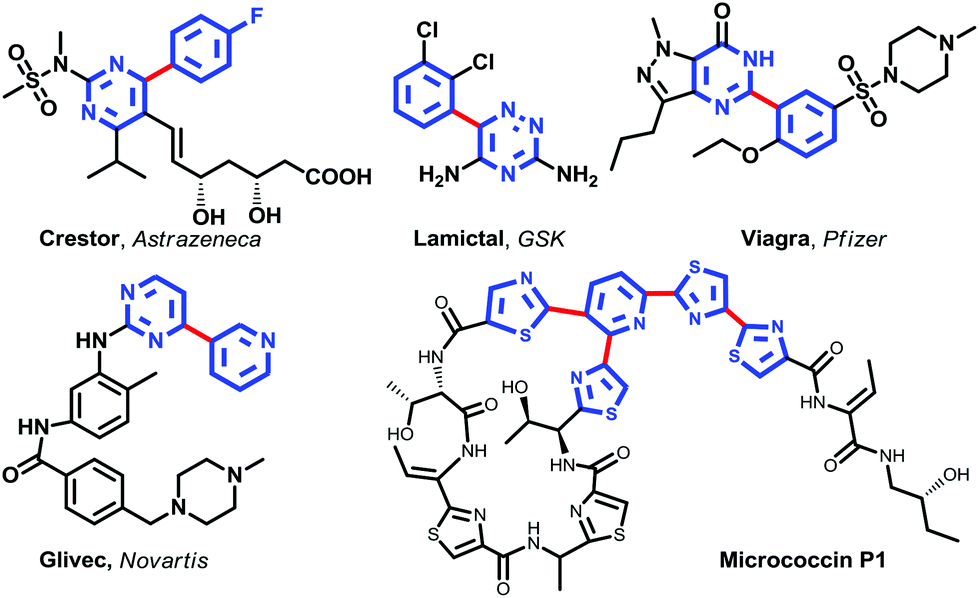 | ||
| Fig. 1 Examples of the heterobiaryl motifs. | ||
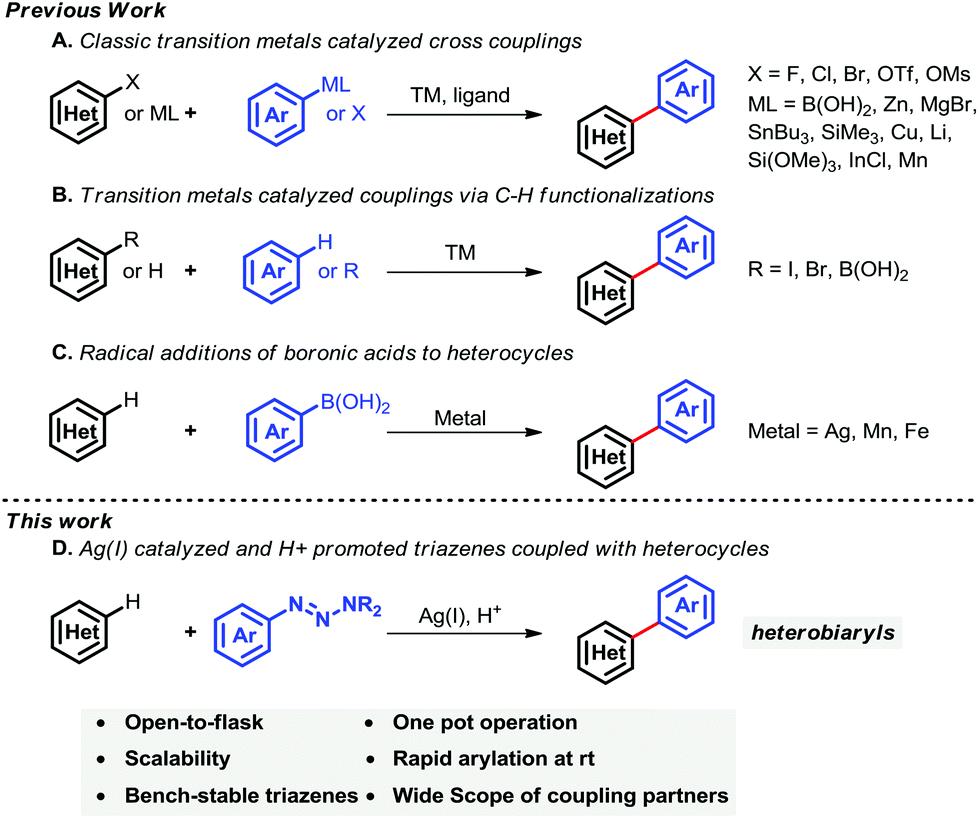
Methodologies2 for synthesizing the core heterobiaryl framework mainly rely upon (i) nucleophilic aromatic substituted reactions, (ii) couplings of pre-activated arenes (i.e., haloarenes) with partners (i.e., phenylboronic acids) (Suzuki–Miyaura cross-coupling reactions),3 (iii) the vibrant C–H activation strategy,4 and (iv) the Minisci reaction (Scheme 1).5
 | ||
| Scheme 1 Major strategies for the construction of heterobiaryls. | ||
Modern arylation methods mainly rely on transition-metal-catalyzed (Pd, Cu, Rh, Ru, Ir, Fe, Zn and Ni) cross-coupling reactions.6 Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reaction has been achieved dramatically over the past few decades. Recent progress in Suzuki–Miyaura cross-couplings which afforded heterobiaryls have been reported (Scheme 1A). For example, Miura,3b Feringa,3c,d Hartwig,3i Buchwald,3k De Meijere et al.3l reported that transition metals (e.g., Pd, Cu, Ni) efficiently catalyzed haloarenes (e.g., aryl iodides, aryl chlorides) with heterocycles. Most recently, transition-metal-catalyzed aromatic C–H bond functionalizations have been of great interest owing to their broad synthetic applications, particularly in the fields of organic synthesis and medicinal chemistry. C–H bond activations and subsequent functionalizations have many advantages compared with classic pre-activated strategies. Direct C–H bond functionalizations7 bypass the use of pre-activated reaction partners, thus leading to a more green process. Pioneering work using C–H functionalization that afforded heterobiaryls was reported by Itami and co-workers7i–n in 2008 (Scheme 1B), the arylation of electron-deficient nitrogen heterocycles with iodoarenes promoted by potassium tert-butoxide was described. Later, they reported that transition-metal-catalyzed (e.g., Pd, Ni, Ir) pre-activated haloarenes (or haloheteroarenes) directly coupled with heteroarenes (or arenes) and a wide range of heterobiaryls were constructed. Unfortunately, most of these cross-coupling reactions suffer from relatively harsh reaction