 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Progress in La-doped SrTiO3 (LST)-based anode materials for solid oxide fuel cells
Xinwen
Zhou
,
Ning
Yan
,
Karl T.
Chuang
and
Jingli
Luo
*
Department of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 2G6. E-mail: Luoj@ualberta.ca; Fax: +1 780 492 2881; Tel: +1 780 492 2232
First published on 25th October 2013
Abstract
Solid oxide fuel cells (SOFCs) have appeared as a promising technology for a wide variety of potential commercial applications to lessen the urgency of energy shortage and environmental pollution associated with using conventional fossil fuels. Among the worldwide SOFCs research activities, the progress of SOFCs fed with hydrocarbon fuels that contain trace amount of H2S is one of the most important research directions. Thereby, it becomes crucial to design novel electrode materials with enhanced catalytic activity, stability and tolerances to carbon deposition and sulfur poisoning. La-substituted SrTiO3 (LST) based perovskite anodes have been widely investigated because of their high electronic conductivity in reducing atmospheres, excellent dimensional and chemical stability upon redox cycling and outstanding sulfur and coking tolerances. In this review paper, we will describe the development of LST-based anode materials for SOFCs in recent years. The synthesis, structure and fuel cell performance of the doped LST and LST-based composite anode materials are summarized in detail. The mechanism of H2S-induced enhancement effect for electrochemical reactions on the LST-based anode materials is explored. The challenges related to the future developments of LST-based anode materials for SOFCs are also discussed.
1. Introduction
Conventional fossil fuels are the main energy sources at present and for the foreseeable future. However, the energy sources of this type are limited in nature and non-renewable, which can lead to serious energy crises in the future. Also, the environmental problems associated with using conventional fossil fuels are causing public concerns and frequently appear as the headlines in various social media. Solid oxide fuel cells (SOFCs), on the other hand, have been considered as one of the most efficient and environmental-friendly technologies to tackle some of challenges that energy industry is currently facing. The distinguishing advantages of SOFCs include high energy conversion efficiency which is projected to be up to 60%1 and great fuel flexibility. In theory, SOFCs can be fueled by essentially any fuels ranging from gaseous hydrogen, hydrocarbons to solid carbon. A typical SOFC single cell is consisted of three major components: a cathode, an anode and a ceramic electrolyte which is usually a solid oxygen ionic conductor. On the cathode side, O2 is reduced and incorporated into the electrolyte defects, namely oxygen vacancies, resulting in the formation of O2− ions. These O2− ions will be driven by the difference of chemical potential, which is established between the two opposite electrodes, to move through the electrolyte to the anode where they can oxidize the fuels. All the electrons generated during the electrochemical oxidation process will be released to the external circuit for power generation.Presently, one of the major concerns associated with utilizing hydrocarbon fuels in SOFCs is the anode catalyst coking and sulfur poisoning since H2S exists in most raw feedstock. The commonly employed Ni-based anode catalyst can be easily deactivated by carbon deposits and suffers irreversible sulfur poisoning. Although steam reforming and desulfurization enable the suppression of coking and H2S removal, these processes inevitably add the extra cost and complexity into the overall system. Therefore, intensive efforts have been devoted to developing novel anode catalysts.
In recent years, many excellent review papers have been published with focusing on cathodes,2–4 electrolytes,5–8 interconnects,9,10 sealants,11,12 current collector materials,13,14 SOFCs models15,16 and other correlative studies17,18 about SOFCs. Also, a number of excellent review papers are available in the literature summarizing the progress of SOFCs anode catalysts.19–21 Thus, in this review, first, we will only very briefly introduce the commonly used anode materials for SOFCs. Then, we will focus on the recent progress of La-substituted SrTiO3 (LST) based anode materials since the doped SrTiO3 has attracted increasing attentions as a promising anode candidate. The development of a variety of LST-based anode materials will be reviewed in details. Finally, the phenomena and mechanisms of the H2S-induced enhancement of the SOFC performance in SOFCs fed by H2S containing fuels will be summarized via using the LST-based anode. Meanwhile, the LST-based anode fabrication process, the relationship between microstructure and the fuel cell performance, and miscellaneous issues regarding to LST-based materials will also be discussed. A complete and comprehensive coverage of all the research activities on the LST-based anode materials may not be an easy task, therefore, this short review paper is meant to be a summary of most of the correlative research in recent years.
2. A brief overview of common anode materials for SOFCs
As described in the earlier review articles,2,20,21 the general requirements for SOFCs anode materials should include: high electronic conductivity, excellent catalytic activity towards electro-oxidation of fuels, high ionic conductivity, chemical and thermal stability, adequate durability and compatibility with other SOFCs components, easy fabrication, and low cost. The other desirable properties are tolerances to carbon deposition or coking when using hydrocarbon fuels,22 and to sulfur poisoning when using fuels containing H2S.23,24In general, the anode materials in SOFCs can be categorized as metal-based cermet, simple oxides, complex oxides and other alternative anode materials.
Advantages of the metal-based anode materials (e.g. Ni–yttria-stabilized-zirconia (Ni–YSZ)) include most requirements for anode materials mentioned earlier. However, the metal-based anode materials are easily poisoned by the trace impurities (e.g. H2S) in fuels, which could lead to serious fuel cell performance degradation. It has been revealed that Ni/YSZ anode supported SOFCs can only tolerate up to 1 ppm H2S and 10 ppm HCl without significant performance degradation.25 Carbon deposition, redox cycling instability and Ni agglomeration upon prolonged usage are the other major problems observed on Ni/YSZ anode. Recently, Ni and Cu-based cermet anode catalysts have been reviewed.20,21,24,26 Some metal-based anode materials such as Ir/Ce0.9Gd0.1O2−δ,27 Ni–Gd0.1Ce0.9O2−δ28,29 have been reported. Zirconia-based (e.g. Y0.2Ti0.18Zr0.62O1.9030) and ceria-based (e.g. CeO2,31 Ce0.8Gd0.2O1.932) fluorites are the common simple oxide anode materials and have been reviewed particularly.20,21,33
Perovskite-based oxide (general stoichiometry ABO3, where A and B are metal cations) is a typical complex oxide catalyst which is known to have excellent thermal and mechanical stability, physical compatibility with typical electrolyte materials, flexibility, and low production cost, which makes it attractive as electrode material in SOFCs. Recently, different perovskite-based materials including chromite (e.g. La0.75BaxSr0.25−xCr0.5Mn0.5O334), vanadate (e.g. La0.7Sr0.3VO3.8535), titanate (e.g. Sr0.895Y0.07TiO3−δ,36 Nb-doped SrTiO337) etc. have been reported to be used as electrode materials in SOFCs. Double perovskites A2BB′O6 containing alternating BO6/2 and B′O6/2 corner-shared octahedral38 are the first on the list of interest due to their reasonable ionic conductivity, redox stability and stability in presence of sulphurous impurities.39,40 Some new double perovskites such as LnBaCo1.6Ni0.4O5+δ (Ln = Pr, Nd, and Sm).41 Sr2MMoO6 (M = Mg, Mn),42 Sr2−xVMoO6−δ,43 La2ZnMnO644 have been explored as electrode materials in SOFCs. Different perovskite-based anode materials applied for SOFCs have been reviewed.20,21,45–47 In addition, other type of complex oxides such as rutile,48 tungsten bronze,49etc. are also explored as anode catalysts.20,21
For perovskite-based materials ABO3, varying A and/or B can generate a large number of different compounds. Among them, strontium titanate (SrTiO3) whose A and B-sites are occupied by divalent Sr2+ and tetravalent Ti4+ (Fig. 1)50 is an excellent example to explain the origin of the increased electronic conductivity upon doping. O-2p orbitals and the empty conduction band from Ti-3d orbitals make the electronic energy band of SrTiO3 exhibit an n-type semiconducting behavior in reducing atmospheres due to the redox couple Ti4+/Ti3+.51 An A-site or a B-site doped SrTiO3 can improve its total electrical conductivity. Lanthanum is an appropriate donor dopant because its ionic radius is similar to that of Sr2+. Its stability in the trivalent state ensures its incorporation into the perovskite lattice as La3+. Due to the difference in valence between La3+ and Sr2+, introduction of La into the lattice requires that the lattice defect structure be modified to maintain electroneutrality.52 At the same time, the introduction of La to Sr sites causes the formation of oxygen-rich planes, which will improve the ionic conductivity of LST.20 La-substituted SrTiO3 (LST) based anode materials have been widely investigated because of their high electronic conductivity in reducing atmospheres, outstanding dimensional and chemical stability upon redox cycling and excellent sulfur and coking tolerances.
 | ||
| Fig. 1 Idealized structure of cubic perovskite SrTiO3. Reprinted from ref. 50, Copyright (2013), with permission from Royal Society of Chemistry. | ||
3. LST-based anode materials
This section reviews the synthesis, structure, thermal and electrical properties, and fuel cell performance of LST-based candidates including LST, further doped LST and LST composite anode materials.3.1 LaxSr1−xTiO3
It is well known that LST materials sintered in reducing atmospheres cause a change in the titanium valence from Ti4+ to Ti3+, thus enhancing the electronic charge carrier concentration.51 Moos etal.53 investigated the solubility of lanthanum in LaxSr1−xTiO3 in oxygen-rich atmospheres. A pure strontium vacancy compensation mechanism was observed for lanthanum contents up to x = 0.3. Above x = 0.4, additional titanium vacancies form, but their concentration remains negligible compared to the predominating strontium vacancies. At x = 0.5 and 0.6 the lattice structure was found to be slightly distorted.LST-based materials used as potential anode for SOFCs were firstly investigated by Irvine et al.54 in 1997. They found that single phase A-site deficient perovskites LaxSr1−3x/2TiO3−δ could be achieved in air at 0 ≤ x ≤ 0.6, and the materials remained at single phase under both high and low oxygen partial pressures at 930 °C. As La content increased, the electrical conductivity also increased to as high as 7 S cm−1 (PO2 = 10−20 atm) at 930 °C for x = 0.6. This kind of A-site deficient LST-based anode materials was further studied by the group.55–57 New family of perovskite oxides such as La2Srn−2TinO3n+158,59 and La4Srn−4TinO3n+260 were also explored as a potential anodes for fuel cells. Due to the high electrical conductivity, LST-based materials were also used as the interconnect materials61 and support layer62 for resisting carbon deposition.
The thermal, electrical, and electrocatalytical properties of LST powders (LaxSr1−xTiO3, x = 0.1, 0.2, 0.3, 0.35, and 0.4) were studied by Marina et al.52 The samples sintered in air exhibited an electrical conductivity in the order of 1–16 S cm−1. But LaxSr1−xTiO3 sintered in hydrogen at 1650 °C showed 80–360 S cm−1 under the typical SOFCs experimental operation conditions. No significant chemical expansion or contraction of LaxSr1−xTiO3 with x < 0.4 was observed when exposed under a wide range of PO2 because the thermal expansion of LST was close to that of YSZ. LaxSr1−xTiO3 was found to be dimensionally and chemically stable when subjected to oxidation–reduction cycling. The structural and electrical properties of La0.3Sr0.7TiO3−α and other donor-doped SrTiO3 based oxides were also investigated by Hashimoto et al.63,64 They found that the conductivity showed a strong dependence on the PO2. When the PO2 was over 100 Pa, the conductivity drastically dropped with increasing PO2. The La1−xSrxTiO3 (x = 0.5, 0.6, 0.7, 0.8) samples have been tested for possible use as a matrix phase for producing composite anodes for SOFCs.65 It was found that La0.5Sr0.5TiO3 demonstrated some ionic conductivity and could be used as a basic phase for producing composite anodes. The thermal expansion mechanism of LaxSr1−xTiO3,66 sintering characteristics of (LaxSr1−x)1−yTi1−zO367 and LaxSr1−xTiO368 were also studied.
