Catalytic activity and selectivity of reusable α-MoO3 nanobelts toward oxidation of olefins and sulfides using economical peroxides†
Maasoumeh
Jafarpour
*,
Mahboube
Ghahramaninezhad
and
Abdolreza
Rezaeifard
*
Catalysis Research Laboratory, Department of Chemistry, Faculty of Science, University of Birjand, Birjand, 97179-414 Iran. E-mail: mjafarpour@birjand.ac.ir; rrezaeifard@birjand.ac.ir; rrezaeifard@gmail.com; Fax: +98 561 2502515; Tel: +98 561 2502516
First published on 4th October 2013
Abstract
The novel catalytic activity of α-MoO3 nanobelts prepared by a new and safe sol–gel method for the epoxidation of olefins and oxidation of sulfides to sulfoxides using H2O2 in ethanol as a safe solvent has been exploited. The reactions also proceeded efficiently in the presence of tert-butyl hydroperoxide (TBHP). Good/high yields and excellent selectivity resulted. The ammonia TPD profile demonstrated strong acidic sites in synthesized α-MoO3 nanobelts, which generated different catalytic activity than the bulk material. The separation and reuse of this heterogeneous nanocatalyst was simple, effective and economical in the presented oxidation methods.
1. Introduction
The oxidative functionalization of organic compounds is of major importance for both organic synthesis and the industrial production of bulk and fine chemicals. Among the different oxidation products, epoxides and sulfoxides are invaluable precursors with a wide variety of applications in organic synthesis, particularly in the preparation of chiral chemicals and drugs.1 Metal-catalyzed oxidation is one of the most elegant and environmentally friendly routes for the production of these compound classes.This is of particular importance, considering that the conservation and management of resources should be the main focus of interest when novel chemical processes are developed. Thus, the innovation and improvement of catalytic epoxidation methods where molecular oxygen or economical peroxides, namely hydrogen peroxide and tert-butyl hydroperoxide (TBHP), are employed as terminal oxidants is highly desirable.2–5
Metal oxides represent one of the most important and widely employed categories of solid catalyst, either as active phases or as supports. They are utilized for both their acid–base and redox properties and constitute the largest family of catalysts in heterogeneous catalysis. Recently, the use of nanomaterials has increased, as their activity is very high under mild conditions due to their very large surface area.6–9
Molybdenum catalyzed oxygen atom transfer (OAT) chemistry is a very important chemical reaction, both in biological systems and on an industrial scale. In biology, molybdenum is found in a large family of enzymes that catalyze important OAT reactions in the living cell.10,11 Several research groups,12,13 including ours,14–18 have developed functional and structural model molybdenum complexes and investigated their OAT reactivity for a better understanding of this important reaction.
Molybdenum oxide is widely used in sensors, lubricants, and fuel cell materials.19–22 Catalysis using MoO3 greatly relies on the crystal phase, surface structure, and particle size.23 Bulk MoO3 alone presented low/moderate catalytic oxidation activity24–26 and a ligand/additive or Lewis acid should be added to reach the desired activity.27–30
The synthesis and structural properties of nanostructured MoO3 have been widely investigated, however, there is only limited work on its catalytic activity.31 Just recently, we have developed a new and safe route to synthesize α-MoO3 nanobelts by a simple sol–gel method.32 An orthorhombic lattice system was established by X-ray diffraction (XRD), Fourier transform infrared (FT-IR) spectroscopy and Raman analyses. The HRTEM (high resolution transmission electron microscopy) images revealed that the nanobelt-form mostly ranged from 20–70 nm in width and 200–400 nm in length. The prepared nanostructured MoO3 exhibited a high efficiency in catalyzing the condensation reaction of various 1,2-diamine and carbonyl compounds for synthesis of heterocyclic compounds.32 We would now like to describe its catalytic potential in the epoxidation of olefins and oxygenation of sulfides to sulfoxides using hydrogen peroxide and TBHP, both of which are industrially and environmentally important oxidants.
2. Experimental
2.1. General remarks
All chemicals were purchased from Merck or Fluka Chemical Companies. Powder XRD was performed on a Bruker D8-advance X-ray diffractometer with Cu Kα (λ = 1.54178 Å) radiation. The FT-IR spectra were recorded on NICOLET system 800 beam splitter KBr SCAL = 800, used with resolution in the range 400–4000 cm. Raman spectra were recorded on a Bruker Senterra (2009) model with spectral range 200–3500 cm−1 and laser wavenumber 785 nm. Thermogravimetric analysis (TGA) of nanopowders was performed in air by Shimadzu 50. Scanning electron microscopy (SEM) micrographs were obtained by SEM instrumentation (SEM, XL-30 FEG SEM, Philips, at 20 kV) and HRTEM images were obtained by TEM instrumentation (Philips CM 10).Temperature-programmed desorption of NH3 adsorbed on a catalyst (NH3-TPD) was carried out in a conventional flow system equipped with a thermal conductivity detector (TCD).