conditions, requirement for specific ligands, and synthesis of pre-activated coupling partners. In contrast, radical reactions have many advantages compared with many transition-metal-mediated processes. For example, many radical reactions can be performed under mild conditions, such as using water as a reaction medium, and are amenable to a wide range of functionalizations. Specifically, synthesis of heterobiaryls by addition of aromatic radicals to heterocycles is a challenging task. This may be primarily due to the difficulties in controlling chemo- and/or regio-selectivities, although the radical method has advanced dramatically compared with many well-established arylation methods. In 1971, the initial work, namely addition of alkyl radicals to protonated heterocycles, was reported by Minisci and co-workers.5 Disadvantages of these processes include harsh reaction conditions and comparably narrow substrate scope (alkyl radicals).8 Recently, Baran et al. reported that arylboronic acids generate aryl radicals, which can be subsequently added to heterocycles at ambient temperature. Consequently, considerable heterobiaryls were synthesized from a wide range of aromatic boronic acids and heterocycles (Scheme 1C).9
Although arylation of heterocycles has been widely achieved in the past few decades, the need for a new synthetic method that is able to address existing shortcomings, e.g. harsh reaction conditions, remains urgent. In recent years, aromatic triazene has gained substantial interest due to its considerable benefits.10 Aromatic triazenes as a kind of activated aryl group are easily and rapidly prepared from bench-available anilines, comparatively stable at ambient temperature, tolerate a broad range of functional groups, and readily scaled-up beyond a hundred gram level. In view of their advantages, aryl triazenes have attracted extensive attention from the chemistry community.10a–e Herein, we disclose the first general protocol to furnish heterobiaryls by mild, open-flask arylations of heteroarenes using triazenes via C–H bond functionalization. Notably, the transformation is amenable to a wide range of functionalities and is readily performed on a multigram scale.