A series of A-site deficient perovskite oxides (La0.3Sr0.7)1−xTiO3−δ (x = 0, 0.03, 0.05, 0.07, 0.10) were investigated by Li et al.69 Standard four terminal DC methods and electron-blocking method were applied to measure the total electrical conductivity and ionic conductivity, respectively. As A-site deficiency level increased, the ionic conductivity increased but the electronic conductivity decreased (Fig. 2). The ionic conductivity of (La0.3Sr0.7)0.93TiO3−δ was as high as 0.2–1.6 × 10−2 S cm−1 at 500–950 °C and 1.0 × 10−2 S cm−1 at 800 °C, more than twice that of La0.3Sr0.7TiO3−δ. Its electrical conductivity was in the range of 83–299 S cm−1 at 50–950 °C and 145 S cm−1 at 800 °C. It was also assumed that A-site deficiency could improve the thermal stability of (La0.3Sr0.7)1−xTiO3−δ. Burnat et al.70 found that the phase stability of A-site deficient La0.2Sr0.7TiO3 could be enhanced significantly by adding some Sr to form La0.2Sr0.706TiO3. The secondary phase such as TiO2 could invoke the loss of electrical performance.
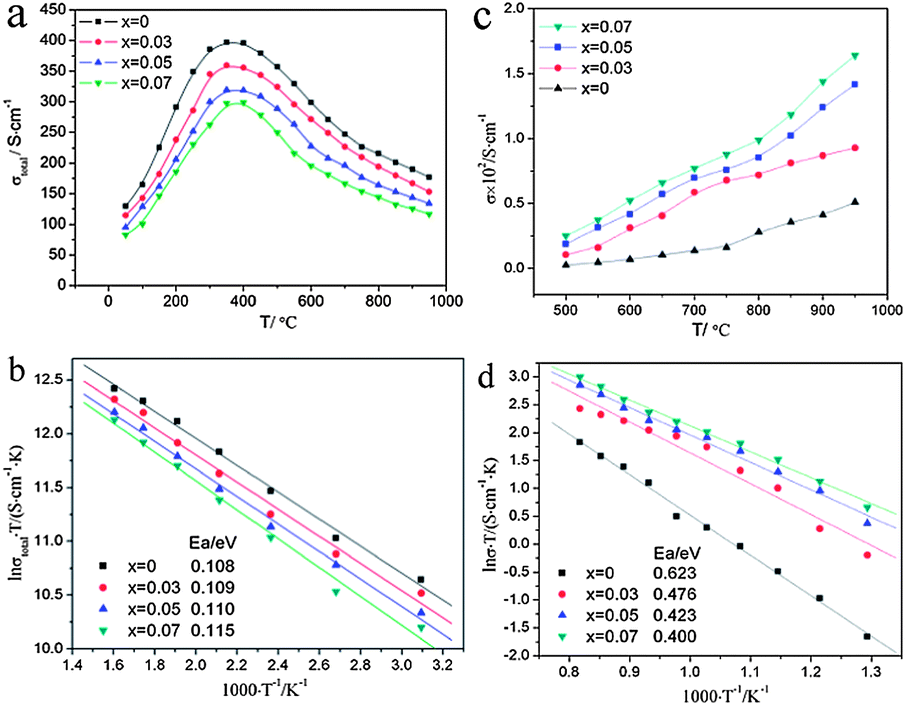 | ||
Fig. 2 Electrical conductivities of (La0.3Sr0.7)1−xTiO3−δ (x = 0, 0.03, 0.05, 0.07, 0.10) as functions of A-site-deficient level and temperature in 50–950 °C (a) and ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σT versus 1000/T in 50–350 °C (b). Ionic conductivities of (La0.3Sr0.7)1−xTiO3−δ (x = 0, 0.03, 0.05, 0.07, 0.10) as functions of A-site-deficient level and temperature (c) and lnσT versus 1000/T (d) in 500–950 °C. Reprinted from ref. 69, Copyright (2010), with permission from International Association for Hydrogen Energy. σT versus 1000/T in 50–350 °C (b). Ionic conductivities of (La0.3Sr0.7)1−xTiO3−δ (x = 0, 0.03, 0.05, 0.07, 0.10) as functions of A-site-deficient level and temperature (c) and lnσT versus 1000/T (d) in 500–950 °C. Reprinted from ref. 69, Copyright (2010), with permission from International Association for Hydrogen Energy. | ||
The performances of fuel cells were compared using LST anode with that using Y doped SrTiO3 (YST).71 It was found that adding the extra electrolyte materials, such as YSZ, into the doped SrTiO3 anode could improve cell performance, suggesting insufficient ionic conductivity of LST. Moreover, the electrical conductivity of YST was lower than that of LST at the same temperature. Recently, A-site deficient Y0.07Sr0.895TiO3 (YST) and La0.2Sr0.7TiO3 (LST) were synthesized through spray pyrolysis method and compared for the electrical conductivity and chemical expansion.72 Under same reducing conditions, YST had better chemical expansion, while LST had better conductivity. To accommodate both conductivity and redox stability, LST was sintered at 1400 °C in air and reduced at around 1000–1200 °C, they then became suitable candidates for full ceramic based SOFCs anodes. Computational prediction was also explored to study the properties of LST-based materials.73
Under the operating conditions of SOFCs, the chemical compatibility between the electrode materials and the electrolyte was a major concern. Burnat et al.74 investigated the chemical interactions between common electrolyte materials and 20 wt% La doped Sr titanates with various A-site occupancy by SEM/EDX microscopy and XRD. It was found that all A-site deficient LSTs promoted a reaction with Sc and YSZ, while stoichiometric LST was more stable.
From the above analysis, we can conclude the following. The maximal solubility of La in SrTiO3 is 0.6. However, the common x in LaxSr1−xTiO3 is 0.3 or 0.4. The structure will be distorted when the x exceeds 0.5. La-doped LST materials with a normal stoichiometry and A-site deficiency are the two main candidates having been examined. Compared to the stoichiometric LST, A-site deficient LST has higher conductivity but lower phase stability and chemical stability.
3.2 Doped LaxSr1−xTiO3
The main disadvantages of LST anode materials are their low ionic conductivity and poor electrocatalytic activity. Presently, the two main methods of increasing the ionic conductivity and improving the fuel cell performance including doping LST with other metal ions and introducing of active second phase to LST to form composite anode.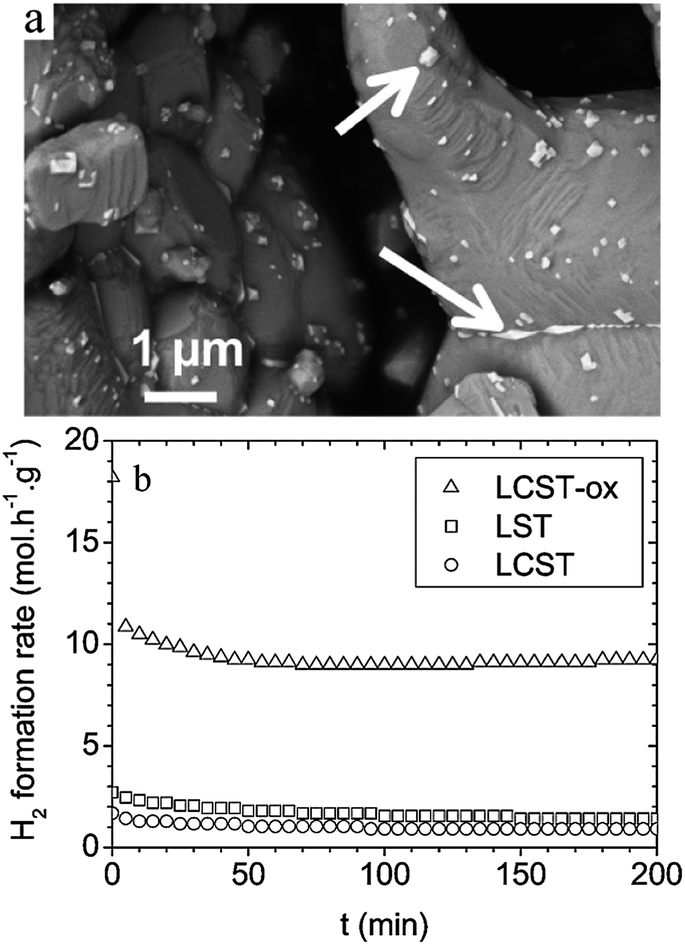 | ||
| Fig. 3 (a) FEG-SEM image of LCST after oxidation at 1200 °C in air. The arrows point at Ce-enriched phases. (b) Hydrogen formation rates vs. time upon reaction of a 10:1:9 CH4–H2O–N2 mixtures over LST, LCST and LCST-ox samples. T = 900 °C. Reprinted from ref. 75, Copyright (2011), with permission from American Chemical Society. | ||
Vincent et al.76 found that Ba partial substitution for Sr in LST (La0.4Sr0.6−xBaxTiO3, 0 < x ≤ 0.2 (LSBT)) could increase its ionic conductivity, catalytic activity to the oxidation of CH4 and improve the stability of the LST structure. Fuel cells were fabricated using commercial YSZ disks (300 μm thick and 25 mm in diameter) as the electrolyte and the composited YSZ/LSM 50/50 wt% as the cathode. It was observed that all fuel cells using LST or LSBT had limited activity for the conversion of hydrogen and methane, and the activity increased with the level of substitution by Ba. Most importantly, the fuel cell performance was significantly enhanced when H2S was present in either CH4 or H2 fuel. The different amount of La0.4Sr0.5Ba0.1TiO3 was impregnated into porous YSZ by Vincent et al.77Fig. 4 compares the performance with the number of times LSBT was impregnated into porous YSZ. The temperature for each test was 850 °C and the sequences of the fuels used were: H2, H2 + 0.5% H2S, CH4 + 0.5% H2S and CH4. The experiments clearly suggested that when the feed was CH4 + 0.5% H2S, the power density was consistently higher than that in pure H2 and pure CH4. Six impregnations provided maximum power density in all the fuels. The maximum power density was 84 mW cm−2 when the feed was CH4 + 0.5% H2S, which was about 3.5 times of that when using pure CH4 as the fuel. Based on the Ba-doped LST materials, Li et al.78 found that pure BaTiO3 anode had higher tolerance to carbon deposition, better electrochemical performance and much higher stability during long term operation compared to SrTiO3 and La2Ti2O7 anodes, especially in H2S-containing atmospheres.
