Fluorescent anion sensing by bisquinolinium pyridine-2,6-dicarboxamide receptors in water†
Alejandro
Dorazco-González
a,
Marcos Flores
Alamo
b,
Carolina
Godoy-Alcántar
c,
Herbert
Höpfl
c and
Anatoly K.
Yatsimirsky
*b
aCentro Conjunto de Investigación en Química Sustentable, UAEM-UNAM, Carretera Toluca-Atlacomulco Km 14.5, C. P. 50200, Toluca, Estado de México, México
bFacultad de Química, Universidad Nacional Autónoma de México, 04510 México D.F., México. E-mail: anatoli@unam.mx; Fax: +52 55 5616 2010; Tel: +52 55 5622 3813
cCentro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001, C.P. 62209 Cuernavaca, México
First published on 4th October 2013
Abstract
Dicationic N-methylated at quinolyl moieties derivatives of three isomers of N,N′-bis(quinolyl)pyridine-2,6-dicarboxamide, and respective N-methyl quinolinium benzamides as reference compounds, have been prepared and characterized by crystal structures, spectral and acid–base properties in water. First pKa values of dicarboxamides between 8.1 and 9.3 determined spectrophotometrically are unusually low for amides. Dicarboxamide derivatives of 3- (1) and 6-aminoquinoline (2) undergo efficient fluorescence quenching by halide, acetate, pyrophosphate and nucleotide anions but the derivative of 5-aminoquinoline (3) shows very small quenching effects. The shape of Stern–Volmer plots for dicarboxamides indicates the existence of ground state complexation with anions, which is absent for related benzamides. Association constants, KA, with anions were calculated from analysis of concentration profiles of the quenching effects on the fluorescence of 1 and 2. Quenching by nucleoside triphosphates is much more efficient than by inorganic anions. Efficient binding of even simple inorganic anions by neutral amide N–H donors in water is attributed to high acidity of amides and preorganized rigid structure of the receptors.
Introduction
Anion recognition by H-bond donor receptors is mostly restricted to non-aqueous media.1,2 This seriously limits their practical applications as sensors or selectors and for this reason the search for receptors capable to function in water is an important current trend in this area.3,4 Recently we reported highly efficient anion recognition by dicationic N-methylated at pyridyl moieties derivatives of N,N′-bis(pyridyl)pyridine-2,6-dicarboxamide in MeCN.5 Tight and selective anion binding by these receptors was achieved by the presence of two convergent and strongly acidified amide NH donors, which in principle may operate also in water. The positive charge of receptors is strongly delocalized and does not contribute significantly to the observed affinity even in low polar acetonitrile solvent,5 but makes the receptors sufficiently water soluble. These features of the system prompted us to test their anion recognition properties in water. In order to have receptors with more advanced optical properties potentially suitable for sensor applications we have prepared for this study the analogous compounds 1–3 with fluorescent quinolinium instead of pyridynium groups, Scheme 1. Also, a series of respective quinolinium benzamides 4–6 was prepared as reference compounds lacking the bisamide potential anion binding site.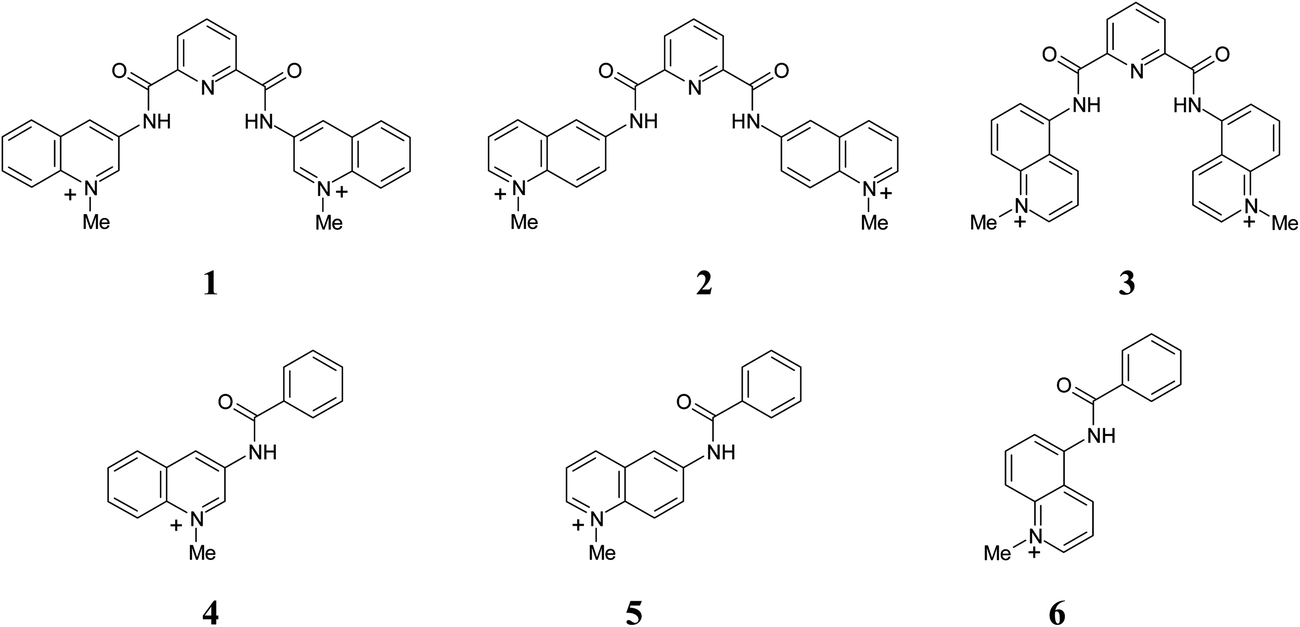 | ||
| Scheme 1 Pyridine-2,6-dicarboxamide receptors and respective N-quinolyl benzamides employed in this study. | ||
Among these receptors compound 1 was reported previously as selective G-quadruplex ligand.6 It also was structurally characterized and studied as an anion receptor in MeCN where it showed high selectivity toward chloride.5 The purpose of preparing additional isomeric compounds 2 and 3 with differently positioned N–Me+ center in respect to the amide group was to modify the electronic and optical properties of the receptors in order to optimize their function.
Quinolinium derivatives find many applications in sensing and molecular recognition. They are used as intercalators,7 fluorescent pH-sensors,8,9 signaling components of receptors for antibiotics,10 proteins,11 heparin,12 saccharides,13 fluoride and cyanide ions.14 Perhaps the most important application is using them as fluorescent indicators for measurement of intracellular chloride concentration based on dynamic quenching of quinolinium fluorescence by chloride.15,16 Both selectivity and sensitivity of this method could be improved by adding to quinolinium ions the receptor properties toward chloride. Recently reported tripodal tris-quinolinium receptors designed for this purpose indeed show markedly increased quenching by anions in MeCN,17,18 but the effect disappears in water,18 and this is what typically happens with hydrogen-bonding receptors when you attempt to use them in aqueous solutions.
In this paper we demonstrate that receptors 1–3 conserve reduced, but still noticeable, affinity to anions in water with the same selectivity to chloride among halide anions as in MeCN and show fairly strong binding to nucleoside triphosphate anions. The positional isomers appeared to possess significantly different conformations strongly affecting both spectral and recognition properties of the receptors.