Results and discussion
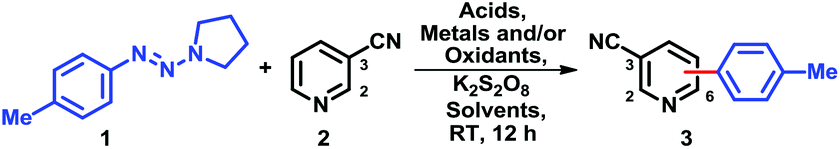
To generate reactive species from triazenes capable of adding to heteroaromatics, we chose a mild oxidizing environment consisting of potassium persulfate (3 equiv.), silver nitrate (20 mol%); as a model system, 4-tolyltriazene (1) and 3-cyanopyridine (2) were chosen.A screen11 of common solvents (Table 1, entries 1–5) revealed that only a two-phase system of CH2Cl2 and water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) afforded a synthetically useful yield of 3 (88%); CHCl3, in sharp contrast, gave 3 in just 45% yield. With the best solvents system in hand, we next sought an inexpensive oxidant (Table 1, entries 6–10). Benzoquinone and DDQ as oxidants gave low conversions. With hypervalent iodine reagents such as iodobenzene diacetate, we observed a moderate conversion. Peroxides TBHP and tBuOOBz also failed to provide any promising results. Actually, a comprehensive oxidant screening was done, all failed.11 Next, a complete list of acids11 was also evaluated, the best result was obtained when trifluoroacetic acid was employed (Table 1, entries 3, 11–13). Among aqueous acids, hydrochloric acid provided low yield (36%) while acetic acid and 65% perchloric acid gave 60% and 70% yields, separately. We coincidentally discovered that an excess of an acid generally improved the yield of 3. Further investigations revealed that excess addition of trifluoroacetic acid (10.5 equiv.) afforded the best result (Table 1, entry 3).11 Polar effect5b,f,12,13 and greater solubility may account for the high transformation efficiency. Interestingly, pure boron trifluoride provided a low yield (Table 1, entry 14).14
:4) afforded a synthetically useful yield of 3 (88%); CHCl3, in sharp contrast, gave 3 in just 45% yield. With the best solvents system in hand, we next sought an inexpensive oxidant (Table 1, entries 6–10). Benzoquinone and DDQ as oxidants gave low conversions. With hypervalent iodine reagents such as iodobenzene diacetate, we observed a moderate conversion. Peroxides TBHP and tBuOOBz also failed to provide any promising results. Actually, a comprehensive oxidant screening was done, all failed.11 Next, a complete list of acids11 was also evaluated, the best result was obtained when trifluoroacetic acid was employed (Table 1, entries 3, 11–13). Among aqueous acids, hydrochloric acid provided low yield (36%) while acetic acid and 65% perchloric acid gave 60% and 70% yields, separately. We coincidentally discovered that an excess of an acid generally improved the yield of 3. Further investigations revealed that excess addition of trifluoroacetic acid (10.5 equiv.) afforded the best result (Table 1, entry 3).11 Polar effect5b,f,12,13 and greater solubility may account for the high transformation efficiency. Interestingly, pure boron trifluoride provided a low yield (Table 1, entry 14).14