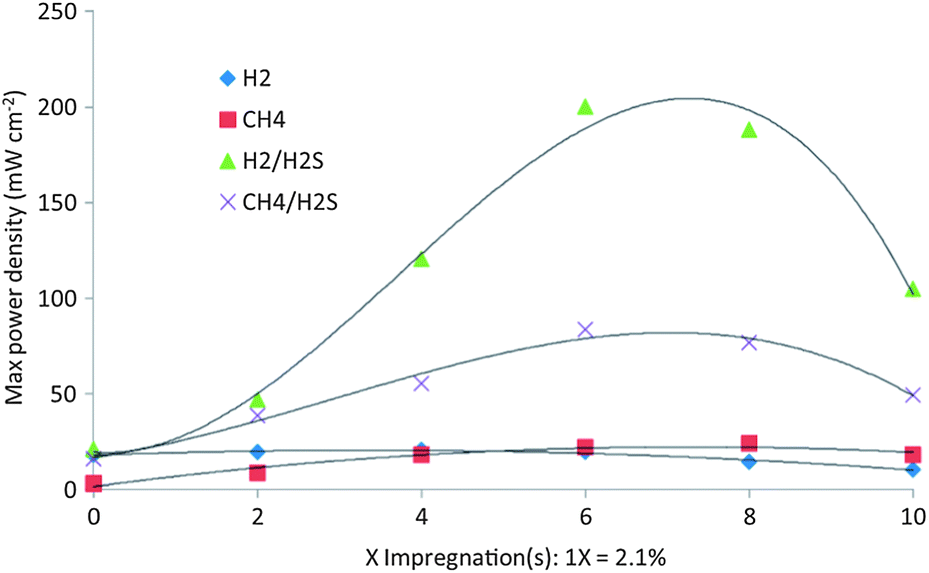 | ||
| Fig. 4 Dependence of compensated maximum power density on the number of times LSBT was impregnated into YSZ obtained at 850 °C. Reprinted from ref. 77, Copyright (2012), with permission from Elsevier. | ||
In addition to the Ba-doping in Sr site, LST with Ca-doping in Sr site was also studied by Verbraeken et al.79 The pure La0.20Sr0.25Ca0.45TiO3 (LSCT) anode initially showed a poor performance. Then the LSCT was impregnated with ceria and nickel oxide to form different anode materials including pure LSCT, LSCT + 10 wt% CeO2, LSCT + 5 wt% Ni, LSCT + 10 wt% CeO2 + 5 wt% Ni, LSCT + 8 wt% CeO2 + 3 wt% Ni. When both nickel and ceria were added as impregnates, a drastic improvement of the anode performance was observed comparing to cases of impregnating with ceria only and pure LSCT. These new anodes also showed excellent stability upon redox cycling, and appeared as a promising alternative to Ni–cermet electrodes. Further studies were done on the structure and conductivity of a series of Ca-doping in A-site deficient perovskite La0.2Sr0.7−xCaxTiO3.80 The results indicated that La0.2Sr0.7−xCaxTiO3 with 0.1 ≤ x ≤ 0.4 were tetragonal and samples with 0.45 ≤ x ≤ 0.7 were orthorhombic at room temperature. A 0.3% increase in lattice volume of the samples was observed after their reduction at 900 °C in 5% H2. The conductivity was definitely improved when compared to La0.2Sr0.7TiO3. By increasing Ca doping content, the conductivity of the reduced samples increased dramatically, reaching the peak of 27.53 S cm−1 in La0.2Sr0.25Ca0.45TiO3. However, the conductivity decreased as x was further increased. Later, a solution method was introduced to synthesize La0.25Sr0.25Ca0.45TiO3 by Yaqub et al.81
The La0.7Sr0.3Cr0.8Ti0.2O3−δ with chromium-rich composition was synthesized using the Pechini method82 and showed a n-type conductivity and high stability under reducing conditions. The perovskite electrodes were electrochemically active towards hydrogen oxidation, although the electronic conductivity (0.1 S cm−1 at 850 °C and 10−20 bar PO2) was not sufficient. A series of perovskite oxides La0.75Sr0.25Cr0.5X0.5O3−δ (X = Co, Fe, Ti, Mn) as SOFCs anode electrocatalysts have been studied by Danilovic et al.83 They concluded that the performance of the perovskite oxides depended on the nature of the substituent element X. Temperature programmed reaction (TPR) of CH4 under O2-free conditions showed that the catalytic activity of the oxides for conversion of CH4 was in the order of Co > Mn > Fe > Ti. Within this series of catalysts, the order of maximum fuel cell power density depended on feed: CH4, X = Fe > Mn > Ti; H2, X = Fe > Mn > Ti; and 0.5% H2S/CH4, X = Ti > Fe > Mn. It was also found that gadolinia doped ceria (GDC) or YSZ combined with La0.75Sr0.25Cr0.5Ti0.5O3−δ could improve the performance. The La0.3Sr0.55Ti1−xCrxO3−δ single-phase perovskite structure can be obtained in air when the dopant content of Cr did not exceed 20 mol%.84 The particle diameter obtained at 800 °C was less than 60 nm and the particles showed an excellent chemical compatibility with YSZ at 1400 °C. At 800 °C, the conductivity of La0.3Sr0.55Ti0.8Cr0.2O3−δ pellet was 1.96 × 10−3 S cm−1 in static air, which needed to be further improved. The Cr-doping decreased the lattice parameter while it increased the sinterability of La0.3Sr0.7Ti1−xCrxO3−δ (LSTC, x = 0, 0.1, 0.2) materials.85 The total electrical conductivity decreased as Ti cations were partially substituted by Cr. However, the electrical conductivity of the LSTC sample with x = 0.2 was still 53 S cm−1 at 800 °C in 5% H2/Ar. The electrical conductivity of Cr-doped samples declined significantly when the testing atmosphere shifted from 5% H2/Ar to air. When the atmosphere was switched back to 5% H2/Ar, the values of the electrical conductivity could mostly be recovered except for the initial cycle. The highest power density obtained with La0.3Sr0.7Ti0.8Cr0.3O3−δ as the anode reached 43.86 mW cm−2 at 900 °C using H2 as the fuel, which was higher than that of undoped La0.3Sr0.7TiO3−δ anode material (∼30 mW cm−2). The better fuel cell performance of LSTC anode materials could be attributed to the better contact between LSTC and YSZ, and the increased electrical conductivity.
The microstructural and electrochemical properties of Mn-doped LST anode material La0.4Sr0.6Ti0.8Mn0.2O3±δ (LSTM) fabricated via liquid-phase impregnation have also been investigated.86 The thermal stability was improved with decreasing Mn content. The electrical conductivity of a 10 wt% CeO2–50 wt% LSTM–YSZ composite anode was higher than that of a 50 wt% LSTM–YSZ anode and was stable under reducing conditions. The maximal power densities of 50 wt% LSTM–YSZ anode were less than 100 mW cm−2 over the entire measured temperature range. The addition of 10 wt% of CeO2 and 1 wt% of Pd as catalysts increased the power density in H2 to 150 and 210 mW cm−2 at 800 and 850 °C, respectively. The performance of A-site-deficient (La1−xSrx)0.95Mn0.5Ti0.5O3−δ and A-site-stoichiometric La1−xSrxMn0.5Ti0.5O3−δ perovskites were studied by Kolotygin et al.87 The total conductivity, thermal and chemical expansion, and steady-state oxygen permeation limited by oxygen surface exchange kinetics increased with increasing x. The presence of H2S traces (5 ppm) in H2-containing gas mixtures did not result in detectable decomposition of the perovskite phases. La0.6Sr0.4Ti1−xMnxO3−δ (LSTM, x = 0.2, 0.4, 0.6, 0.8) were also studied.88 The increase in electrical conductivity as well as oxygen vacancy concentration in a reducing atmosphere resulted in an increase in the higher H2 and CH4 oxidation rate in SOFCs. The single cell with a La0.6Sr0.4Ti0.2Mn0.8O3−δ anode, Ba0.6Sr0.4Co0.5Fe0.5O3−δ–GDC cathode and GDC electrolyte reached maximum power densities of 0.29 W cm−2 and 0.24 W cm−2 in humidified H2 and CH4 at 800 °C, respectively. The recent study on the electrochemical and thermomechanical properties of La0.5Sr0.5Mn0.5Ti0.5O3−δ and (La0.55Sr0.45)0.95Mn0.5Cr0.3Ti0.2O3−δ89 confirmed the suitability of LSTM as an alternative anode material.
The ionic transference numbers of La0.4Sr0.5Ti0.6Fe0.4O3−δ were determined to be between 2 × 10−4 to 6 × 10−4 in air by faradaic efficiency measurements, indicating that the substitution of Sr for La would reduce the ionic and total conductivity.90 Nonetheless the La containing compositions showed sufficient stability and a matched thermal expansion coefficient with YSZ. A series of candidate dopants X (X = Al3+, Ga3+, Fen+, Mg2+, Mnn+ and Sc3+) have been investigated in their search for La0.33Sr0.67Ti0.92X0.08O3+δ anodes by Miller et al.91 The impedance results for the electrical half cell tests in H2–3% H2O at 900 °C are shown in Fig. 5a. The La0.33Sr0.67TiO3+δ had the lowest polarisation resistances; doped Sc and Ga gave had the second lowest polarisation resistances followed by doped Al, Fe, Mn and Mg. Fig. 5b shows the over-potential versus current density curves corresponding to the La0.33Sr0.67Ti0.92X0.08O3+δ–YSZ anodes. A reduction in polarisation resistance was observed when the anode was under the load compared to that at the open circuit potential. Ti site Fe doped LST was also explored as solid oxide electrolysis cells (SOECs) cathodes for hydrogen production.92
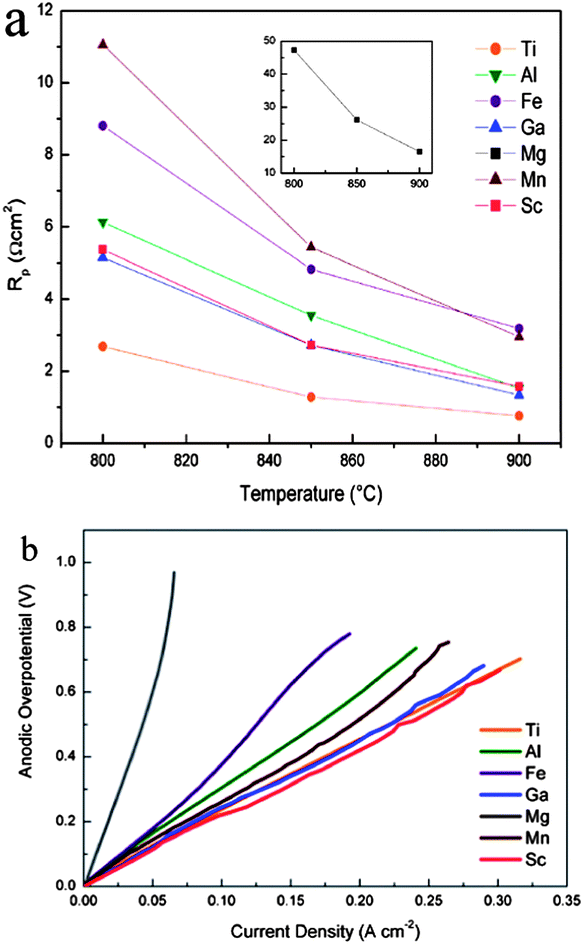 | ||
| Fig. 5 (a) The variation of polarization resistance with temperature for all LSTX–YSZ anodes. (b) Anodic overpotentials of La4Sr8Ti11XO36+ε–YSZ anodes operating in H2–3% H2O at 900 °C. Reprinted from ref. 91, Copyright (2011), with permission from Elsevier. | ||
It is well known that catalytic activities of transition metals generally follow the order of Co > Mn > Ni > Fe > Cr on the oxidation–reduction process.93 Thus, various amount of Co was doped into Ti-site in La0.2Sr0.8TiO3 (LSTC) by Yoo et al.94 The stability of LST anode was greatly improved with the Co doping. A single cell with Ni-impregnated LSTC–GDC composite as the anode, Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) as the cathode and La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM) as the electrolyte exhibited a maximum power density of ∼250 mW cm−2 at 800 °C. The Rp value of anode (∼0.9 Ω cm2) was responsible for most of the total Rp value (∼0.95 Ω cm2). Li et al.95 also considered LSTC as a promising anode candidate and found that at 1500 °C, the solid solution limits of La in LaxSr1−xTiO3−δ and Co in La0.3Sr0.7CoyTi1−yO3−δ were about 40 mol% and 7 mol%, respectively. Co-doping into La0.3Sr0.7TiO3−δ increased the oxygen vacancy concentration and decreased the migration energy for oxygen ions, leading to a significant increase in ionic conductivity but at the expense of some electrical conductivity loss. The electrical and ionic conductivities of La0.3Sr0.7Co0.07Ti0.93O3−δ at 700 °C were 63 S cm−1 and 6 × 10−3 S cm−1 respectively. At the same time, both La0.3Sr0.7TiO3−δ and La0.3Sr0.7Co0.07Ti0.93O3−δ showed relatively stable electrical conductivities under oxygen partial pressure of 10−14 to 10−19 atm at 800 °C. Recently, Cui et al.96 found that the doped Co in La0.3Sr0.7Co0.07Ti0.93O3−δ could exsoluted to the LSCT surface after reduction in H2 at 900 °C. The active Co nanoparticles could reduce the anode polarization resistance. A maximum power density of 300 mW cm−2 was achieved when the cell was fueled with H2 containing 5000 ppm H2S at 900 °C. Also, the LSCT exhibited high sulfur resistance and redox stability. The performance of La0.2Sr0.8Ti0.98Co0.02O3 anode material was reported by Yoo et al.97 The maximum power density of the cell with Ni-impregnated LSTC–GDC anode increased from 638 to 924 mW cm−2 at 800 °C for 30 h in H2. This value was very high compared to other reported values using LSTC anode. In CH4 fuel, while the cell performance with Ni–GDC anode decreased ∼44% for 30 h, the cell performance with Ni-impregnated LSTC–GDC anode was relatively stable for 30 h. The study further proved that LST anode exhibited high tolerance to carbon induced deactivation in the internal utilization of hydrocarbon fuels while sustained a reasonable performance in H2 fuel compared to the conventional Ni-based anode. The reproducibility of crystallographic information of La0.4Sr0.6Ti1−xCoxO3+δ (0.0 ≤ y ≤ 0.5) was also studied by XRD and TEM techniques.98
A new La0.5Sr0.5Ti0.75Ni0.25O3−δ (LSTN) compound was synthesized using a nitrate–citrate gel technique.99 The LSTN presented a metallic behavior after reduction at 1200 °C in H2. The high-temperature treatment led to the precipitation of Ni nanoparticles on the surface and an improved electrical conductivity that may be due to the formation of excess Ti3+. The obtained polarization resistance was 0.55 Ω cm2 at 800 °C under H2–H2O (97/3) using symmetrical cells.