Results and discussion
Structures and acid–base properties of receptors
Iodide salts of 1–3 were prepared by reacting 2,6-pyridinedicarbonyl dichloride with respective isomers of aminoquinoline and subsequent prolonged treatment with MeI under reflux. The respective triflates were obtained by treatment with silver triflate in MeCN. Compounds 4–6 were prepared similarly with benzoyl chloride instead of 2,6-pyridinedicarbonyl dichloride as the acylating reagent.Crystal structures were determined for the triflates of 2, 3, 4, 6, and iodide of 5 (Table 1S, ESI†). The crystal structures of dicationic receptors 1–3 are shown in Fig. 1. In all dications amide groups are coplanar with the central pyridine ring and their N–H bonds are turned inside the cleft toward each other. The distances between the protons increases from 2.953 Å in 3 to 3.131 Å in 1 and to 3.207 Å in 2. Counter-ion binding involves multiple short contacts/H-bonds with N–H and C–H donors in the case of 1, only N–H donors for 3, and in the case of 2, the cleft between amide groups is filled with a cluster of 3 water molecules to which the counter-ions are bound. The properties of amide group are essentially the same in all three isomers with average C–N bond length 1.356 ± 0.008 Å corresponding to the length of Csp2–Nsp2 bond 1.355 Å.19 The most significant structural difference in this series of receptors is the progressive loss of co-planarity between the central pyridine dicarboxamide structure and N-methyl quinolinium rings. In compound 1 the angles between the pyridine plane and the planes of left and right quinolinium rings are 9.6° and 11.52° respectively; they become 38.61° and 11.47° in 2 and finally 39.48° and 31.12° in 3. The binding of anions in these receptors is expected to occur via hydrogen bonding to N–H proton donors of amide groups and to ortho-C–H donors of cationic aromatic rings.5 Obviously the C–H bonds have the optimum orientation for intracavity anion binding when the quinolinium rings are co-planar with the pyridine dicarboxamide fragment as in 1, which is therefore the most preorganized receptor for small spherical anions like chloride.
 | ||
| Fig. 1 Perspective views of the molecular structures of triflates of 1–3. The structure of 1 was reported previously5 and is shown for comparison with other isomers. | ||
This loss of co-planarity seems to be principally electronic rather than steric effect. This conclusion is supported by comparison with crystal structures of simple benzamides 4–6 as shown in Fig. 2. Like in the case of dicarboxamides, quinolinium rings are progressively less co-planar with amide groups on going from 3- to 6- and to 5-aminoquinoline isomers. Thus the angles between the planes of amide and quinolinium groups are 11.21° for 4, 24.50° for 5 and 32.66° for 6. Such variability is not unusual for benzamides: dihedral angles between aromatic groups in reported structures of N-aryl benzamides vary from nearly 0 to as large as 80°, although no general trend has been established yet.20
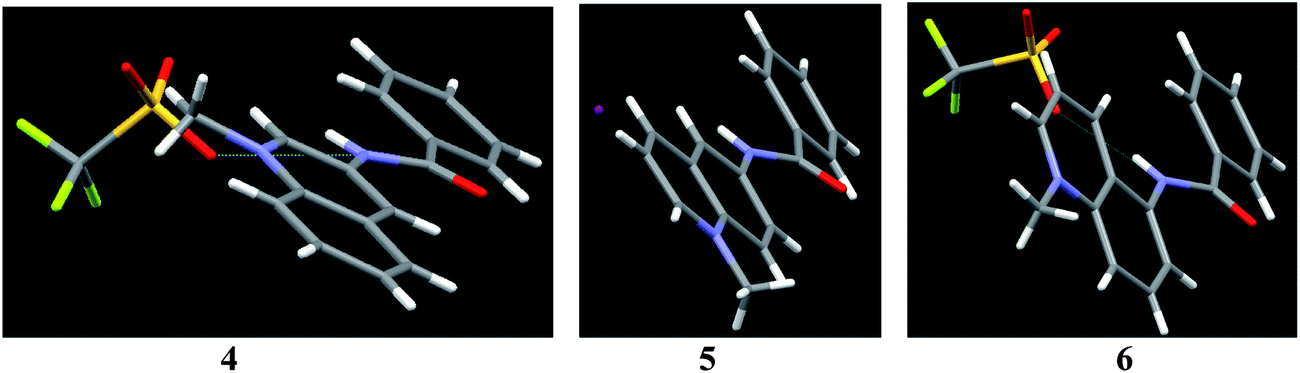 | ||
| Fig. 2 Perspective views of the molecular structures of triflates of 4 and 6 and of iodide of 5. | ||
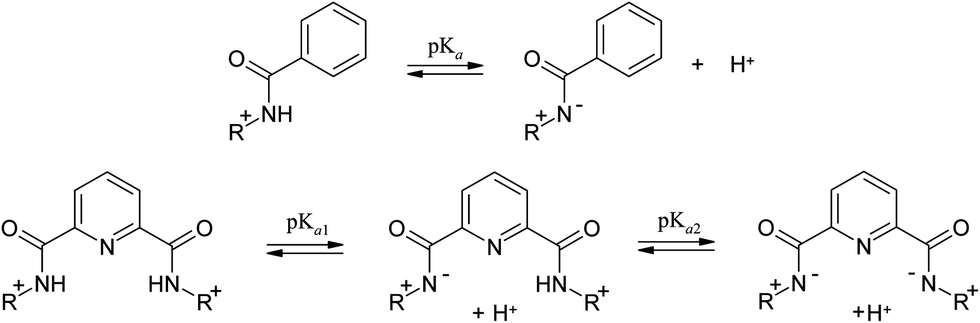
The acid dissociation constants of 1–6 were determined by UV-vis and fluorometric pH-titrations. Absorption maxima of 1–6 at pH 6.5 collected in Table 1 (Fig. 1S in ESI† shows the complete spectra) are in the range typically observed for quinolinium salts.8 A long wavelength absorption band around 340–350 nm observed in some of compounds 1–6 as a shoulder is attributed to n → π* transition.8 On increasing pH the absorbance at shorter wavelengths decreases while that at longer wavelengths it increases for all compounds. Fig. 3 shows as an example of the results for 1 and 4. In the case of 4 all spectra pass through four clear isosbestic points indicating the co-existence of only two differently absorbing species, initial cationic amide and its deprotonated zwitter-ionic form (Scheme 2). In the case of 1 there are no strict isosbestic points indicating more than two differently absorbing species at equilibrium. Indeed, for a bisamide one expects two consecutive deprotonation processes (Scheme 2) generating differently absorbing mono- and diprotonated forms.