|
|
||||
|---|---|---|---|---|
| Entry | Solvent(s) | Oxidant | Acid | Yield (%) |
| a 3-Cyanopyridine 2 (0.2 mmol), 4-tolyltriazene 1 (0.3 mmol), TFA (0.16 mL), CH2Cl2–H2O (0.75 mL/1.25 mL), AgNO3 (20 mol%), K2S2O8 (3.0 equiv.), RT, 12 h. b C2/C6 ratio approx. 1.1/1. | ||||
| 1 | DMF–H2O | AgNO3 | TFA | 20 |
| 2 | THF–H2O | AgNO3 | TFA | 29 |
| 3 | CH2Cl2–H2O | AgNO3 | TFA | 80 |
| 4 | CHCl3–H2O | AgNO3 | TFA | 45 |
| 5 | CH3NO2 | AgNO3 | TFA | Trace |
| 6 | CH2Cl2–H2O | BQ | TFA | 0 |
| 7 | CH2Cl2–H2O | DDQ | TFA | 0 |
| 8 | CH2Cl2–H2O | PhI(OAc)2 | TFA | 0 |
| 9 | CH2Cl2–H2O | TBHP | TFA | 0 |
| 10 | CH2Cl2–H2O | t BuOOBz | TFA | 0 |
| 11 | CH2Cl2–H2O | AgNO3 | 36% HCl | 0 |
| 12 | CH2Cl2–H2O | AgNO3 | AcOH | 60b |
| 13 | CH2Cl2–H2O | AgNO3 | 65% HClO4 | 70b |
| 14 | CH2Cl2–H2O | AgNO3 | BF3·OEt2 | 0 |
With the optimized reaction conditions in hand, we next screened the triazene scopes combined with 4-cyanopyridine (5) (Chart 1). Triazenes with both electron-withdrawing and electron-donating substituents in the para-position provided good to excellent yields. 4-Tolyltriazene provided a high yield of coupled adduct 6a, but there was no regioselectivity (C2/C3-arylation = 1.2/1). Electron-deficient substrates such as 4-fluoro- and 4-chlorophenyltriazenes showed high conversions and isolated yields of 6b and 6c, respectively. This reflected that the increased nucleophilic character of the radical intermediates induced by the electron withdrawing groups on the phenyl rings and consequently were more efficiently coupled with protonated heterocycles.5b,12 Not surprisingly, these reactions were both highly regioselective, favoring couplings at the more electrophilic and sterically accessible ortho positions on the protonated pyridines. Protonated heteroaromatics are electron-poor substrates, which reacted with nucleophilic radicals with high regioselectivities to yield the arylated heterobiaryls. Functional groups such as an ether, carbonyl, and ester were also tolerated, yielding 6d–g in moderate to good yields, respectively. Likewise, triazenes with electron-withdrawing substituents in the 3-position favored better yields, e.g., 6i–l, while a methoxy in this position led to adduct 6m in modest yield; despite having an electron-withdrawing group, the 3-formyl example 6n was inexplicably obtained in low yield accompanied by several minor by-products. The presence of substituents, either electron-rich or deficient, adjacent to the triazenes on the phenyl rings was detrimental (see 6o–q).15 Polysubstituted systems were more complicated. For instance, the combination