Ruiz-Morales et al.100 has demonstrated the concept of inducing functionality through disorder of extended defects in LST. Mn was known to accept lower coordination numbers in perovskites and thus it may facilitate oxide-ion migration. Similarly Ga was well known to adopt lower coordination than octahedral in perovskite-related oxides. They further studied the Ga and Mn doped in Ti site for La4Sr8Ti12O38−z. The maximum power density in wet H2 was close to 0.5 W cm−2 and the power density in wet CH4 was two times higher than that in 5% H2, reaching the value of about 0.35 W cm−2. After running for two days, cycling from 950 °C to 850 °C, in wet 5% H2, wet H2 and wet methane, no trace of carbon could be detected visually or by thermogravimetric analysis. It was thought that the materials design concept could lead to the devices that enable more-efficient energy extraction from fossil fuels and carbon-neutral fuels. Based on the research of the La0.33Sr0.67Ti0.92X0.08O3+δ,91 La0.4Sr0.4GaxTi1−xO3−x/2−δ (0 ≤ x ≤ 0.15) were chosen to study the improvement of n-type conductivity in perovskite oxides by enhancing bulk oxide ion mobility.101 The study suggested that both oxygen deficiency (δ) and conductivity (σ) of the reduced La0.4Sr0.4GaxTi1−xO3−x/2−δ were increased significantly with Ga doping. The oxygen deficiency was increased from 0.035 to 0.060 when Ga stoichiometry was enhanced from x = 0 to x = 0.15 after reduction. The corresponding conductivity values were also increased with doping and showed a maximum conductivity of 50 S cm−1 for x = 0.05. They also found that the Ga doping could promote a fast reduction of the samples and significantly increase the stability of the reduced phase in oxidizing conditions at high temperatures.
La1−xSrxSc1−yTiyO3−d (LSST) was synthesized through a glycine nitrate process by Hatchwell et al.102 A-site deficiency led to an increased ionic conductivity over LSST. The highest total conductivity achieved for (La0.8Sr0.2)0.99Sc0.9Ti0.1O3−d in air with ∼1% H2O at 800 °C was 23 mS cm−1, in which 6 mS cm−1 was ionic. Li et al.103 found that the Sc-doping in LST increased the oxygen vacancy concentration and decreased the oxygen migration energy. The ionic conductivity of La0.3Sr0.7Sc0.1Ti0.9O3−δ was 1 × 10−2 S cm−1 at 800 °C at the oxygen partial pressure of 10−19 atm. This value increased about 230% compared with pure La0.3Sr0.7TiO3−δ.
Gorte et al.104 thought that the morphologies design and control synthesis of anode were particularly crucial because a larger triple phase boundary (TPB) and the thermal compatibility were required to obtain high fuel cell performance. Based on this viewpoint, Chang et al.105 reported a novel core–shell anode material, which was prepared with Nb-doped LST perovskite core of (La0.3Sr0.7)(Ti1−xNbx)O3 (LSTNx) and the shell of multiple elements doped ceria (La0.75Sr0.2Ba0.05)0.175Ce0.825O1.891 (LSBC) by a citric acid-based combustion coating process. The obtained LSTNs powders morphologies were shown in Fig. 6a and c. The elemental analysis on the core–shell SLTN0.1–12 mol% LSBC powders was shown in Fig. 6b. The shell LSBC nanoparticles adhered on the larger calcined SLTN0.1 core particles were distinguished, as shown in Fig. 6c. It indicated the element of Ce existed on the nanoparticles of the shell (Fig. 6e), which further proved the core–shell formation. The Sr and Ti distributed over core–shell main body (Fig. 6d and f). The Sr and Ti mapping images indicated the core SLTN also existed below the nanoparticles except the large core as seen at the center of Fig. 6c. The power density increased three times by using the core–shell structural anode than that without using the core–shell anode in a half-cell test. This can be attributed to the increased the effective TPB sites and the reduced the interface thermal expansion and lattice matching, as well as extended the ionic conduction path from the LSBC electrolyte to the core–shell anode. The authors thought that the benefits of core–shell structure include simple anode material and structure preparation for the anode without the tedious mixing or impregnation of secondary functional particles. It was also suggested that the core–shell structured anode could be applied in the design of other anode materials.
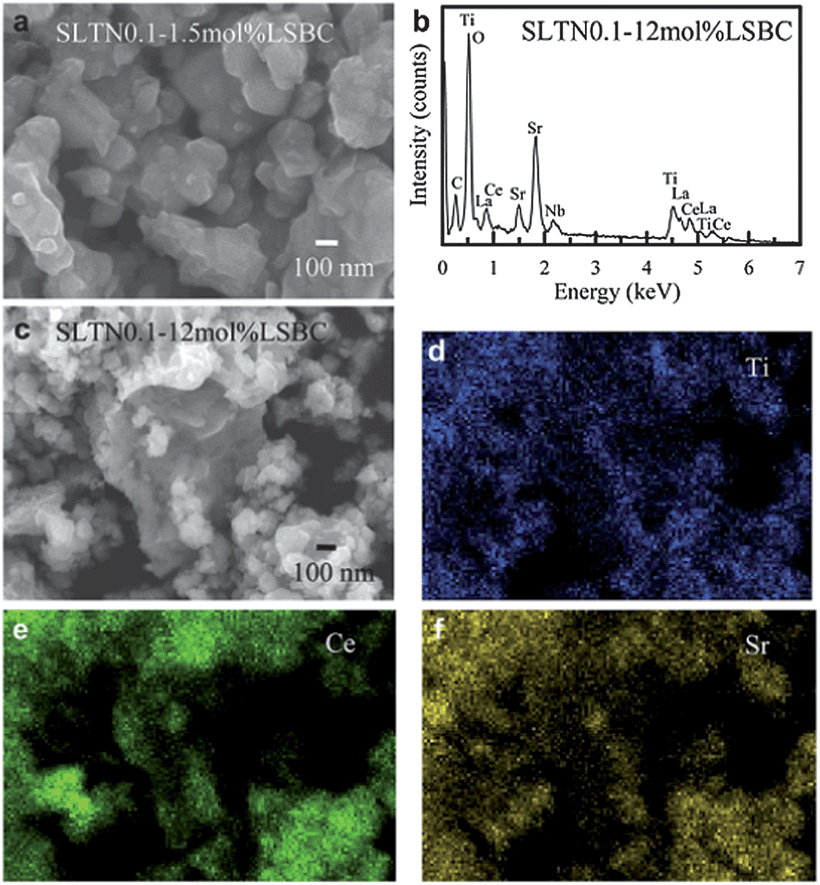 | ||
| Fig. 6 FESEM images and EDS elemental analysis of the prepared core–shell anode powders, (a) FESEM image of LSTN0.1–1.5 mol% LSBC, (b) EDS pattern of LSTN0.1–12 mol% LSBC, (c) FESEM image of LSTN0.1–12 mol% LSBC, and the EDS elemental mappings on the LSTN0.1–12 mol% LSBC, (d) Ti, (e) Ce as well as (f) Sr. Reprinted from ref. 105, Copyright (2012), with permission from International Association for Hydrogen Energy. | ||
3.3 LST-based composite anode materials
Combining LST with other conductor or active catalyst to form a composite anode is an effective way to improve the conductivity and electrocatalytic performance. In this part of the review, the developments of the LST-based composite anode materials are summarized.Ahn et al.62 have studied YSZ and La0.3Sr0.7TiO3 (LST) composite support layer for SOFCs. Excellent performance of the SOFCs was obtained using an anode having a functional layer made from Pd-doped CeO2 in YSZ and a current collector from LST.107 The maximum power densities were 208 mW cm−2 at 973 K and 539 mW cm−2 at 1073 K using humidified CH4 as fuel. At the same time, the anode showed no evidence for carbon formation during an overnight exposure to CH4. They also found that the functional layer could be prepared by impregnation of 1 wt% Pd and 10 wt% CeO2 into porous LST and YSZ.108 This strategy allowed much greater flexibility in electrode design to include different layers. Furthermore, the performance of the LST + Pd-doped CeO2 in YSZ was considered to be only modest. The conductivities of composites with 35 vol% LST were only 0.1 S cm−1 at 1173 K in humidified H2, more than 200 lower than the value reported for bulk LST.109 LST was added to porous YSZ by infiltration method to form LST–YSZ composite anode materials.110 The conductivity of this composite depended strongly on the pretreatment conditions but was greater than 0.4 S cm−1 after being heated to 1173 K in humidified H2. With the addition of 0.5 wt% Pd and 5 wt% CeO2, the maximum power density of LST–YSZ composite anode materials increased from less than 20 to 780 mW cm−2 for operating in humidified H2 at 1073 K. Hill et al.111 investigated the electrochemical conversion pathway of CH4 on Ni/YSZ and La0.3Sr0.7TiO3 bi-layer anode materials in details. They found that the pathway include the CH4 decomposition and subsequent oxidation C and H2, which strongly depended on the anode polarization.
Based on the LST–CeO2 composite anode materials, different doped CeO2 including GDC (Gd-doped CeO2), YDC (Y-doped CeO2), SDC (Sm-doped CeO2), LDC (La-doped CeO2) etc. were further explored as the additive to improve the performance of LST-based anode materials.