λ
abs (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
λ em | UV-vis titrations | Fluorometric titrations | |||
|---|---|---|---|---|---|---|
| pKa1 | pKa2 | pKa1 | pKa2 | |||
| a Ref. 5. | ||||||
| 1 | 275 (4.59); 344 (4.10) | 403 | 9.17a | 11.5a | 7.61(4) | 9.41(6) |
| 2 | 270 (4.54); 342 sh (3.85) | 453 | 8.1(1) | 11.18(2) | 7.55(2) | 10.25(5) |
| 3 | 239 (4.71); 319 (4.08); 340 sh (3.64) | 515 | 9.29(8) | 11.40(5) | 7.31(4) | 9.59(8) |
| 4 | 243 (4.50); 270 (4.52); 347 (3.87) | 405 | 10.56(3) | 10.33(2) | ||
| 5 | 268 (4.54); 324 (3.76); 350 sh (3.62) | 461 | 12.25(2) | 7.22(8) | 11.82(9) | |
| 6 | 239 (4.54); 319 (3.86); 340 sh (3.63) | 533 | 10.65(3) | 10.63(2) | ||
 | ||
| Fig. 3 UV-vis pH titration of 0.02 mM 1 (a) and 4 (b). | ||
 | ||
| Scheme 2 Acid dissociation equilibria for mono and bisamide compounds. | ||
Insets in Fig. 3a and b illustrate the fitting of the profiles of absorbances at a single wavelength vs. pH to the respective theoretical equations and Table 1 collects the pKa values calculated from the fitting procedure. Monoamides 4–6 are more acidic than ordinary amides due to strong electron accepting effect of the quinolinium group. Calculated with ACD software,21 pKa values 8.89; 10.84 and 10.14 for 4, 5 and 6, respectively, are not too far from the experimental values and follow a similar trend 4 < 6 < 5. First pKa values of dicarboxamides 1–3 are even lower due to stronger electron accepting properties of pyridyl as compared to phenyl group and to a statistical factor. Second pKa values expectedly are higher, but still are rather low for amide groups.
Fluorescence spectra of 1–6 are shown in Fig. 4a and the respective emission maxima are given in Table 1. In the series of benzamides the emission maxima are shifted to longer wavelengths with concomitant decrease in the fluorescence intensity on going from 4 to 5 and to 6. The latter can be attributed to the progressive increase in the dihedral angle between quinoline and amide groups causing increased intramolecular charge transfer quenching. A similar although less pronounced trend is observed for dicarboxamides 1–3.
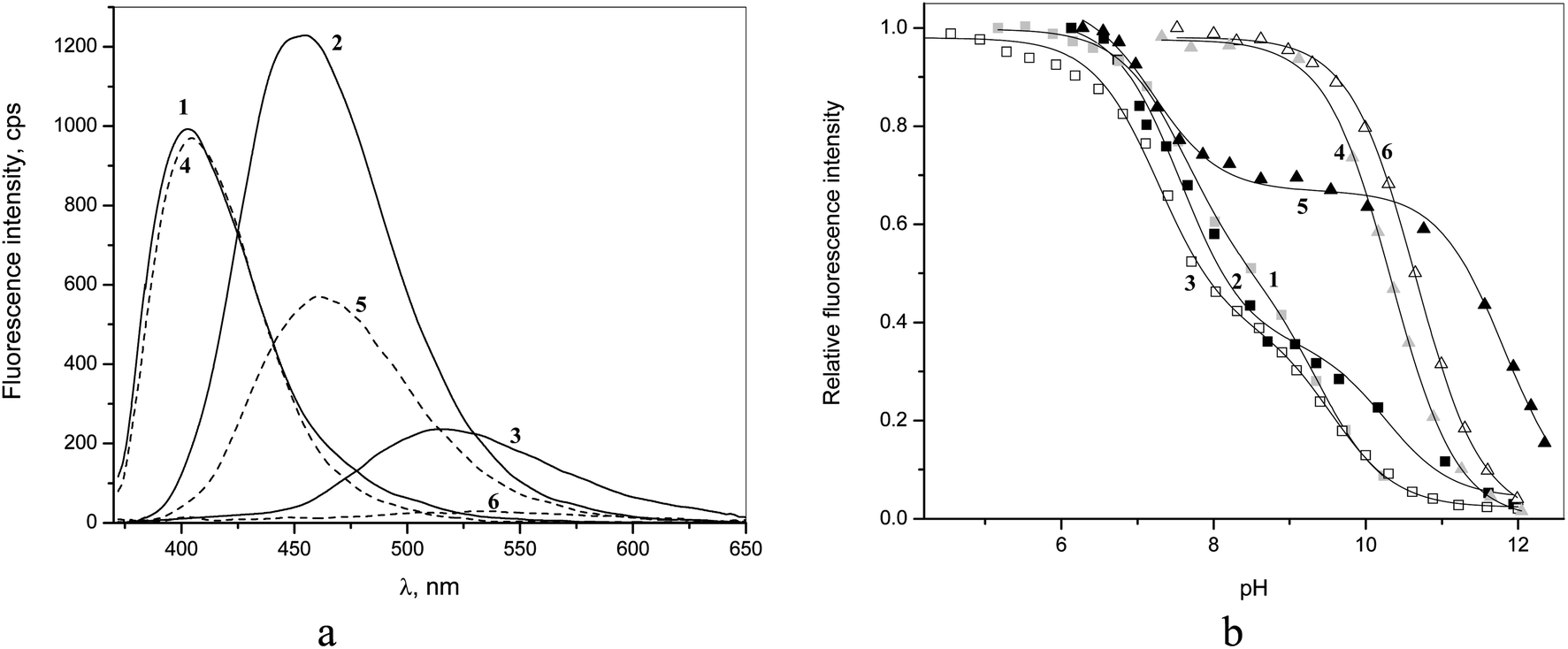 | ||
| Fig. 4 (a) Fluorescence emission spectra of 5 μM 1–6 at pH 6.5 (excitation at 350 nm); (b) pH-titration profiles observed at emission maximum of each compound. | ||
The pH-profiles of fluorescence intensity are shown in Fig. 4b. For monoamides 4 and 6 one observes essentially the same pKa values as from spectrophotometric titrations (Table 1), but in the case of 5 besides one pKa in basic range of pH, which is close to spectrophotometrically determined pKa, one observes an additional 4.6 units lower pKa in a neutral solution (Table 1). Such a situation indicates deprotonation of the excited state of the fluorophore, which is a stronger acid than its ground state.22 This second pKa cannot be considered as a true excited state pKa* however because in neutral solutions the concentration of protons is too low to protonate back the deprotonated form of the excited state during its lifetime and the equilibrium cannot be achieved.22 Thus the observation of a second reduced pKa in fluorometric titration profile just indicates a significant acidification of 5 in the excited state leading to its partial quenching due to the excited state proton transfer. In all cases the deprotonated forms of the amides are not fluorescent.
The fluorometric titrations of bisamides 1–3 are consistent with two consecutive pKa values, which are approximately 2 units smaller than those determined by spectrophotometric titrations (Table 1) indicating the excited state acidification of amide bonds for all isomers. The assignment of these apparent pKa values is rather uncertain. The only clear conclusion is that below pH 7 all receptors are in dicationic non-dissociated form both in ground and excited states and for this reason their interactions with anions were studied at pH 6.5.