of 4-fluoro-3-chloro-substituents afforded just 54% yield of 6r, whereas only 35% of 6s was formed. In the latter case, the 2-chloro-substituent clearly overshadowed the otherwise favorable effect of the 4-trifluoromethyl group. On the other hand, 3,4-dimethoxy reinforced the negative influence of electron-donating groups and allowed a mere 13% of 6t. Unfortunately, heteroaryl triazenes failed to provide arylated heterobiaryls, whether at ambient temperature or elevated temperature (60 °C). Their ability to act as good radical acceptors is a likely factor.
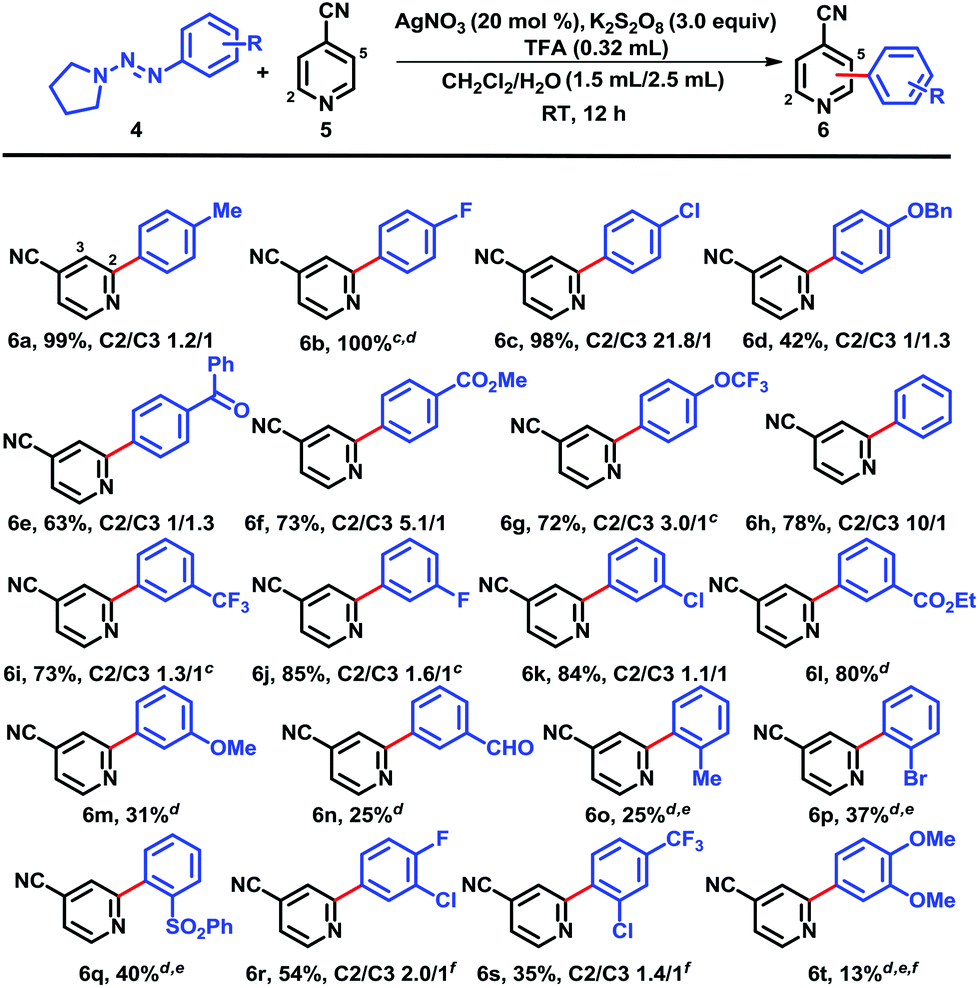 | ||
| Chart 1 Scope of aryltriazene (4) coupling with 4-cyanopyridine (5).a,b aUnless otherwise specified, all reactions were performed on the following scale: 4-cyanopyridine (5) (0.4 mmol), aryltriazene (0.6 mmol), TFA (0.32 mL), CH2Cl2–H2O (3:4, 3.75 mL), AgNO3 (20 mol%), K2S2O8 (3.0 equiv.), RT, 12 h. bAll yields and ratios were isolated results. cReaction time ≤3 h. dOne isomer only. eAddition of a second round of AgNO3 (20 mol%), K2S2O8 (3.0 equiv.) was needed. fReactions were run twice and crude products were purified by SiO2 flash chromatography, and then prep TLC. | ||
Next, we explored the structural varieties in the heterocyclic coupling partners (Chart 2). Electron-withdrawing groups on the para-position of pyridines gave excellent yields, for example, cyano (6a) and trifluoromethyl (8a); while the moderate electron-donating tert-butyl group provided a modest 54% isolated yield of 8b and the strongly donating substituent methoxy failed to give any arylation product 8c that may also be due to the polar effects.5b,f,12,13meta-Substituted pyridine derivatives including cyano (7d), acetyl (7e), carboethoxy (7f), phenyl (7g), and diethylamide (7h) gave adducts 8d–h in moderate yields as expected. These substituents cannot interact directly with the pyridyl nitrogens that may account for their low conversion, and addition of a second round of AgNO3/K2S2O8 in order to completely consume all of the starting pyridines was required. The regioselectivities appeared to favor regions of low electron density. Pyridine itself gave 61% yield of 8i with a regioselectivity ratio 1.6/1. Notably, ortho-substituents (e.g., fluoro, chloro, bromo and methyl) completely suppressed couplings and the unreacted pyridines could be recovered. Other heteroarenes, such as 5-bromopyrimidine (8j), pyrimidine (8k), pyridazine (8l), pyrazine (8m), phthalazine (8n), qunioxaline (8o), quinoline (8p, 8q), and isoquinoline (8r, 8s), were all acceptable coupling partners. In contrast, 1H-benzimidazole led to coupling product 8t in less than 20% yield. Interestingly, 3,6-dichloro-pyridazine gave arylated product 8u in only 5% isolated yield.