The addition of GDC to LST has been proved to be an efficient way to improve the performance of the cell in the above mentioned work.83,94,97 Dense and porous A-site deficient La0.2Sr0.7TiO3 were prepared by Savaniu et al.112 Two types of fuel cells, electrolyte supported and anode supported, were produced using YSZ as the electrolyte, porous LST impregnated with Ce0.8Gd0.2O2 and Cu as the anode, and La0.6Sr0.4CoO3 as the cathode. Maximum power densities of 0.22 and 0.5 W cm−2 at 750 °C were achieved using dense and porous La0.2Sr0.7TiO3 in humidified H2, respectively. In another work, La0.2Sr0.8TiO3 powers were mixed with Ce0.8Gd0.2O2−δ (GDC) powders in 50:50 weight ratio to form LST–GDC composite in which Ni was impregnated afterwards.113 GDC could provide the ionic conduction path whereas the small amount of Ni would enhance the catalytic activity. The polarization resistance (Rp) of LST–GDC anode was greatly reduced relative to that of pure LST anode. When Ni was impregnated into LST–GDC composite, the Rp value was further reduced to 1 Ω cm2 at 800 °C in humidified H2 (Fig. 7), which was ∼10% of that for pure LST anode. The maximum power density of the single cell with LST–GDC + Ni anode was about 275 mW cm−2 at 800 °C, much larger than 60 mW cm−2 for the cell with LST–GDC anode. A similar study was conducted in ref. 114 in which La0.2Sr0.8TiO3–GDC–Ni anodes of various composition ratios were prepared as the anode. With LSGM as the electrolyte and La0.6Sc0.4Co0.2Fe0.8O3 (LSCF) as the cathode material at 800 °C in humidified H2, the performance of single cell was about 300 mW cm−2, increasing drastically from 67 mW cm−2 for the cell without Ni impregnation. It was suggested that in order to further reduce the polarization resistance, the optimization of anode microstructure and the content and distribution of Ni particles might be helpful. A-site deficient LST backbone impregnated with GDC and Ni115 or Cu116 was also investigated by Savaniu et al. A-site deficient La0.2Sr0.7TiO3 was impregnated using solutions with 20 mol% GDC and a metal Cu. YSZ and La0.6Sr0.4CoO3 (LSC) thin layer were used as the electrolyte and the cathode material, respectively. Fuel cells tests demonstrated that remarkable power densities about 0.5 W cm−2 at 750 °C could be achieved using pure, humidified H2 as fuel.
 | ||
| Fig. 7 The polarization resistance values of symmetrical LSGM electrolyte-supported cell with LST, LST–GDC and Ni-impregnated LST–GDC anodes as a function of time. All data were extracted from the impedance spectra obtained under open circuit conditions at 800 °C. All values increased with time. Reprinted from ref. 113, Copyright (2009), with permission from Elsevier. | ||
Y0.2Ce0.8O2−δ (YDC) was also explored as the additive to LST-based composite because of its high ionic conductivity and catalytic performance. It was found that La0.4Sr0.6TiO3±δ (LST) and YDC composite was a stable, high performance material for use as anode in SOFCs fed by 0.5% H2S-containing syngas, whereas pure CeO2 was not chemically stable using the same fuel.117 Moreover, both LST and YDC were synthesized using citrate–nitrate gel combustion method.118 The obtained LST and YDC powders were ball-milled and calcined to obtain the LST–YDC composite anode. The maximum power densities at 850 °C were 169, 102 and 39 mW cm−2 when the cell was fueled with syngas having 0.5% H2S, pure syngas and 0.5% H2S balanced with Ar, respectively. Gas chromatographic and mass spectrometric analyses revealed that the presence of H2S improved the rate of fuel electrochemical oxidation while H2S itself was not converted and its oxidation was not the source of the enhanced performance. The presence of H2S in different feeds was evidenced to have negligible influence over the fuel cell stability.
Pillai et al.119 studied the SOFCs with La0.2Sr0.8TiO3 anode-side supports, along with NiO–Ce0.8Sm0.2O2 (SDC) (1:1 by weight) adhesion layer, NiO–YSZ (1:1 by weight) anode active layer, YSZ electrolyte, and LSM–YSZ cathode. The colloidal solutions of NiO–SDC, NiO–YSZ, and YSZ were sequentially dropped and coated onto the LST pellets. The obtained LST-supported SOFCs showed improved stability against coking in natural gas, compared with conventional Ni–YSZ anode-supported SOFCs. There was no long-term degradation in H2 fuel with 50–100 ppm H2S, and no degradation during redox cycling. However, the maximum power density was decreased from 850 mW cm−2 to about 700 W cm−2 when the fuel was changed from dry H2 to dry H2 containing 100 ppm H2S, which further indicated the poor sulfur tolerance of Ni-based catalyst.
Besides the CeO2-based oxide, the addition of Bi2O3 into LST was also reported to be able to significantly reduce the fuel cell's polarization, refine the grains, increase the triple phase boundary, and then to improve the electrochemical performance.120
Although the nanostructured electrocatalytic active composition exsoluted from the doped LST matrix is a potential way to form high performance LST composite anode. The effective control of the exsoluted particle quantity and the prevention of their aggregation at high temperature are crucial and remain to be solved in future research. In addition, the LST-based composite anode materials including Ni nanoparticles could not be used directly for the hydrocarbon fuel because of their poor carbon resistance character.
3.4 LST synthesis methods
Overview of all the LST materials covered in this review suggests that conventional solid state method53–60,62–67,71,76,78–80,85,90–96,100,101,108,115,116,119,120 was still the main route to prepare LST materials due to its simplicity and relatively low processing cost. However, the solid state method needs a long period of time to facilitate the diffusion at high temperature (usually >1200 °C). The obtained products were often coarse and agglomerated, exhibiting limited purity and homogeneity. Therefore, the new approaches for the development of nano-structured electrodes are needed and some soft ways have drawn increasing attentions in recent years.125 The other different routes to synthesize LST-based materials include Pechini route,61,75,82 impregnation or infiltration method,77,86,107,110 combustion synthesis,83,88 citric acid–nitrate process,68,84,98,99,105,118 glycine–nitrate and citrate synthesis routes,52,87,102 spray pyrolysis method,72 and nanoparticle-based spray pyrolysis70,74 to synthesize LST-based materials. The LST powders obtained through the above soft methods have small particle size and narrow size distribution or porous structure which would enlarge the TPBs and then improve the performance. Recently, Birol et al.126 reviewed the cellulose-assisted glycine nitrate process of preparing ceramic nanoparticles. Some other soft methods such as co-precipitation method,127 sol–gel route128 and rheological phase reaction129,130 were the potential techniques to synthesize LST-based anode materials.For the synthesis of LST composite anode materials, the common way was to mix pure LST with other materials automatically.62,74,106,108,113,114,120,137 Impregnation or infiltration technology107,110,112,116 was another commonly used and effective way to add the active components to the LST materials. Combination of the combustion method and automatical mix,118 colloidal deposition technique119 were also applied to blend LST and other materials. Exsolution method was a compellent way to obtain LST-based anode materials76,92,96,99,121–123 and would be further explored in the future work.
3.5 Conductivity and performance of LST-based anode materials
Table 1 summaries the conductivities of selected LST-based anode materials. We can see that the pure SrTiO3 has a low ionic conductivity. The co-doped La and other metal ions in A-site and B-site doping can obviously improve the conductivity. The type of doping element and the doping quantity should be experimentally optimized in the future work.| Anode composition | Electronic conductivity (S cm−1) | Ionic conductivity (S cm−1) | Tem. (°C) | Fuel | Ref. |
|---|---|---|---|---|---|
| La0.6Sr0.1TiO3−δ | 7 | — | 930 | 5% H2/Ar | 54 |
| La2Sr4Ti6O19−δ | 40 | — | 950 | 5% H2/Ar | 58 |
| La0.4Sr0.4TiO3 | ∼100 | — | 900 | 5% H2/Ar | 60 |
| La0.3Sr0.7TiO3−α | ∼100 | — | 1000 | 9% H2/N2 | 63 |
| La0.1Sr0.9TiO3 | 12 | — | 1000 | 30% H2/N2 | 68 |
| (La0.3Sr0.7)0.93TiO3−δ | 145 | 1.0 × 10−2 | 800 | 5% H2/Ar | 69 |
| La0.2Sr0.7TiO3 | 0.3 | — | 800 | 5% H2/Ar | 72 |
| La0.2Sr0.7TiO3 | 30–40 | — | 1000 | 5% H2/Ar | |
| La0.2Sr0.7TiO3 | ∼100 | — | 1200 | 5% H2/Ar | |
| La0.2Sr0.7TiO3 | 600 | — | 600 | 5% H2/N2 | 70 |
| La0.2Sr0.25Ca0.45TiO3 | 27.53 | — | 900 | 5% H2/Ar | 80 |
| La0.3Sr0.55Ti0.8Cr0.2O3−δ | 1.96 × 10−3 | — | 800 | Static air | 84 |
| La0.3Sr0.7Ti0.8Cr0.2O3−δ | 53 | — | 800 | 5% H2/Ar | 85 |
| La0.33Sr0.67Ti0.92Al0.08O3+δ | 1.5 | — | 900 | 5% H2/Ar | 91 |
| La0.33Sr0.67Ti0.92Sc0.08O3+δ | 5.5 | — | 900 | 5% H2/Ar | |
| SrTiO3 | 0.03 | — | 700 | 5% H2/Ar | 95 |
| La0.3Sr0.7TiO3−δ | 247 | 1 × 10−3 | 700 | 5% H2/Ar | |
| La0.3Sr0.7Co0.07Ti0.93O3−δ | 63 | 6 × 10−3 | 700 | 5% H2/Ar | |
| La0.5Sr0.5Ti0.75Ni0.25O3−δ | 2.2 × 10−3 | — | 800 | Air | 99 |
| La0.5Sr0.5Ti0.75Ni0.25O3−δ | 4.9 × 10−4 | — | 800 | 2% H2/Ar | |
| La0.5Sr0.5Ti0.75Ni0.25O3−δ | 2.3 | — | 800 | 2% H2/Ar | |
| La0.4Sr0.4Ga0.05Ti0.95O2.95/2−δ | ∼50 | — | 880 | 5% H2/Ar | 101 |
| (La0.8Sr0.2)0.99Sc0.9Ti0.1O3−d | 6 × 10−3 | — | 800 | 1% H2O/air | 102 |
| La0.3Sr0.7Sc0.1Ti0.9O3−δ | 49 | 1 × 10−2 | 800 | 5% H2/Ar | 103 |
| La0.3Sr0.7TiO3 | >0.4 | — | 900 | 3% H2O/H2 | 110 |
Table 2 lists the fuel cell performances of selected LST-based anode materials. It can be seen that the pure LST materials have a poor electrocatalytic performance for H2 oxidation. The common methods to improve the performance include: (1) exploring new methods to synthesize LST-based anode materials with a nanostructure; (2) mixing, impregnation or infiltration of high ionic conductor (CeO2-based materials, Bi2O3, etc.) and high catalytic materials (Pd, Cu, Ni, etc.) into porous LST; (3) exsolution method is a potential technology to form LST-based composite anode materials.