Anion quenching and complexation studies
Interactions of receptors 1–6 with anions were studied by fluorometric titrations because the spectrophotometric response to anion binding was very weak. As a first step simple inorganic anions were tested. No interaction was observed with sulfate, nitrate, phosphate and perchlorate anions at concentrations up to 0.05 M. Halide anions, pyrophosphate (PPi) and acetate in most cases induced noticeable quenching.Quenching effects of inorganic anions and acetate on the fluorescence of 1 and 4 (inset) are shown in Fig. 5a in Stern–Volmer coordinates (I0/I vs. quencher concentration, where I0 and I are the fluorescence intensities in the absence and in the presence of the quencher, respectively). For the monoamide 4, simple linear plots with the Stern–Volmer constant KSV given in Table 2 were observed. The KSV values follow the typical for quinolinium salts in the order F− ≪ Cl− < Br− < I− > AcO−.23 The plot for iodide shows a very small upward curvature (see Fig. 2S in ESI† for an extended plot) observed when both dynamic and static quenching contribute simultaneously to the observed effect.24 The respective equation for the quenching effect takes in this case the form (1) where [Q] is the quencher concentration and KA is the ground state association constant of the quencher with the fluorophore.
| I0/I = (1 + KSV[Q])(1 + KA[Q]) | (1) |
| Anion | 1 | 2 | 3 | 4 | 5 | 6 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| K A | K SV | ρ | K A | K SV | ρ | K SV | K SV | K SV | K SV | |
| a K A = 7.2 M−1. b K A = 106 M−1, ρ = 0.81. | ||||||||||
| F | 0.3(1) | 4.4 | 3.6 | 0 | 6.1 | 2.5 | ||||
| Cl | 120 | 12 | 0.64 | 110 | 6.3 | 0.57 | 0 | 17 | 2.4 | 0 |
| Br | 52 | 1000 | 150 | 0.68 | 18 | 34 | 57 | 0.93 | ||
| I | 18 | 87 | 180 | 130 | 36a | 100 | 21 | |||
| PPi | 300 | 11 | 0.62 | 690 | 6 | 0.65 | 0 | 21 | 4.9b | 0 |
| AcO | 210 | 4.8 | 0.82 | 510 | 13 | 0.68 | 7.4 | 5.0 | 12 | 3.0 |
 | ||
| Fig. 5 Stern–Volmer plots for quenching of fluorescence by inorganic anions and acetate at pH 6.5 (a) for 1 and 4 (inset) (excitation at 350 nm, emission at 400 nm), (b) for 2 and 5 (inset) (excitation at 350 nm, emission at 400 nm). | ||
Fitting of the plot in Fig. 5a for 4 and I− gives a small association constant KA = 7.2 M−1, which can be attributed to weak ion pairing of iodide with the quinolinium cation by analogy with known weak association of voluminous anions with quaternary ammonium cations in water driven by the hydrophobic effect.25 In addition there may be some contribution from a charge-transfer interaction reported for I− and N-alkylpyridinium cations.26
The quenching of 1 by Br− and I− has the same characteristics as that of 4. With Br− one observes a simple linear plot with increased KSV and with I− an upwardly curved plot, which follows eqn (1) with also increased KSV and KA values. All constants increase by a factor of about 2, which can be attributed to a statistical factor since 1 contains two quinolinium groups. Interactions with pyrophosphate (PPi), Cl− and AcO− show different features, however (Fig. 2S in ESI† shows superimposed plots for 1 and 4, which makes the differences more evident). The quenching plots are downwardly curved at low concentrations, but become linear at higher concentrations of anions. The profiles of this type are often observed for compounds possessing fluorophores of different accessibility to the quencher, e.g., tryptophan residues in different positions in a protein,24 but for a single fluorophore it indicates simultaneous dynamic and static quenching when the ground state complexation does not quench the fluorescence completely and the fluorophore–quencher complex still possesses some reduced by a factor ρ fluorescence intensity.17 The respective equation takes the form of eqn (2).17 Parameters found by fitting the results to eqn (2) are given in Table 2.
 | (2) |
Eqn (2) is approximate because it assumes similar quenching with a single KSV value for both free and complexed forms of the fluorophore as well as equal rates of photoexcitation of free and complexed forms. A more exact equation can be found in ref. 27 but it contains larger number of adjustable parameters, which cannot be reliably determined from the fitting of plots like those shown in Fig. 5. Another limitation of eqn (2) is that when ρ = 1 it takes the form of classical Stern–Volmer eqn (3) and the association constant cannot be calculated. In practice we found that already with ρ > 0.9 the fitting to eqn (2) gives very large relative errors in KA and KSV exceeding 100% and in such cases (e.g., quenching of 1 by Br−) the results were fitted to eqn (3). Thus the association constant can be reliably calculated only if the static quenching is sufficiently strong, that is more than by 10%.
| I0/I = (1 + KSV[Q]) | (3) |
The dynamic quenching of quinolinium salts by anions reflected in the Stern–Volmer constant takes place through various mechanisms, such as intersystem crossing and charge-transfer quenching.28,23 The static quenching is related to complex formation of anions with dicarboxamide receptors, which occurs via hydrogen bonding to NH and aromatic CH proton donors,5 as outlined in Scheme 3. It has been proposed for several analogous systems that bind in such a way that anions induce quenching by promoting the intramolecular photoinduced electron transfer (PET) from the electron donor atoms of a substituent involved in hydrogen bonding to the anion to the excited state of the fluorophore.29 The effect can be interpreted as a consequence of increased negative charge on the amide groups due to the shift of the proton inside the anion-HN hydrogen bond toward the anion and is in line with the observation of total fluorescence quenching induced by complete deprotonation of the receptor (see above). Also typical for this mechanism is a very small complexation-induced change in the absorption spectrum of the receptor.29 The efficiency of this complexation-induced quenching is expressed in terms of parameter ρ in eqn (2): the smaller ρ values correspond to a stronger quenching. A general mechanism of complexation and quenching phenomena in this system is outlined in the Scheme 3.
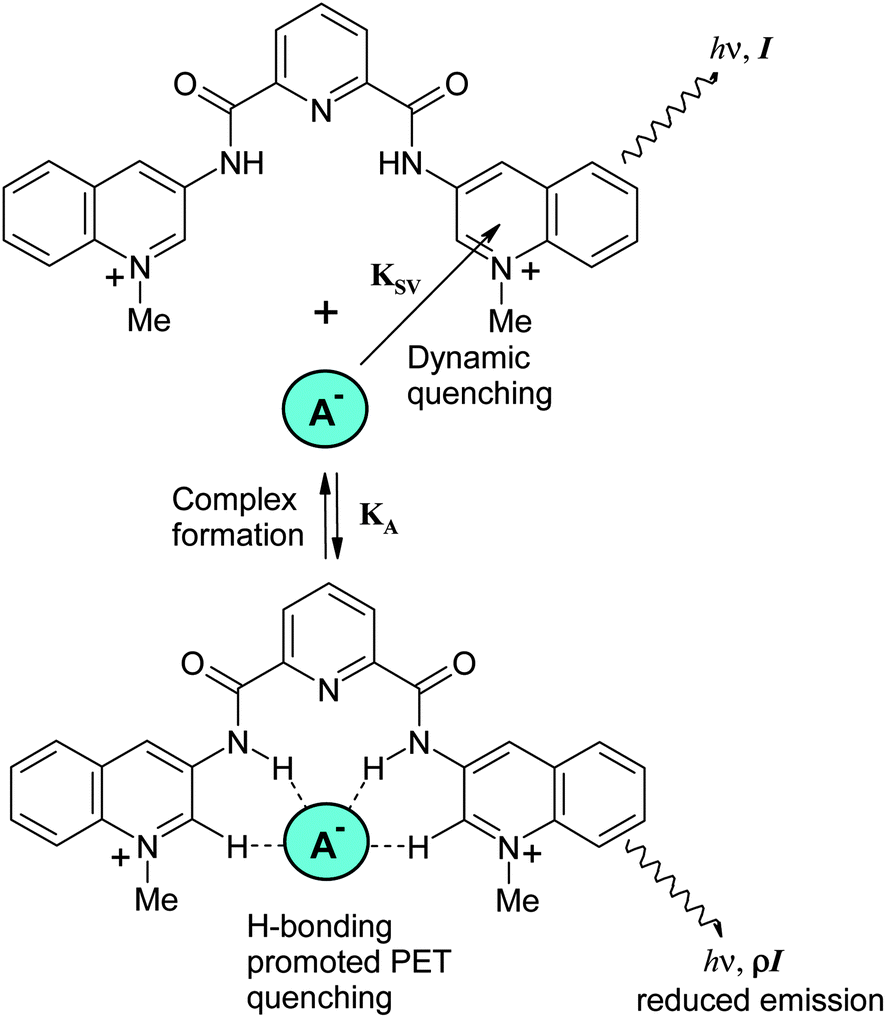 | ||
| Scheme 3 Quenching mechanism of dicarboxamide receptors by anions. | ||
Fig. 5b shows similar data for quenching of 2 and 5. Again quenching of the monoamide 5 follows simple linear dependence, but for the dicarboxamide 2 one observes with Cl−, Br−, AcO− and PPi non-linear plots indicative of complexation with these anions. With the most basic PPi some curvature is observed even for 5. Receptor 2 differs from 1 in two aspects: it is more acidic (see Table 2) and has a larger and more open cavity (see Fig. 1). In line with these features it forms more stable complexes with basic acetate and pyrophosphate anions and among halides prefers larger bromide over smaller chloride anion.