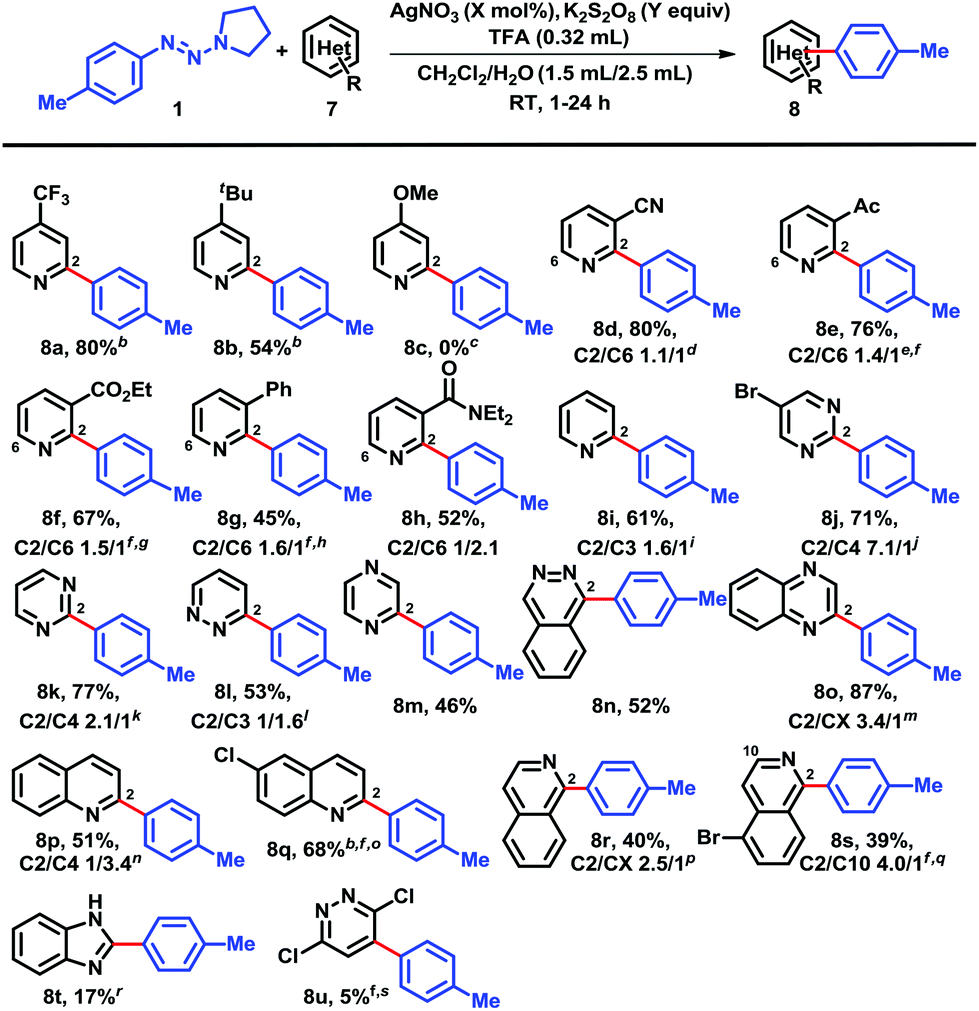 | ||
| Chart 2 Scope of heteroarenes (7) coupling with 4-tolyltriazene (1).a aUnless otherwise specified, all reactions were performed on the following scale: heteroarenes (0.4 mmol), 4-tolyltriazene (0.6 mmol), TFA (0.32 mL), CH2Cl2–H2O (3:4, 3.75 mL), AgNO3 (20 mol%), K2S2O8 (3.0 equiv.), RT, 12 h. All yields were based on isolated material and brsm. bOne isomer only. cStarting material was recovered. dTrace amount of starting material was detected. e25% starting material was recovered after 48 h. fAddition of a second round of AgNO3 (20 mol%) and K2S2O8 (3.0 equiv.) was needed. g40% starting material was recovered after 24 h. h48% starting material was recovered after 24 h. iOnly two isomers were detected. jC2 was isolated alone; other positional isomers were collected as mixtures. kC5 isomer was not detected. l1 mmol scale. mC2 arylation product along with acid-catalyzed hydrolysis product. nTwo isomers were isolated. oOnly one isomer was detected through second round addition of AgNO3 (20 mol%) and K2S2O8 (3.0 equiv.). p30% starting material was recovered along with a C1 arylation product and a mixture of other positional isomers. qC9 arylation product was not found. rIsolated yield after 48 h. s5% isolated product; most of the starting material was recovered. | ||
Encouraged by the results above, we subsequently conducted the practical arylation reaction, it was deemed important to validate the practicality of the methodology on a gram-scale (Scheme 2).16 When substrates 5 and 9 were primary arylated with triazene 4b under our standard reaction conditions, the corresponding heterobiaryls 6b and 10 were obtained in similar chemical yields.