| Anode composition | Electrolyte (thickness)/cathode | Tem. (°C) | Fuel | MPDa (mW cm−2) | Ref. |
|---|---|---|---|---|---|
| a MPD = maximal power density. | |||||
| La0.4Sr0.6TiO3 | YSZ (160 μm)/LSM | 1000 | Humidified H2 | 260 | 52 |
| La2Sr4Ti6O19−δ | YSZ (2 mm)/LSM | 900 | Humidified H2 | 76 | 59 |
| La0.3Sr0.7TiO3 + CeO2 + Cu | YSZ (60 μm)/LSM | 700 | H2 | 200 | 62 |
| La0.2Sr0.8TiO3 | YSZ (280 μm)/Pt | 900 | 5% H2 + 95% Ar | 12 | 71 |
| La0.4Sr0.45Ba0.15TiO3 | YSZ (300 μm)/LSM | 850 | H2 | ∼39 | 76 |
| La0.4Sr0.5Ba0.1TiO3 | YSZ (300 μm)/LSM | 850 | H2 | 20 | 77 |
| La0.20Sr0.25Ca0.45TiO + CeO2 + Ni | ScSZ (310 μm)/LSM | 900 | Humidified H2 | ∼300 | 79 |
| La0.3Sr0.7TiO3−δ | YSZ (400 μm)/LSM | 900 | Humidified H2 | ∼31 | 85 |
| La0.3Sr0.7Ti0.8Cr0.2O3−δ | 44 | ||||
| La0.4Sr0.6Ti0.8Mn0.2O3±δ | YSZ (50 μm)/LSM | 800 | Humidified H2 | 30 | 86 |
| La0.4Sr0.6Ti0.8Mn0.2O3±δ + Pd | ∼70 | ||||
| La0.4Sr0.6Ti0.8Mn0.2O3±δ + CeO2 | 100 | ||||
| La0.4Sr0.6Ti0.8Mn0.2O3±δ + Pd + CeO2 | 150 | ||||
| La0.6Sr0.4Ti0.2Mn0.8O3−δ | GDC (500 μm)/Ba0.6Sr0.4Co0.5Fe0.5O3−δ | 800 | Humidified H2 | 290 | 88 |
| La0.2Sr0.8Ti0.98Co0.02O3 + GDC + Ni | LSGM (500 μm)/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 800 | Humidified H2 | 250 | 94 |
| La0.3Sr0.7TiO3−δ | YSZ (300 μm)/LSM | 900 | Humidified H2 | 90 | 96 |
| La0.3Sr0.7Ti0.93Co0.07O3−δ | 230 | ||||
| La0.2Sr0.8Ti0.98Co0.02O3 + GDC + Ni | LSGM (250 μm)/La0.6Sr0.4Co0.2Fe0.8O3−δ | 800 | Humidified H2 | 638 | 97 |
| La4Sr8Ti11Mn0.5Ga0.5O37.5 | YSZ (2 mm)/LSM | 950 | Humidified H2 | 500 | 100 |
| La0.4Sr0.6TiO3 + CeO2 | YSZ (300 μm)/ScSz + LSM | 900 | Humidified H2 | 172 | 106 |
| La0.3Sr0.7TiO3 | YSZ (60 μm)/LSF | 800 | Humidified H2 | 20 | 110 |
| La0.3Sr0.7TiO3 + CeO2 + Pd | 780 | ||||
| La0.3Sr0.7TiO3 + Ni/YSZ | YSZ (300 μm)/Pt | 750 | Humidified H2 | 102 | 111 |
| Dense La0.2Sr0.7TiO3 + GDC + Cu | YSZ (150 μm)/La0.6Sr0.4CoO3 (LSC) | 750 | Humidified H2 | 220 | 112 |
| Porous La0.2Sr0.7TiO3GDC + Cu | 500 | ||||
| La0.2Sr0.8TiO3 + GDC | LSGM (500 μm)/Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 800 | Humidified H2 | 60 | 113 |
| La0.2Sr0.8TiO3 + GDC + Ni | (BSCF) | 275 | |||
| La0.2Sr0.8TiO3 + GDC | LSGM (150 μm)/La0.6Sc0.4Co0.2Fe0.8O3 | 800 | Humidified H2 | 67 | 114 |
| La0.2Sr0.8TiO3 + GDC + Ni | (LSCF) | 300 | |||
| La0.44Sr0.56TiO3 + GDC + Ni | YSZ (32 μm)/LSC64 | 850 | Humidified H2 | 185 | 115 |
| La0.2Sr0.7TiO3 + GDC + Cu | YSZ (50–75 μm)/La0.6Sr0.4CoO3 | 750 | Humidified H2 | 500 | 116 |
| La0.4Sr0.6TiO3±δ + YDC | YSZ (300 μm)/LSM | 850 | 40% H2 + 60% CO | 102 | 118 |
| La0.2Sr0.8TiO3 + NiO–SDC + NiO–YSZ | YSZ/LSM | 800 | Dry H2 | 850 | 119 |
| La0.4Sr0.6TiO3 | YSZ (500 μm)/LSM | 1000 | Humidified H2 | 175 | 136 |
4. H2S-induced enhancement for electrochemical reaction on LST-based anode materials
H2S is a common impurity in many industrial feedstocks such as coal derived syngas and raw natural gas. Although H2S may serve as an increasingly significant source for sulfur production, it is a major industrial pollutant that is toxic and corrosive. Direct electrochemical conversion of H2S in SOFCs would offer both economical and environmental benefits.131–135 It is known that even trace amount of H2S could lead to severe performance degradation of the fuel cells when using Ni-based anode materials.24–26 Therefore, a series of LST-based anode materials have been developed in our lab, which showed not only a high H2S poisoning resistance and high chemical stability but also a H2S-induced enhancement effect for electrochemical reaction when using a fuel containing H2S.The sulfur tolerance of La1−xSrxBO3/YSZ anodes (B = Mn, Cr, and Ti) was examined at 1273 K in a H2/H2O fuel using YSZ electrolyte, LSM cathode.136 The study indicated that the La0.4Sr0.6TiO3 anode showed no degradation in the presence of up to 5000 ppm of H2S in H2. It was found that the performance of the fuel cell improved significantly in the presence of 5000 ppm of H2S especially at the higher current densities. But the reason for this improvement was not clear in this work. The sulfur tolerance of the perovskite-based anodes decreased in the order La0.4Sr0.6TiO3 (LST) > La1−xSrxCrO3 (LSC) > La1−xSrxMnyCr1−yO3 (LSCM).
Table 3 compares the fuel cell performances of LST-based anode materials when the fuel contained trace amount of H2S. It is clearly shown that the performance was dramatically improved when H2S was present in the fuels such as CH4,76,77,83,137 H2,76,77,136 syngas (H2 + CO)117,118. This can also be observed clearly in Fig. 8. In ref. 119, the performance was decreased when using the dry H2 containing 100 ppm H2S, which could be attributed to the poisoning of Ni-based active catalyst in LST-based composite anode.
| Anode composition | Electrolyte/cathode | Tem. (°C) | Fuel | MPDa (mW cm−2) | Ref. |
|---|---|---|---|---|---|
| a MPD = maximal power density. | |||||
| La0.4Sr0.45Ba0.15TiO3 | YSZ (300 μm)/LSM | 850 | H2 | ∼39 | 76 |
| H2 + 0.5% H2S | ∼50 | ||||
| CH4 | ∼1.5 | ||||
| CH4 + 0.5% H2S | ∼7.5 | ||||
| La0.4Sr0.5Ba0.1TiO3 | YSZ (300 μm)/LSM | 850 | H2 | 20 | 77 |
| H2 + 0.5% H2S | 137 | ||||
| CH4 | 24 | ||||
| CH4 + 0.5% H2S | 75 | ||||
| La0.3Sr0.55Ti1−xCrxO3−δ | YSZ (300 μm)/Pt | 900 | CH4 | ∼2 | 83 |
| CH4 + 0.5% H2S | ∼140 | ||||
| La0.3Sr0.7Ti0.93Co0.07O3−δ | YSZ (300 μm)/LSM | 900 | H2 | 230 | 96 |
| H2 + 0.5% H2S | 300 | ||||
| La0.4Sr0.6TiO3±δ + YDC | YSZ (300 μm)/LSM | 850 | 40% H2 + 60% CO | 102 | 118 |
| 40% H2 + 60% CO + 0.5% H2S | 169 | ||||
| La0.2Sr0.8TiO3 + NiO–SDC + NiO–YSZ | YSZ/LSM | 800 | Dry H2 | 850 | 119 |
| Dry H2 + 100 ppm H2S | 700 | ||||
| La0.4Sr0.6TiO3 | YSZ (500 μm)/LSM | 1000 | Humidified H2 | 175 | 136 |
| Humidified H2 + 0.5% H2S | ∼210 | ||||
| La0.4Sr0.6TiO3−δ | YSZ/Pt | 850 | CH4 | ∼2 | 137 |
| CH4 + 5% H2S | 320 | ||||
| CH4 + 20% H2S | 450 | ||||
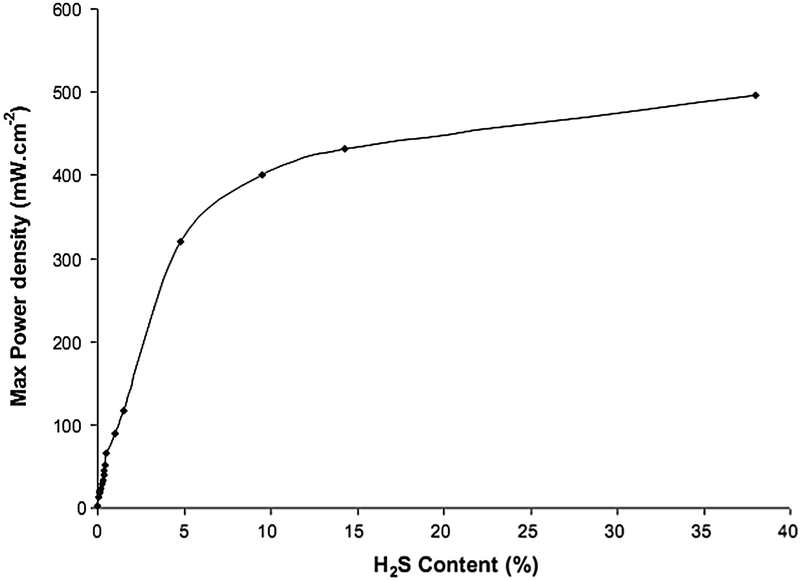 | ||
| Fig. 8 Maximal power density (after compensation) as a function of H2S concentration in CH4 (after flow corrections) at 850 °C. Fuels were fed at 50 mL min−1. Reprinted from ref. 137, Copyright (2011), with permission from Elsevier. | ||
A series of experiments were designed to systematically investigate the electrochemical performance and compositions of anode gases to explore the mechanism of the significant performance enhancement with H2S presence in either CH4 or H2 fuel. La0.4Sr0.6TiO3−δ was mixed with commercial YSZ power with a 50/50 weight ratio to form the anode materials.137 Platinum paste was used as the cathode and the current collector. Electrolyte was the commercial YSZ dicks. The power density improved dramatically from 2 mW cm−2 with pure CH4 as fuel to more than 450 mW cm−2 for CH4 containing 20% H2S (Fig. 8). From thermodynamic calculations and mass spectroscopic data obtained at different potential stages during a regular potentiodynamic run, it was concluded that H2S was not the only fuel converted, and its interaction with CH4 was the key factor for the enhancement of performance. The anode effluent gas mixture included H2S, SO2, H2 and CS2, which was consistent with thermodynamic predictions. The conclusion was that H2S had a synergistic effect on CH4 oxidation, i.e., CH4 and H2S must not be considered as two separate fuels. H2S actually ran as a powerful promoter for fuel electro-oxidation. At low current density, there was a rapid conversion of surface species, whereas at high current density there was a mass transfer blocking effect from build-up of the surface intermediates. The predominant form of sulfur in the gas phase was H2S previously presented. There was at least 3% H2 in the mixture. CH4 conversion proceeded via rapid formation of syngas, and the reaction path probably included CS2 and other sulfur species. The selectivity of the obtained products could be controlled by adjusting the applied voltage. In order to further explore the mechanism of the H2S enhancement effect in details, in situ technologies such as in situ Raman,138in situ XPS139etc. were recommended to be applied in the field in the future work.