The behavior of the last isomer 3 was rather unexpected. It does not show any quenching by Cl− and PPi, very weak quenching by Br− and no indication of complex formation with anions (Fig. 3Sa, Table 2S, ESI†). Since its monoamide analog 6 also shows very weak anion quenching (Fig. 3Sb, Table 2S, ESI†), this behavior seems to be caused by electronic rather than possible conformational effects.
Previously reported anion complexation by 1 in MeCN shows a similar selectivity (F− < Cl− > Br− ∼ I− < AcO−), but much higher affinity with KA values in the range of 104–106 M−1.5 However, being rather modest the association constants of monoanions with 1 and 2 in water are significantly larger and show different trends than those reported for these anions with simple organic dications such as diprotonated polyamines.30 For instance, KA values or F−, Cl− and AcO− with diprotonated ethylenediamine, 1,4-diaminobutane and spermidine extrapolated to zero ionic strength show little variation with the polyamine structure and are equal on average to 100, 4 and 20 M−1. Binding of Br− and I− was not detected. These data correspond to a “normal” order of affinity F− > AcO− > Cl− > Br− > I− determined by size and basicity of anions, but with receptors 1 and 2 we see much higher binding constants with clear peak selectivity for Cl− and Br− among halides, which have a similar affinity as acetate. More basic and highly negatively charged pyrophosphate forms expectably very stable complexes with dications of polyamines with KA values about 105 M−1,30 but has just a bit larger affinity to 1 and 2 than acetate clearly illustrating the minor contribution of simple Coulombic attraction to association free energy with these receptors. Thus binding of anions to dicarboxamide receptors is governed by the amount to which they complement the receptor cleft rather than by electrostatics.
Relatively high affinity of receptors 1 and 2 to PPi prompted us to study interaction of these receptors with nucleotide triphosphates. For comparison also di- and monophosphates were included. The results are shown in Fig. 6a and b.
 | ||
| Fig. 6 Stern–Volmer plots for quenching of the fluorescence by nucleotides and adenosine at pH 6.5 (a) for 1 and 4 (inset), (b) for 2 and 5 (inset). | ||
The quenching efficiency of nucleotides toward dicarboxamide receptors 1 and 2 is much larger than that of similarly charged pyrophosphate and also it is much larger than that toward monoamides 4 and 5. Another important aspect is that for quenching of 1 and 2 one observes non-linear Stern–Volmer plots indicative of complex formation, but the quenching of 4 and 5 follows a linear dependence. These qualitative observations clearly show the importance of both dicarboxamide structure of the receptor and the presence of a nucleobase for nucleotide recognition. For the quantitative analysis the results for 1 and 2 were fitted to the eqn (2) and the fitting parameters are collected in Table 3 together with KSV values for receptors 4 and 5.
Analyzing the results for monoamides 4 and 5 one observes that quenching by adenine nucleotides is approximately twice as strong as that by adenosine. This effect can be attributed to electrostatic attraction of nucleotides to cationic fluorophore. The quenching efficiency for nucleotides with different nucleobases follows the order G > A > C. Quenching by nucleobases proceeds via PET mechanism31 and the relative quenching efficiency of nucleobases depends on the direction of the electron transfer:31 when the nucleobase serves as the electron acceptor the order is C > A > G, but when the excited state of the fluorophore serves as the electron acceptor, which is more likely for quinolinium fluorophores, the order is G > A > C as observed in our case. The ground-state association often observed between nucleosides or nucleotides with aromatic compounds is not detected at concentrations below 10 mM employed in these experiments.
The dicarboxamide receptor 1 undergoes much stronger quenching by nucleotides as compared to monoamide 4. For instance, 5 mM ATP reduces the fluorescence intensity of 4 by a factor of 1.4, but the same effect with 1 is observed already with 0.1 mM ATP. However, the dynamic quenching of 1 occurs with approximately the same efficiency, as is evident from comparison of KSV values for both receptors. The enhanced quenching can be attributed therefore to association between 1 and nucleotides inducing strong static quenching. It follows from eqn (2) that the initial slope of the plot of I0/I vs. [Q] at low quencher concentration is given by eqn (4).
| d(I0/I)/d[Q] = KSV + KA(1 − ρ) | (4) |
Therefore with KA = 5400 M−1 and ρ = 0.25 for ATP the additional contribution to quenching will be equal to 4000 M−1, which represents a 40-fold increase in the quenching efficiency as compared to that for 4. The nucleobase selectivity both in dynamic and static quenching of 1 is the same as in the case of 4, G > A > C. In the series of adenine nucleotides the quenching efficiency decreases in the order ATP > ADP > AMP. For AMP the Stern–Volmer plot is linear, but significantly larger KSV for AMP than for adenosine as well as for AMP and 4 indicates that some binding between AMP and 1 should occur.