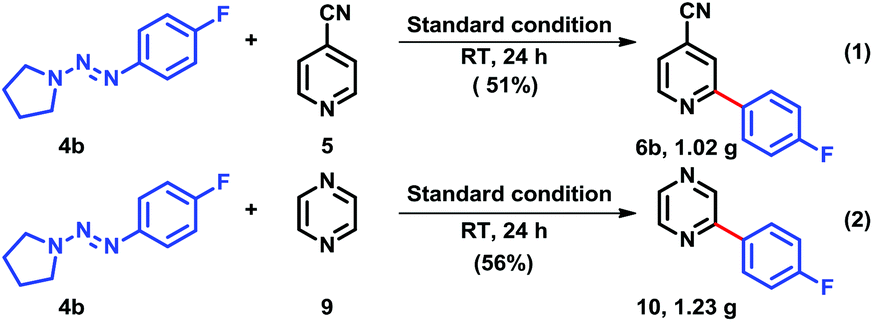 | ||
| Scheme 2 Scale-up of heterocycles coupling with aryltriazene (4b). | ||
To seek its further application in synthetic community, we were delighted to find that 4-tolyltriazene 1 was able to react with benzoquinone 11 with formation of arylated adduct 12 under the standard reaction conditions in 73% isolated yield (Scheme 3). This result indicated that benzoquinone derivatives were also good radical acceptors under the current reaction conditions, which provided an interesting synthetic approach to constructions of arylated benzoquinone derivatives.
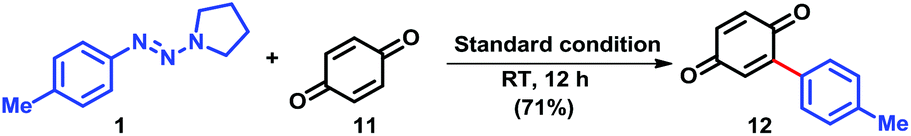 | ||
| Scheme 3 Arylation of benzoquinone derivatives (11). | ||
To demonstrate the usage of this method, we also found that under the optimized reaction conditions, triazenes 4b could be coupled with (−)-Nicotine, a naturally-occurring compound, with a pyrrolidine functional group, providing di-arylated heterocycle 13 in 38% isolated yield (Scheme 4).17
 | ||
| Scheme 4 Synthetic application. | ||
A preliminary experiment to evaluate the plausible reaction pathway is provided in Scheme 5. We were delighted to isolate N-(p-tolyl)acetamide (14) in 42% isolated yield under the reaction conditions,9c which was involved in the radical process.18
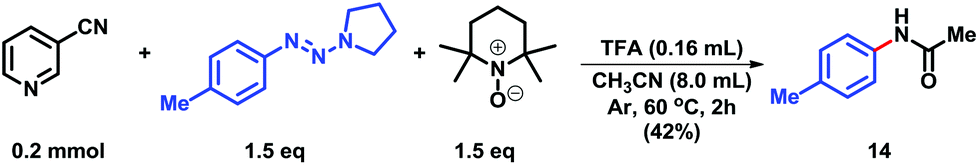 | ||
| Scheme 5 Mechanistic investigation. | ||
Based on our results and relevant investigations by other groups,5,9,11,19 we provided a plausible reaction pathway as shown in Scheme 6.
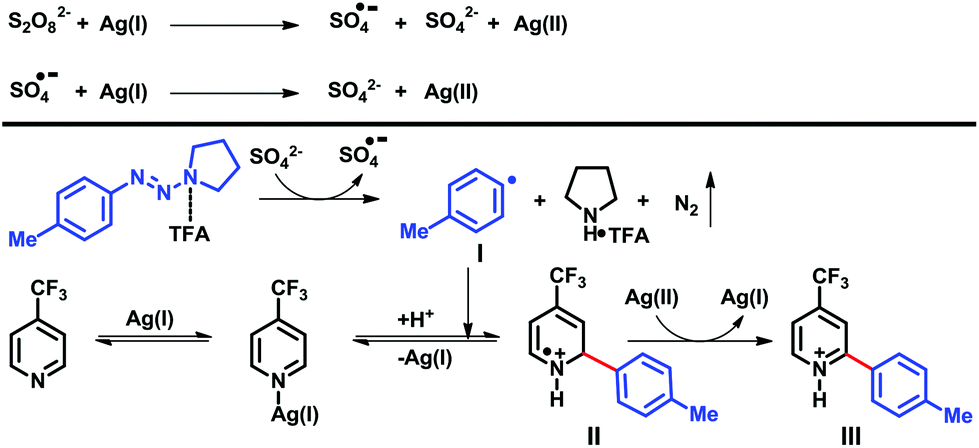 | ||
| Scheme 6 Plausible reaction pathway. | ||
It is known20 that in the presence of Ag(I)-salts, a persulfate anion disproportionates into a sulfate dianion and a sulfate radical anion. The Ag(I) salt is oxidized to a Ag(II) species by peroxydisulfate or a sulfate radical anion.