5. Conclusions
The recent progress in the field of LST-based anode materials in SOFCs are summarized. Although pure LST had very limited activity for oxidation of H2S-free fuels, it exhibited superior balance between high chemical stability in sulfur/carbon environment and adequate electrical conductivity compared to other anode candidates. Thus it could be used as an anode matrix material and its properties could be further tailored depending on the fuel selected. Doping and composting of LST are the most effective ways to increase the ionic conductivity and the electrocatalytical activity of LST-based anode.Future work on the LST-based anode materials in SOFCs can focus on the following aspects: (1) Exploring new methods to synthesize LST materials, doping LST with appropriate elements in different site, impregnating, mixing or coating nano-structured ionic conductor or exsoluted electrocatalytical active nanoparticles to LST anode. (2) Combining LST-based materials with other catalytic active material to form LST-based composite anode materials using impregnation, infiltration technique or some other new methods. The added secondary phases should sustain sufficient thermal stability at elevated temperature. (3) Exploring the fuel conversion mechanism of LST-based anode especially the H2S-induced enhancement effect for electrochemical reaction using in situ experimental technologies.
Acknowledgements
This research was supported through funding to the Solid Oxide Fuel Cell Canada Strategic Research Network and Strategic Grant from the Natural Science and Engineering Research Council (NSERC).Notes and references
- K. Onda, T. Iwanari, N. Miyauchi, K. Ito, T. Ohba, Y. Sakaki and S. Nagata, J. Electrochem. Soc., 2003, 150, A1569 CrossRef CAS PubMed.
- A. J. Jacobson, Chem. Mater., 2010, 22, 660 CrossRef CAS.
- C. Sun, R. Hui and F. Roller, J. Solid State Electrochem., 2010, 14, 1125 CrossRef CAS PubMed.
- Z. Jiang, C. Xia and F. Chen, Electrochim. Acta, 2010, 55, 3595 CrossRef CAS PubMed.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935 CrossRef CAS PubMed.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355 RSC.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Adv. Mater., 2012, 24, 195 CrossRef CAS PubMed.
- M. Ni, Int. J. Hydrogen Energy, 2013, 38, 2846 CrossRef CAS PubMed.
- L. Wu and X. Liu, J. Mater. Sci. Technol., 2010, 26, 293 Search PubMed.
- N. Shaigan, W. Qu, D. G. Ivey and W. Chen, J. Power Sources, 2010, 195, 1529 CrossRef CAS PubMed.
- M. K. Mahapatra and K. Lu, J. Power Sources, 2010, 195, 7129 CrossRef CAS PubMed.
- D. Coillot, F. O. Méar, H. Nonnet and L. Montagne, Int. J. Hydrogen Energy, 2012, 37, 9351 CrossRef CAS PubMed.
- A. Sarikaya, V. Petrovsky and F. Dogan, J. Electrochem. Soc., 2012, 159, F665 CrossRef CAS PubMed.
- N. Yan, X. Z. Fu, J. L. Luo, K. T. Chuang and A. R. Sanger, J. Power Sources, 2012, 198, 164 CrossRef CAS PubMed.
- K. Wang, D. Hissel, M. C. Péra, N. Steiner, D. Marra, M. Sorrentino, C. Pianese, M. Monteverde, P. Cardone and J. Saarinen, Int. J. Hydrogen Energy, 2011, 36, 7212 CrossRef CAS PubMed.
- S. A. Hajimolana, M. A. Hussain, W. M. A. W. Daud, M. Soroush and A. Shamiri, Renewable Sustainable Energy Rev., 2011, 15, 1893 CrossRef CAS PubMed.
- A. Choudhury, H. Chandra and A. Arora, Renewable Sustainable Energy Rev., 2013, 20, 430 CrossRef CAS PubMed.
- L. Barelli, E. Barluzzi and G. Bidini, Int. J. Hydrogen Energy, 2013, 38, 5060 CrossRef CAS PubMed.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. Mcevoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed.
- P. I. Cowin, C. T. G. Petit, R. Lan, J. T. S. Irvine and S. Tao, Adv. Energy Mater., 2011, 1, 314 CrossRef CAS.
- X. M. Ge, S. H. Chan, Q. L. Liu and Q. Sun, Adv. Energy Mater., 2012, 2, 1156 CrossRef CAS.
- K. Gironaa, J. Laurencinb, J. Fouletierc and F. Lefebvre-Joud, J. Power Sources, 2012, 210, 381 CrossRef PubMed.
- K. T. Chuang, J. L. Luo and A. R. Sanger, Chem. Ind. Chem. Eng. Q., 2008, 14, 69 CrossRef CAS.
- Z. Cheng, J. H. Wang, Y. Choi, L. Yang, M. C. Lin and M. Liu, Energy Environ. Sci., 2011, 4, 4380 CAS.
- M. Btesznowski, J. Jewulski and A. Zieleniak, Cent. Eur. J. Chem., 2013, 11, 960 CrossRef PubMed.
- M. Ettler, H. Timmermann, J. Malzbender, A. Weber and N. H. Menzler, J. Power Sources, 2010, 195, 5452 CrossRef CAS PubMed.
- G. Postole, K. Girona, J. Toyir, A. Kaddouri and P. Gélin, Fuel Cells, 2012, 12, 275 CrossRef CAS.
- Y. Chen, Q. Liu, Z. Yang, F. Chen and M. Han, RSC Adv., 2012, 2, 12118 RSC.
- L. Mlamar, B. Colldeforns, L. Yedra, S. Estradé, F. Peiró, A. Morata, T. Andreu and A. Tarancón, J. Mater. Chem. A, 2013, 1, 4531 Search PubMed.
- A. Kelaidopoulou, A. Siddle, A. L. Dicks, A. Kaiser and J. T. S. Irvine, Fuel Cells, 2001, 1, 219 CrossRef CAS.
- P. R. L. Keating, D. O. Scanlon and G. W. Watson, J. Mater. Chem. C, 2013, 1, 1093 RSC.
- C. Goulart and E. Djurado, J. Eur. Ceram. Soc., 2013, 33, 769 CrossRef CAS PubMed.
- J. W. Fergus, Solid State Ionics, 2006, 177, 1529 CrossRef CAS PubMed.
- E. Lay, L. Dessemond and G. Gauthier, J. Power Sources, 2013, 221, 149 CrossRef CAS PubMed.
- J. S. Park, J. Luo, L. Adijanto, J. M. Vohs and R. J. Gorte, J. Power Sources, 2013, 222, 123 CrossRef CAS PubMed.
- Q. Ma, F. Tietz, A. Leonide and E. Ivers-Tiffée, J. Power Sources, 2011, 196, 7308 CrossRef CAS PubMed.
- A. M. Hussain, J. V. T. Høgh, T. Jacobsen and N. Bonanos, Int. J. Hydrogen Energy, 2012, 37, 4309 CrossRef PubMed.
- S. W. Tao and J. T. S. Irvine, J. Mater. Chem., 2002, 12, 2356 RSC.
- Y. H. Huang, R. I. Dass, Z. L. Xing and J. B. Goodenough, Science, 2006, 312, 254 CrossRef CAS PubMed.
- H. Li, Y. Tian, Z. Wang, F. Qie and Y. Li, RSC Adv., 2012, 2, 3857 RSC.
- X. Che, Y. Shen, H. Li and T. He, J. Power Sources, 2013, 222, 288 CrossRef CAS PubMed.
- L. Troncoso, M. J. Martínez-Lope, J. A. Alonso and M. T. Fernández-Díaz, J. Appl. Phys., 2013, 113, 023511 CrossRef.
- A. J. Weisentein, N. Childs, R. Amendola, D. Driscoll, S. W. Sofie, P. Gannon and R. Smith, Mater. Chem. Phys., 2013, 139, 706 CrossRef CAS PubMed.
- R. Martínez-Coronado, A. Aguadero, J. A. Alonso and M. T. Fernández-Díaz, Solid State Sci., 2013, 18, 64 CrossRef PubMed.
- J. B. Goodenough, Rep. Prog. Phys., 2004, 67, 1915 CrossRef CAS.
- Y. Zheng, W. Zhou, R. Ran and Z. Shao, Prog. Chem., 2008, 20, 413 CAS.
- I. Z. Rahman, M. A. Raza and M. A. Rahman, Adv. Mater. Res., 2012, 445, 497 CAS.
- P. I. Cowin, C. T. G. Petit, R. Lan, C. J. Schaschke and S. Tao, Solid State Ionics, 2013, 236, 48 CrossRef CAS PubMed.
- L. Adijanto, R. Küngas, J. Park, J. M. Vohs and R. J. Gorte, Int. J. Hydrogen Energy, 2011, 36, 15722 CrossRef CAS PubMed.
- K. Singh, J. Nowotny and V. Thangadurai, Chem. Soc. Rev., 2013, 42, 1961 RSC.
- K. Uematsu, O. Sakurai, N. Mizutani and M. Kato, J. Mater. Sci., 1984, 19, 3671 CrossRef CAS.
- O. A. Marina, N. L. Canfield and J. W. Stevenson, Solid State Ionics, 2002, 149, 21 CrossRef CAS.
- R. Moos, T. Bischoff, W. Menesklou and K. H. Härdtl, J. Mater. Sci., 1997, 32, 4247 CrossRef CAS.
- P. R. Slater, D. P. Fagg and J. T. S. Irvine, J. Mater. Chem., 1997, 7, 2495 RSC.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2010, 22, 5042 CrossRef CAS.
- A. Tesfai, C. Savaniu and J. T. S. Irvine, ECS Trans., 2011, 35, 557 CrossRef CAS PubMed.
- G. Tsekouras and J. T. S. Irvine, J. Mater. Chem., 2011, 21, 9367 RSC.
- J. Canales-Vánzquez, W. Zhou and J. T. S. Irvine, Ionics, 2002, 8, 252 CrossRef.
- J. Canales-Vázquez, S. W. Tao and J. T. S. Irvine, Solid State Ionics, 2003, 159, 159 CrossRef.
- J. Canales-Vázquez, M. J. Smith, J. T. S. Irivine and W. Zhou, Adv. Funct. Mater., 2005, 15, 1000 CrossRef.
- B. K. Park, J. W. Lee, S. B. Lee, T. H. Lim, S. J. Park, P. H. Song, W. B. Im and D. R. Shin, Int. J. Hydrogen Energy, 2012, 37, 4319 CrossRef CAS PubMed.
- K. Ahn, S. Jung, J. M. Vohs and R. J. Gorte, Ceram. Int., 2007, 33, 1065 CrossRef CAS PubMed.
- S. Hashimoto, L. Kindermann, P. H. Larsen, F. W. Poulsen and M. Mogensen, J. Electroceram., 2006, 16, 103 CrossRef CAS.
- S. Hashimoto, F. W. Poulsen and M. Mogensen, J. Alloys Compd., 2007, 439, 232 CrossRef CAS PubMed.
- Y. Tsvetkova and V. Kozhukharov, Mater. Des., 2009, 30, 206 CrossRef CAS PubMed.
- Z. Wang, M. Mori and T. Itoh, J. Electrochem. Soc., 2010, 157, B1783 CrossRef CAS PubMed.
- M. Mori, Z. Wang, T. Itoh, S. Yabui, K. Murai and T. Moriga, J. Fuel Cell Sci. Technol., 2011, 8, 051014 CrossRef.
- Z. Wang and M. Mori, J. Fuel Cell Sci. Technol., 2011, 8, 051018 CrossRef.
- X. Li, H. Zhao, X. Zhou, N. Xu, Z. Xie and N. Chen, Int. J. Hydrogen Energy, 2010, 35, 7913 CrossRef CAS PubMed.