To prove that the observed association is a true ground state reaction and KA found from fluorescence data is not affected by possible excited state association,27 the binding constant of ATP to 1 was determined also spectrophotometrically (Fig. 4S, ESI†) and a reasonably close value of KA = 6100 ± 400 M−1 was obtained. The binding constant for ATP is ca. 20 times larger and ρ value is 2.5 times smaller than those for pyrophosphate. Both stronger binding and stronger static quenching effect can be explained if adenine nucleobase can interact with quinolinium rings of receptor simultaneously with trapping of nucleotide phosphate by dicarboxamide cleft as sketched in Scheme 4. A bent conformation of ATP with terminal phosphate positioned near adenine required for this binding mode was found in some ATP–protein complexes.32 In terms of this model higher quenching efficiency of ATP as compared to ADP and AMP can result from better fit of terminal phosphate and nucleobase to the respective sites of the receptor allowed by the longer polyphosphate chain of ATP.
 | ||
| Scheme 4 Proposed binding mode of ATP to dicarboxamide receptors. | ||
Similar behavior was observed with 2. Spectrophotometrically determined KA = 7200 ± 600 M−1 for ATP is again reasonably close to KA found from fluorescence results (Table 3). We used this more soluble in water receptor also for 1H and 31P NMR studies of interaction with ATP. Fig. 7a shows the aromatic region of 1H NMR spectra of 2, ATP and their mixture. Evidently signals of both components undergo significant up-field shifts by 0.2–0.6 ppm in the mixture indicating mutual contact of aromatic groups of host and guest. A similar picture was observed, e.g., in 1H NMR spectra of ATP complexes with acridine bearing macrocyclic polyamine receptors.33
 | ||
| Fig. 7 (a) The aromatic region of 1H NMR spectra of 2 mM 2 (top), and ATP (bottom, red peaks) and their 1:1 mixture at pH 6.5 in D2O; (b) 31P NMR spectra of 1 mM ATP alone (bottom) and in the presence of 1 equivalent of 2 in D2O at pH 6.5. | ||
Fig. 7b illustrates titration of ATP by 2 monitored by 31P NMR. The signal, which belongs to terminal γ-phosphate group of ATP undergoes a characteristic down-field shift observed in ATP complexes with polycations34,35,36a and attributed to a decreased degree of protonation of the phosphate group (at pH 6.5 ATP is present in a monoprotonated form) in the complex.34b Thus results of NMR titrations confirm the binding mode proposed in Scheme 4.
There are many examples of receptors for nucleotide recognition containing aromatic groups linked to protonated polyazamacrocyles36 or other polycations37 operating through simultaneous stacking interactions of nucleobases and electrostatic binding of phosphate moiety. A new feature of receptors 1 and 2 is that they provide for the binding of phosphate moiety neutral amide N–H donors working in water. Association constants of about 5 × 103 M−1 found with these receptors are of the same order of magnitude as reported for, e.g., tetracationic anthracene derivatives37 or protonated azamacrocycles with incorporated dipyridine or terpyridine fragments.36d
Experimental section
Materials
Solvents were purified and dried using standard procedures. Other reagents and components of buffer solutions MES, MOPS and CHES all from Aldrich, were used as supplied. Buffer solutions were prepared by adjusting pH of 0.01 M free acids with concentrated NaOH to desired values.N,N′-Di(3-N′′-methylquinolinium)pyridine-2,6-dicarboxamide trifluoromethanesulfonate (1)
The compound was synthesized as was reported previously.5N,N′-Di(6-N′′-methylquinolinium)pyridine-2,6-dicarboxamide trifluoromethanesulfonate (2)
A mixture of 6-amino-quinoline (1.4 g, 9.50 mmol) and 2,6-pyridinedicarbonyl dichloride (1.00 g, 4.75 mmol) in 40 mL of dry toluene was stirred under reflux for 5 h. The yellow precipitate was collected by filtration and washed with acetone and 5% NaHCO3 solution to give N,N-bis-(6-quilonyl)pyridine-2,6-dicarboxamide (1.86 g) in 90% yield, which was reacted with 10 equiv. of CH3I in DMF–acetone (1:1 v/v, 50 mL) for 1 day. The resulting yellow powder was filtered and washed with cold MeOH to give the iodide salt. The iodide salt (0.30 g, 0.42 mmol) was dissolved in 200 mL of hot H2O then 2 equiv. of silver triflate (0.22 g, 0.85 mmol) were added. The mixture was stirred overnight at room temperature. The precipitate was filtered off and the solvent was removed under reduced pressure to produce the product (0.28 g) in 90% yield. 1H NMR (300 MHz, DMSO-d6) δ 11.64 (s, 2H), 9.42 (d, J = 5.6 Hz, 2H), 9.34 (d, J = 8.4 Hz 2H), 9.22 (s, 2H), 8.67 (m, 4H), 8.55 (d, J = 7.6 Hz, 2H), 8.44 (t, J = 7.6 Hz, 1H); 8.18 (dd, J = 5.7 Hz, 2H), 4.67 (s, 6H); 13C NMR (75 MHz, DMSO-d6) δ 162.5, 148.6, 148.2, 146.4, 140.6, 138.8, 135.4, 130.2, 129.7, 126.2, 122.8, 122.5, 120.1, 117.6, 45.3; MS (FAB, m/z) 598 [M + Tf]+; IR (KBr) 3330, 3088, 1685, 1539, 1251, 1223, 1154 cm−1; anal. cal. for C29H23F6N5O8S2 (747.643): C, 46.59; H, 3.10; N, 9.37. Found: C, 46.51; H, 3.12; N, 9.34.
N,N′-Di(5-N′′-methylquinolinium)pyridine-2,6-dicarboxamide trifluoromethanesulfonate (3)
It was obtained following the same procedure as for 2 from 5-amino-quinoline instead of 6-amino-quinoline with 70% yield. 1H NMR (300 MHz, DMSO-d6) δ 11.78 (s, 2H), 9.57 (d, J = 5.5 Hz, 2H), 9.46 (d, J = 8.6 Hz, 2H), 8.53 (m, 4H), 8.42 (m, 3H), 8.25 (m, 4H), 4.69 (s, 6H); 13C NMR (75 MHz, DMSO-d6) δ 163.0, 150. 7, 148.0, 143.7, 140.4, 138.9, 135.4, 135.1, 127.0, 126.1, 125.5, 122.7, 121.4, 118.5, 117.5, 45.9; MS (FAB, m/z) 598 [M + Tf]+; IR (KBr) 3275, 3097, 1684, 1517, 1241, 1160, 1027 cm−1; anal. cal. for C29H23F6N5O8S2 (747.64): C, 46.59; H, 3.10; N, 9.37. Found: C, 46.57; H, 3.11; N, 9.35.N-(3-N′-methylquinolinium)benzamide trifluoromethanesulfonate (4)
A mixture of 3-aminoquinoline (1.0 g, 6.9 mmol) and benzoyl chloride (0.49 g, 3.45 mmol) in 30 mL of dry toluene was stirred under reflux for 3 h. The white precipitate was collected by filtration and washed with acetone and 5% NaHCO3 to give N-(3-quinolinyl)benzamide in 92% yield, which was reacted with 3 equiv. of CH3I in DMF–acetone (1:1 v/v, 50 mL) for 1 day. The resulting yellow powder was filtered and washed with cold MeOH–H2O to give the iodide salt. The iodide salt (0.39 g, 1.0 mmol) was dissolved in 100 mL of hot H2O then one equiv. of silver triflate (0.26 g, 1.0 mmol) was added, the mixture was stirred overnight at room temperature. The precipitate was filtered off and the solvent was removed under reduced pressure to produce the product (0.38 g) in 92% yield. 1H NMR (300 MHz, DMSO-d6) δ 11.32 (s, 1H), 9.87 (s, 1H), 9.38 (s, 1H), 8.48 (d, J = 8.7 Hz, 2H), 8.18 (t, J = 6.2 Hz, 1H), 8.08 (d, J = 7.0 Hz, 2H), 8.04 (m, 1H), 7.67 (m, 3H), 4.7 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 166.2, 144.8, 135.4, 134.1, 133.7, 133.3, 133.0, 132.8, 130.2, 129.8, 129.1, 128.8, 127.9, 122.8, 119.0, 118.5, 46.2; MS (FAB, m/z) 263 [M]+; IR (KBr) 3343, 3098, 1681, 1550, 1373, 1262, 1151 cm−1; anal. cal. for C18H15F3N2O4S (412.4): C, 52.43; H, 3.67; N, 6.79. Found: C, 52.38; H, 3.69; N, 6.75.