Consequently, the sulfate dianion reduces the TFA-activated triazene, providing an aryl radical I with release of pyrrolidine salt, nitrogen and a sulfate radical anion. Further addition of this aryl radical I to the protonated heterocycle, provided radical cation II, which is oxidized by Ag(II), affording the arylated product III and regenerating the Ag(I) catalyst.
Conclusions
In conclusion, we reported the first example of a rapid, open-flask, single-pot, and scalable process in which aryl radicals were added to pyridine derivatives and a benzoquinone using readily available triazenes as coupling partners. In this transformation, a wide range of substrates and functional groups were compatible. Electron-withdrawing and donating groups on para, meta and ortho positions of triazenes and pyridine derivatives were tolerated and the reaction proceeded smoothly to produce heterobiaryls in moderate to good yields. Triazenes are readily synthesized from anilines and can be easily amenable to scale up, thus the strategy provides an efficient and practical tool for the production of useful heterobiaryls.Notes and references
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS
and poster by E. Vitaku, E. A. Ilardi and J. T. Njarðarson, Top 200 Pharmaceutical Products by US Retail Sales in 2011, 2011 Search PubMed
- Heterobiaryls synthesis, see: photochemical pathway:
(a) M. Julliard, C. Siv, G. Vernin and J. Metzger, Helv. Chim. Acta, 1978, 61, 2941–2948 CrossRef CAS PubMed
- For Suzuki reaction, see:
(a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed
- C–H strategies for arylation of heterocycles, see typical reviews:
(a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC
- For selected reviews on the Minisci reaction, see:
(a) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489–519 CrossRef CAS
-
L. Ackermann, Modern Arylation Methods, Wiley-VCH, Weinheim, 2009 Search PubMed
- For selected reviews on C–H bond functionalizations, see:
(a) C. Zhu, R. Wang and J. R. Falck, Chem. – Asian J., 2012, 7, 1502–1514 CrossRef CAS PubMed
- For recent examples, see:
(a) F. Minisci, F. Recupero, C. Punta, C. Gambarotti, F. Antonietti, F. Fontana and G. F. Pedulli, Chem. Commun., 2002, 2496–2497 RSC
-
(a) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed
- For recent references, see:
(a) C.-M. Wang, H. Sun, Y. Fan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 7242–7246 CrossRef PubMed
- For a comprehensive conditions screening, see ESI.†.
- See ref. 6, pp. 492–493 and references cited therein.
-
A. Pross, Theoretical and Physical Principles of Organic Reactivity, John Wiley & Sons, Inc., 1996 Search PubMed
- C. Zhu and M. Yamane, Org. Lett., 2012, 14, 4560–4563 CrossRef CAS PubMed
- 2-Iodine, 2-acetyl and other substituents in triazenes were also investigated, all led to low yields.
- A. A. Desai, H. Ren, M. Mukherjee and W. D. Wulf, Org. Process Res. Dev., 2011, 15, 1108–1115 CrossRef CAS
- We tried to synthesize arylated 2-substituted pyridines, i.e., 2-Me pyridine was coupled with triazene under our standard conditions, it failed. However, in Scheme 4, we show di-arylated product 13, which may be generated simultaneously.
-
(a) N. Satyamurthy, J. R. Barrio, D. G. Schmidt, C. Kammerer, G. T. Bida and M. E. Phelps, J. Org. Chem., 1990, 55, 4560–4564 CrossRef CAS
- For mechanistic studies, see:
(a) N. R. Patel and R. A. Flowers II, J. Am. Chem. Soc., 2013, 135, 4672–4675 CrossRef CAS PubMed
-
(a) J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651–1659 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: General procedure, 1H and 13C NMR spectrum, mechanism studies, triazenes synthesis. See DOI: 10.1039/c4qo00213j |
| This journal is © the Partner Organisations 2014 |