- D. Burnat, A. Heel, L. Holzer, D. Kata, J. Lis and T. Graule, J. Power Sources, 2012, 201, 26 CrossRef CAS PubMed.
- X. Huang, H. Zhao, W. Qiu, W. Wu and X. Li, Energy Convers. Manage., 2007, 48, 1678 CrossRef CAS PubMed.
- Q. Ma and F. Tietz, Solid State Ionics, 2012, 225, 108 CrossRef CAS PubMed.
- S. Suthirakun, S. C. Ammal, G. Xiao, F. Chen, K. Huang, H. C. Loye and A. Heyden, Solid State Ionics, 2012, 228, 37 CrossRef CAS PubMed.
- D. Burnat, A. Heel, L. Holzer, E. Otal, D. Kata and T. Graule, Int. J. Hydrogen Energy, 2012, 37, 18326 CrossRef CAS PubMed.
- C. Périllat-Merceroz, G. Gauthier, P. Roussel, M. Huvé, P. Gélin and R. N. Vannier, Chem. Mater., 2011, 23, 1539 CrossRef.
- A. Vincent, J. L. Luo, K. T. Chuang and A. R. Sanger, J. Power Sources, 2010, 195, 769 CrossRef CAS PubMed.
- A. L. Vincent, A. R. Hanifi, J. L. Luo, K. T. Chuang, A. R. Sanger, T. H. Etsell and P. Sarkar, J. Power Sources, 2012, 215, 301 CrossRef CAS PubMed.
- J. H. Li, X. Z. Fu, J. L. Luo, K. T. Chuang and A. R. Sanger, J. Power Sources, 2012, 213, 69 CrossRef CAS PubMed.
- M. C. Verbraeken, B. Iwanschitz, A. Mai and J. T. S. Irvine, J. Electrochem. Soc., 2012, 159, F757 CrossRef CAS PubMed.
- A. D. Aljaberi and J. T. S. Irvine, J. Mater. Chem. A, 2013, 1, 5868 CAS.
- A. Yaqub, C. Savaniu, N. K. Janua and J. T. S. Irvine, J. Mater. Chem. A, 2013 10.1039/c3ta12860a.
- M. González-Cuencal, W. Zipprich, B. A. Boukamp, G. Pudmich and F. Tietz, Fuel Cells, 2001, 1, 256 CrossRef.
- N. Danilovic, A. Vincent, J. L. Luo, K. T. Chuang, R. Hui and A. R. Sanger, Chem. Mater., 2010, 22, 957 CrossRef CAS.
- F. Yi, H. Li, H. Chen, R. Zhao and X. Jiang, Ceram. Int., 2013, 39, 347 CrossRef CAS PubMed.
- Z. Du, H. Zhao, X. Zhou, Z. Xie and C. Zhang, Int. J. Hydrogen Energy, 2013, 38, 1068 CrossRef CAS PubMed.
- J. H. Kim, D. Miller, H. Schlegl, D. McGrouther and J. T. S. Irvine, Chem. Mater., 2011, 23, 3841 CrossRef CAS.
- V. A. Kolotygin, E. V. Tsipis, A. I. Ivanov, Y. A. Fedotov, I. N. Burmistrov, D. A. Agarkov, V. V. Sinitsyn, S. I. Bredikhin and V. V. Kharton, J. Solid State Electrochem., 2012, 16, 2335 CrossRef CAS.
- M. K. Rath, B. G. Ahn, B. H. Choi, M. J. Ji and K. T. Lee, Ceram. Int., 2013, 39, 6343 CrossRef CAS PubMed.
- V. A. Kolotygin, E. V. Tsipis, M. F. Lü, Y. V. Pivak, S. N. Yarmolenko, S. I. Bredikhin and V. V. Kharton, Solid State Ionics, 2013, 251, 28–33 CrossRef CAS PubMed.
- D. P. Fagg, V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, E. N. Naumovich and J. R. Frade, J. Eur. Ceram. Soc., 2001, 21, 1831 CrossRef CAS.
- D. N. Miller and J. T. S. Irvine, J. Power Sources, 2011, 196, 7323 CrossRef CAS PubMed.
- G. Tsekouras, D. Neagu and J. T. S. Irvine, Energy Environ. Sci., 2013, 6, 256 CAS.
- S. Hui and A. Petric, Mater. Res. Bull., 2002, 37, 1215 CrossRef CAS.
- K. B. Yoo and G. M. Choi, ECS Trans., 2009, 25, 2259 CrossRef CAS PubMed.
- X. Li, H. Zhao, N. Xu, X. Zhou, C. Zhang and N. Chen, Int. J. Hydrogen Energy, 2009, 34, 6407 CrossRef CAS PubMed.
- S. H. Cui, J. H. Li, X. W. Zhou, G. Y. Wang, J. L. Luo, K. T. Chuang, Y. Bai and L. J. Qiao, J. Mater. Chem. A, 2013, 1, 9689 CAS.
- K. B. Yoo, B. H. Park and G. M. Choi, Solid State Ionics, 2012, 225, 104 CrossRef CAS PubMed.
- F. Napolitano, D. G. Lamas, A. Soldati and A. Serquis, Int. J. Hydrogen Energy, 2012, 37, 18302 CrossRef CAS PubMed.
- C. Arrivé, T. Delahaye, O. Joubert and G. Gauthier, J. Power Sources, 2013, 223, 341 CrossRef PubMed.
- J. C. Ruiz-Morales, J. Canales-Vázquez, C. Savaniu, D. Marrero-López, W. Zhou and J. T. S. Irvine, Nature, 2006, 439, 568 CrossRef CAS PubMed.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2011, 23, 1607 CrossRef CAS.
- C. Hatchwell, N. Bonanos and M. Mogensen, Solid State Ionics, 2004, 167, 349 CrossRef CAS PubMed.
- X. Li, H. Zhao, F. Gao, N. Chen and N. Xu, Electrochem. Commun., 2008, 10, 1567 CrossRef CAS PubMed.
- R. J. Gorte and J. M. Vohs, Curr. Opin. Colloid Interface Sci., 2009, 14, 236 CrossRef CAS PubMed.
- H. Y. Chang, S. H. Wang, Y. M. Wang, C. W. Lai, C. H. Lin and S. Y. Cheng, Int. J. Hydrogen Energy, 2012, 37, 7771 CrossRef CAS PubMed.
- X. Sun, S. Wang, Z. Wang, X. Ye, T. Wen and F. Huang, J. Power Sources, 2008, 183, 114 CrossRef CAS PubMed.
- M. D. Gross, J. M. Vohs and R. J. Gorte, Electrochem. Solid-State Lett., 2007, 10, B65 CrossRef CAS PubMed.
- G. Kim, M. D. Gross, W. Wang, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2008, 155, B360 CrossRef CAS PubMed.
- T. Kolodiazhnyi and A. Petric, J. Electroceram., 2005, 15, 5 CrossRef CAS PubMed.
- S. Lee, G. Kim, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2008, 155, B1179 CrossRef CAS PubMed.
- M. A. Buccheri and J. M. Hill, J. Electrochem. Soc., 2012, 159, B361 CrossRef CAS PubMed.
- C. D. Savaniu and J. S. Irvine, J. Mater. Chem., 2009, 19, 8119 RSC.
- K. B. Yoo and G. M. Choi, Solid State Ionics, 2009, 18, 867 CrossRef PubMed.
- K. B. Yoo and G. M. Choi, Solid State Ionics, 2011, 192, 515 CrossRef CAS PubMed.
- C. D. Savaniu, D. N. Miller and J. T. S. Irvine, J. Am. Ceram. Soc., 2013, 96, 1718 CrossRef CAS.
- C. D. Savaniu and J. T. S. Irvine, Solid State Ionics, 2011, 192, 491 CrossRef CAS PubMed.
- M. Roushanafshar, J. L. Luo, K. T. Chuang and A. R. Sanger, ECS Trans., 2011, 35, 2799 CrossRef CAS PubMed.
- M. Roushanafshar, J. L. Luo, A. L. Vincent, K. T. Chuang and A. R. Sanger, Int. J. Hydrogen Energy, 2012, 37, 7762 CrossRef CAS PubMed.
- M. R. Pillai, I. Kim, D. M. Bierschenk and S. A. Barnett, J. Power Sources, 2008, 185, 1086 CrossRef CAS PubMed.
- X. H. Zhang, J. Zhang, C. Yuan and E. J. Liang, Adv. Mater. Res., 2012, 347–353, 3325 Search PubMed.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, M. Shah, L. D. Marks and S. A. Barnett, J. Electrochem. Soc., 2010, 157, B279 CrossRef CAS PubMed.
- B. D. Madsen, W. Kobsiriphat, Y. Wang, L. D. Marks and S. A. Barnett, J. Power Sources, 2007, 166, 64 CrossRef CAS PubMed.
- L. Adijanto, V. B. Padmannabhan, R. Küngas, R. J. Gorte and J. M. Vohs, J. Mater. Chem., 2012, 22, 11396 RSC.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 17731, DOI:10.1038/NCHEM.
- S. P. Jiang, Int. J. Hydrogen Energy, 2012, 37, 449 CrossRef CAS PubMed.
- H. Birol, C. R. Rambo, M. Guiotoku and D. Hotza, RSC Adv., 2013, 3, 2873 RSC.
- R. Pelosato, C. Cristiani, G. Dotelli, M. Mariani, A. Donazzi and I. N. Sora, Int. J. Hydrogen Energy, 2013, 38, 480 CrossRef CAS PubMed.
- H. Okuyucu, H. Cinici and T. Konak, Ceram. Int., 2013, 39, 903 CrossRef CAS PubMed.
- X. Zhou, D. Zhan, C. Cong, G. Guo, L. Wang and K. Zhang, J. Mater. Sci., 2005, 40, 2577 CrossRef CAS PubMed.
- Y. J. Zhong, J. T. Li, Z. G. Wu, X. D. Guo, B. H. Zhong and S. G. Sun, J. Power Sources, 2013, 234, 217 CrossRef CAS PubMed.
- M. Liu, P. He, J. L. Luo, A. R. Sanger and K. T. Chuang, J. Power Sources, 2001, 94, 20 CrossRef CAS.
- P. He, M. Liu, J. L. Luo, A. R. Sanger and K. T. Chuang, J. Electrochem. Soc., 2002, 149, A808 CrossRef CAS PubMed.
- V. Vorontsov, W. An, J. L. Luo, A. R. Sanger and K. T. Chuang, J. Power Sources, 2008, 179, 9 CrossRef CAS PubMed.
- X. Zhu, Q. Zhong, D. Xu, H. Yan and W. Tan, J. Alloys Compd., 2013, 555, 169 CrossRef CAS PubMed.
- D. Xu, Y. Bu, W. Tan and Q. Zhong, Appl. Surf. Sci., 2013, 268, 246 CrossRef CAS PubMed.
- R. Mukundan, E. L. Brosha and F. H. Garzon, Electrochem. Solid-State Lett., 2004, 7, A5 CrossRef CAS PubMed.
- A. L. Vincent, J. L. Luo, K. T. Chuang and A. R. Sanger, Appl. Catal., B, 2011, 106, 114 CAS.
- J. D. Kirtley, D. M. Halat, M. D. McIntyre, B. C. Eigenbrodt and R. A. Walker, Anal. Chem., 2012, 84, 9745 CrossRef CAS PubMed.
- A. K. Huber, M. Falk, M. Rohnke, B. Luerβen, L. Gregoratti, M. Amati and J. Janek, Phys. Chem. Chem. Phys., 2012, 14, 751 RSC.
| This journal is © The Royal Society of Chemistry 2014 |