N-(6-N′-methylquinolinium)benzamide trifluoromethanesulfonate (5)
It was obtained following the same procedure as for (4) from 6-aminoquinoline instead 3-aminoquinoline. The yields were 93%. 1H NMR (300 MHz, DMSO-d6) δ 10.97 (s, 1H), 9.35 (d, J = 5.7 Hz, 1H), 9.24 (d, J = 8.5 Hz, 1H), 9.02 (s, 1H), 8.51 (dd, J = 9.0 Hz, 2H), 8.11 (t, J = 5.9 Hz, 1H), 8.04 (d, J = 6.7 Hz, 2H), 7.61 (m, 3H), 4.60 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 166.4, 148.2, 146.3, 140.0, 132.3, 130.2, 129.3, 128.6, 127.9, 122.8, 122.3, 120.0, 118.5, 116.9, 45.2, MS (FAB, m/z) 263 [M]+; IR (KBr) 3345, 3074, 1678, 1555, 1251, 1237, 1226, 1160 cm−1, anal. cal. for C18H15F3N2O4S (412.4): C, 52.43; H, 3.67; N, 6.79. Found: C, 52.43; H, 3.68; N, 6.75.N-(5-N′-methylquinolinium)benzamide trifluoromethanesulfonate (6)
It was obtained following the same procedure as for (4) from 5-aminoquinoline instead 3-aminoquinoline. The yields were 87%. 1H NMR (300 MHz, DMSO-d6) δ 10.97 (s, 1H), 9.51 (d, J = 5.5 Hz, 1H), 9.27 (d, J = 8.7 Hz, 1H), 8.51 (d, J = 9.0 Hz, 1H), 8.32 (t, J = 7.6 Hz, 1H), 8.13 (m, 4H), 7.64 (m, 3H), 4.66 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 166.7, 150.4, 143.5, 138.8, 136.4, 135.0, 133.6, 132.3, 128.5, 128.1, 126.2, 125.2, 122.8, 121.1, 118.5, 116.6, 45.8; MS (FAB, m/z) 263 [M]+; IR (KBr) 3298, 3114, 1675, 1517, 1358, 1247, 1224, 1148 cm−1; anal. cal. for C18H15F3N2O4S (412.4): C, 52.43; H, 3.67; N, 6.79. Found: C, 52.48; H, 3.69; N, 6.76.X-ray crystallography
Crystals of the triflate salts of all compounds suitable for X-ray diffraction were grown by slow evaporation from the MeOH–water solutions (1:1 vol) except for the iodide salt 5 that was grown from DMF solution.
Single-crystals of 2, 3 and 4 were studied with Oxford Diffraction Gemini “A” diffractometer with a CCD area detector (λCuKα = 1.5418 Å, monochromator: graphite) source equipped with a sealed tube X-ray source. Unit cell constants were determined with a set of 15/3 narrow frame/runs (1° in ω) scans. Data sets consisted of 1201, 671 and 1193 frames of intensity data collected for 2, 3 and 4, respectively, with a frame width of 1° in ω, and a crystal-to-detector distance of 55.00 mm. The double pass method of scanning was used to exclude any noise. The collected frames were integrated by using an orientation matrix determined from the narrow frame scans.
CrysAlisPro RED software packages38 were used for data collection and data integration. Analysis of the integrated data did not reveal any decay. Final cell constants were determined by a global refinement of 4291, 5594 and 6907 reflections (θ < 67.99°) for 2, 3 and 4, respectively. Collected data were corrected for absorbance by using analytical numeric absorption correction39 using a multifaceted crystal model based on expressions upon the Laue symmetry using equivalent reflections.
Structure solution and refinement were carried out with the program SHELXS97.40 Full-matrix least-squares refinement was carried out by minimizing (Fo2 − Fc2)2. All non-hydrogen atoms were refined anisotropically. The H atoms of the water group (H–O) and amine group (H–N) were located in a difference map and refined isotropically with Uiso (H) = 1.5 and 1.2 Ueq for (O) and (N), respectively. H atoms attached to C atoms were placed in geometrically idealized positions and refined as riding on their parent atoms, with C–H = 0.93–0.98 Å with Uiso (H) = 1.2 Ueq (C) for aromatic groups, and Uiso (H) = 1.5 Ueq (C) for methyl groups.
For single-crystals of 5 and 6, X-ray diffraction studies were performed on a Bruker-APEX diffractometer with a CCD area detector (λCuKα = 0.71073 Å, monochromator: graphite). Frames were collected at T = 100 K via ω/φ-rotation at 10 s per frame (SMART).41a The measured intensities were reduced to F2 and corrected for absorption with SADABS (SAINT-NT).41b Corrections were made for Lorentz and polarization effects. Structure solution, refinement, and data output were carried out with the SHELXTL-NT program package.41c,d
Non-hydrogen atoms were refined anisotropically. C–H hydrogen atoms were placed in geometrically calculated positions, using a riding model. O–H and N–H hydrogen atoms have been localized by difference Fourier maps and refined fixing the bond lengths to 0.84 and 0.86 Å, respectively; the isotropic temperature factors have been fixed to a value 1.5 times that of the corresponding oxygen/nitrogen atoms.
Fluorometric titrations
Fluorescence titrations experiments were performed by addition aliquots of stock solutions of anions as sodium salts to a buffered aqueous solution (0.01 M MOPS at pH = 6.5) of the respective compound 1–3 (10−6 M) or 4–5 (10−5 M). The emission spectra (excited at 350 nm) were recorded after additions of aliquots of anions using a Quartz cuvette placed in a Lumina Fluorescence Spectrometer thermostated at 25 ± 0.1 °C with a recirculating water bath. Non-linear least-squares fits of the experimental results to the binding isotherms for 1:1 complexation equilibria were performed by using the Microcal Origin version 7.5 program.