The sample was pretreated in nitrogen at 550 °C for 1 h before adsorbing NH3. The NH3-TPD experiments were carried out at 100–600 °C in a flow of dry He (19 mL min−1). The rate of heating was 10 °C min−1.
The progress of the reactions was monitored by thin layer chromatography (TLC) using silica-gel SIL G/UV 254 plates and also by gas chromatography (GC) on a Shimadzu GC-16A instrument using a 25 m CBP1-S25 (0.32 mm ID, 0.5 μm coating) capillary column. NMR spectra were recorded on a Brucker Avance DPX 250 and 400 MHz instruments.
2.2. Catalyst preparation
To a solution of (NH4)6Mo7O24·4H2O (0.003 mol) in distilled water was added citric acid (0.126 mol) and ethylene glycol (0.126 mol) subsequently. The resulting mixture was stirred at 80 °C for 30 min to give a clear solution. The solution was refluxed at 100 °C for 12 h, which resulted in a metal–citrate homogeneous complex with a small color change from clear to light blue. After cooling, in order to bring about the required chemical reactions for the development of polymerization and evaporation of the solvent, the sol was further heated at 80 °C for 3 h in an open bath until a black-blue wet gel was obtained. During continuous heating at this temperature, the polymerization between citric acid, ethylene glycol and the complex develops and ultimately the sol became more viscous as a wet gel. In the final stage of the sol–gel process, the wet gel was fully dried by direct heating on the hot plate at 120 °C for 3 h. The resulting product was a black-blue powder. This was then calcined in a furnace at 600 °C for 6 h at a rate of 10 °C min−1. Finally, MoO3 nanocrystals with a light green color were obtained (see Fig. S1in ESI† for the synthesis flow chart). The XRD (Fig. S2†), Raman spectra (Fig. S3†) and FT-IR spectra (Fig. S4†) given in the ESI† confirmed the structure of the prepared α-MoO3. Its morphology and properties were studied by HRTEM, (Fig. S5A and B†) SEM, (Fig. S5C†), TPD (Fig. S6†) and TGA (Fig. S7†).322.3. General procedure for epoxidation of olefins
To a mixture of olefin (1 mmol) and nano MoO3 (0.01 mmol) in ethanol (1 mL) was added H2O2 (2 mmol), and the reaction mixture was stirred in air at 70 °C for the required time. The reaction progress was monitored by GC, and the yields of the products were determined by GC and NMR analysis. After completion of the reaction, nano MoO3 (solid phase) was separated by centrifuging followed by decantation (3 × 5 mL ethanol). The desired product (liquid phase) was then extracted by plate chromatography eluted with n-hexane–EtOAc (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Assignment of the products was made by IR, 1H NMR and MS spectral data in comparison with control samples. [Note: oxidation of olefins using TBHP in dichloroethane (DCE) was performed by the same procedure].
2.4. General procedure for oxidation of sulfides
To a mixture of the sulfide (1 mmol) and nano MoO3 (0.01 mmol) in ethanol (1 mL) was added H2O2 (2 mmol), and the reaction mixture was stirred in air at 70 °C for 1 h. The reaction progress was monitored by TLC, and the yields of the products were determined by GC analysis. After the completion of the reaction, nanoMoO3 (solid phase) was separated by centrifuging followed by decantation (3 × 5 mL ethanol). The desired product (liquid phase) was then extracted by plate chromatography eluted with n-hexane–EtOAc (10:3).
Assignment of the products was made by IR, 1H NMR and MS spectral data in comparison with control samples. [Note: oxidation of sulfides using TBHP in DCE was performed with the same procedure].
2.5. Reusability of catalyst
To a mixture of cyclooctene (10 mmol) and nano MoO3 (0.1 mmol) in ethanol (10 mL) was added H2O2 (20 mmol), and the reaction mixture was stirred in air at 70 °C for 10 h. After completion of the reaction, nano MoO3 was separated by centrifuging followed by decantation (3 × 5 mL ethanol). The isolated solid phase (nano MoO3) was dried under reduced pressure and reused for the next run. Catalyst recovery was also investigated in the oxidation of olefins with TBHP in DCE, as well as in the oxidation of sulfides with both H2O2 and TBHP, according to the above mentioned procedures.3. Results and discussion
3.1. Epoxidation of olefins with H2O2
Preliminary experiments were initiated with oxidation of cyclooctene (1 mmol) using H2O2 (2 mmol) as a model reaction under neat conditions, as this did not proceed in the absence of the catalyst under any conditions. To find the optimum reaction conditions, the influence of different factors that may affect the conversion and selectivity of the cyclooctene epoxidation was investigated.A systematic examination of the solvent nature was performed in various solvents, namely chloroform, DCE, acetonitrile, methanol, ethanol and water, using 1 mol% of the nano-MoO3 catalyst (Fig. 1) at different temperatures (Fig. 2). The best yield and conversion rate were obtained in ethanol at 70 °C.