Spectrophotometric, 1H NMR and 31P NMR titrations
The absorption spectra were recorded after additions of aliquots of anions stock solutions to a buffered (10 mM, MOPS at pH = 6.5) aqueous solution of 1–3 (10−5 M) and 4–6 (10−4 M) in a quartz cuvette placed in a compartment of a diode array spectrophotometer thermostated at 25 ± 0.1 °C with a recirculating water bath. NMR titrations were performed on a 300 and 400 MHz spectrometer with more concentrated stock solutions of anions in D2O adding aliquots of them to 5–20 mM receptor solutions directly to NMR tubes. In the cases of 31P NMR titrations, the spectra were recorded at 121.5 MHz using H3PO4(aq) (δ(31P) = 0) as external standard. Aliquots of concentrated stock solution of ATP were added to 10−3 M 1 and 2 solutions directly to NMR tubes.Conclusion
Dicationic pyridine-2,6-dicarboxamide receptors containing N-methylated quinolinium groups attached to amide nitrogens retain significant affinity to halide, acetate and pyrophosphate anions in water and efficiently bind nucleotides. The whole set of results indicates that affinity of these receptors to small anions is dominated by complementarity of anions to the bisamide cleft rather than by electrostatics. The efficient binding of anions by neutral N–H amide donors in water can be attributed to unusually high acidity of amide groups and preorganization of the receptors. This observation points to the possibility to design highly efficient hydrogen bonding receptors in water by using strongly acidified by, e.g., suitable substituents and properly preorganized proton donor groups. The sensitivity of pyridine-2,6-dicarboxamide receptors to nucleoside triphosphates is sufficient to detect them fluorometrically in the micromolar range of concentrations.Acknowledgements
We gratefully acknowledge the CONACyT (project no.101699) for support of this work. Alejandro Dorazco-González thanks CONACyT for the repatriation fellowship.References
- Reviews:
(a) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC
; (b) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971–2985 RSC
- Recent examples in non-aqueous media:
(a) N. G. White and P. D. Beer, Org. Biomol. Chem., 2013, 11, 1326–1333 RSC
- Reviews for anion sensing in water:
(a) S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC
- Selected examples in aqueous media:
(a) L. Feng, H. Li, X. Li, L. Chen, Z. Shen and Y. Guan, Anal. Chim. Acta, 2012, 743, 1–8 CrossRef CAS PubMed
- A. Dorazco-Gonzalez, H. Höpfl, F. Medrano and A. K. Yatsimirsky, J. Org. Chem., 2010, 75, 2259–2273 CrossRef CAS PubMed
-
(a) G. Pennarun, C. Granotier, L. R. Gauthier, D. Gomez, F. Hoffschir, E. Mandine, J.-F. Riou, J.-L. Mergny, P. Mailliet and F. D. Boussin, Oncogene, 2005, 24, 2917–2928 CrossRef CAS PubMed
-
(a) P. E. Vivas-Mejía, J. L. Rodríguez-Cabán, M. Díaz-Velázquez, M. G. Hernández-Pérez, O. Cox and F. A. Gonzalez, Mol. Cell. Biochem., 1997, 177, 69–77 CrossRef
-
(a) W. F. Jager, T. S. Hammink, O. van den Berg and F. C. Grozema, J. Org. Chem., 2010, 75, 2169–2178 CrossRef CAS PubMed
- R. Badugu, J. R. Lakowicz and C. D. Geddes, Talanta, 2005, 66, 569–574 CrossRef CAS PubMed
- L. Zeng, W. Liu, X. Zhuang, J. Wu, P. Wang and W. Zhang, Chem. Commun., 2010, 46, 2435–2437 RSC
- S. Hong, S. S. Yoon, C. Kang and M. Suh, Bull. Korean Chem. Soc., 2004, 25, 345–346 CrossRef CAS
- J. C. Sauceda, R. M. Duke and M. Nitz, ChemBioChem, 2007, 8, 391–394 CrossRef CAS PubMed
- R. Badugu, J. R. Lakowicz and C. D. Geddes, Bioorg. Med. Chem., 2005, 13, 113–119 CrossRef CAS PubMed
-
(a) R. Badugu, J. R. Lakowicz and C. D. Geddes, Sens. Actuators, B, 2005, 104, 103–110 CrossRef CAS
-
(a) A. S. Verkman, Am. J. Physiol., 1990, 259, 376–388 Search PubMed
- A. Graefe, S. E. Stanca, S. Nietzsche, L. Kubicova, R. Beckert, C. Biskup and G. J. Mohr, Anal. Chem., 2008, 80, 6526–6531 CrossRef CAS PubMed
- V. Amendola, L. Fabbrizzi and E. Monzani, Chem.–Eur. J., 2004, 10, 76–82 CrossRef CAS PubMed
- N. Swinburne, M. J. Paterson, A. Beeby and J. W. Steed, Org. Biomol. Chem., 2010, 8, 1010–1016 Search PubMed
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1–19 RSC
-
(a) Y. Ren, Y. Zuo, Y. Xiang and R. Zhu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o3158 CAS
-
ACD/pKa Database, Version 10.01, Advanced Chemistry Development, Inc., Toronto ON, Canada, 2006 Search PubMed
-
B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Verlag GmbH, Weinheim, 2001 Search PubMed
- S. Jayarama and A. S. Verkman, Biophys. Chem., 2000, 85, 49–57 CrossRef
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, LLC, 233 Spring Street, New York, NY, 2006, 3rd edn Search PubMed
- R. Fernandez-Prini, Trans. Faraday Soc., 1968, 64, 2146 RSC
- A. Ray, J. Am. Chem. Soc., 1971, 93, 7146 CrossRef CAS
-
(a) A. Kowalczyk, N. Boens, V. Van den Bergh and F. C. De Schryver, J. Phys. Chem., 1994, 98, 8585–8590 CrossRef CAS
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS
-
(a) T. Gunnlaugsson, H. D. P. Ali, M. Glynn, P. E. Kruger, G. M. Hussey, F. M. Pfeffer, C. M. G. dos Santos and J. Tierney, J. Fluoresc., 2005, 15, 287–299 CrossRef CAS PubMed
-
(a) A. De Robertis, C. De Stefano, C. Foti, O. Giuffre and S. Sammartano, Talanta, 2001, 54, 1135–1152 CrossRef CAS PubMed
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541–5553 CrossRef CAS
-
(a) C. R. Simmons, J. M. Stomel, M. D. McConnell, D. A. Smith, J. L. Watkins, J. P. Allen and J. C. Chaput, ACS Chem. Biol., 2009, 4, 649–658 CrossRef CAS PubMed
- H. Fenniri, M. W. Hosseini and J.-M. Lehn, Helv. Chim. Acta, 1997, 80, 786–803 CrossRef CAS
-
(a) M. W. Hosseini and J.-L. Lehn, Helv. Chim. Acta, 1987, 70, 1312–1319 CrossRef CAS
- Y. Fuentes-Martínez, C. Godoy-Alcántar, F. Medrano, A. Dikiy and A. K. Yatsimirsky, Supramol. Chem., 2010, 22, 212–220 CrossRef
-
(a) E. Arturoni, C. Bazzicalupi, A. Bencini, C. Caltagirone, A. Danesi, A. Garau, C. Giorgi, V. Lippolis and B. Valtancoli, Inorg. Chem., 2008, 47, 6551–6563 CrossRef CAS PubMed
- H. N. Kim, J. H. Moon, S. K. Kim, J. Y. Kwon, Y. J. Jang, J. Y. Lee and J. Yoon, J. Org. Chem., 2011, 76, 3805–3811 CrossRef CAS PubMed
-
Oxford Diffraction, CrysAlis Pro Red, Version 1.171.33.34d, Oxford Diffraction Ltd., Abingdon, Oxfordshire, England, 2009 Search PubMed
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
(a)
SMART: Bruker Molecular Analysis Research Tool, Versions 5.057 and 5.618, Bruker Analytical X-ray Systems, 1997 and 2000 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for compounds 2–6; absorption spectra of compounds 1–6 at pH 6.5; superimposed Stern–Volmer plots for 1 and 4 at pH 6.5; quenching data and Stern–Volmer constants for 3 and 6; spectrophotometric titration of 1 with ATP; 1H and 13C NMR spectra of compounds 2–6. CCDC 941790–941794. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c3ra44363a |
| This journal is © The Royal Society of Chemistry 2014 |