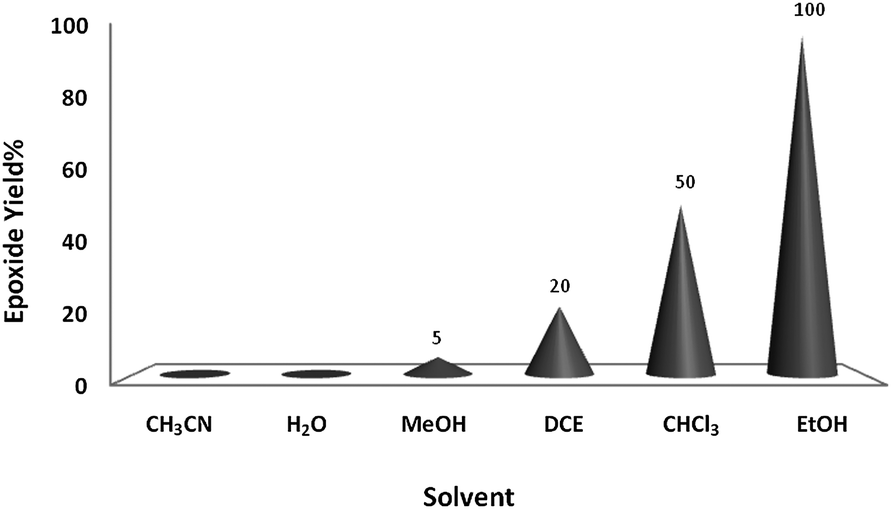 | ||
| Fig. 1 The effect of solvent (1 mL) nature on oxidation of cyclooctene (1 mmol) using H2O2 (2 mmol) catalyzed by nanobelts of α-MoO3 (0.01 mmol) at 70 °C, except for solvents having lower boiling points, which were performed at reflux temperature. | ||
 | ||
| Fig. 2 The effect of temperature on oxidation of cyclooctene (1 mmol) using H2O2(2 mmol)–EtOH (1 mL) catalyzed by nanobelts of α-MoO3 (0.01 mmol). | ||
The reaction was further optimized with respect to catalyst loading (Fig. 3) and oxidant amount (Fig. 4). It was observed that full conversion of cyclooctene required 1 mol% of nanocatalyst and two equivalents of H2O2 over 10 h and an increase in any of these ratios did not noticeably affect the reaction rate.
 | ||
| Fig. 3 Effect of quantity of catalyst on the oxidation of cyclooctene (1 mmol) using H2O2 (2 mmol)–EtOH (1 mL) catalyzed by nanobelts of α-MoO3 at 70 °C. | ||
 | ||
| Fig. 4 Effect of H2O2–olefin molar ratios on oxidation of cyclooctene (1 mmol) in EtOH (1 mL) catalyzed by nanobelts of α-MoO3 (0.01 mmol) at 70 °C. | ||
To evaluate the oxidizing potential of other common oxidants, cyclooctene was subjected to the oxidation protocol using TBHP, NaIO4 and Oxone® under the catalytic influence of α-MoO3 in ethanol at 70 °C (Fig. 5). Only small amounts of the oxidation products were observed.
 | ||
| Fig. 5 Effect of different oxidants (2 mmol) on oxidation of cyclooctene (1 mmol) in EtOH (1 mL) catalyzed by nanobelts of α-MoO3 (0.01 mmol) at 70 °C. | ||
Under the optimized conditions (100:200:1 molar ratio for olefin–H2O2–catalyst in ethanol at 70 °C), cyclooctene converted completely within 10 h and 95% of the corresponding epoxide was secured as the sole product.
It should be mentioned that, when the nano-MoO3 was replaced by its bulk counterpart, cyclooctene remained completely intact under the same conditions. Enhanced acid strength and surface area of nanostructured MoO3 (Fig. S6†) may be a plausible reason for this great improvement in catalytic activity.
In order to establish the general applicability of the method, various olefins were subjected to the oxidation protocol under the catalytic influence of α-MoO3 nanobelts (Table 1).
| Entry | Olefin | Conversion % (isolated yield) | Productb | Time (h) |
|---|---|---|---|---|
|
a The molar ratio of substrate–H2O2–catalyst was 100:200:1. The reactions were run under air at 70 °C.
b The products were identified by 1H NMR or by comparison with control sample retention times using GC analysis.35–39 The selectivity of the products was >99%.
|
||||
| 1 |

|
100 (92) |

|
12 |
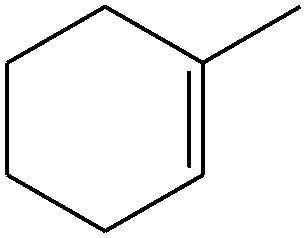
| 2 |
|
100 (95) |

|
10 |
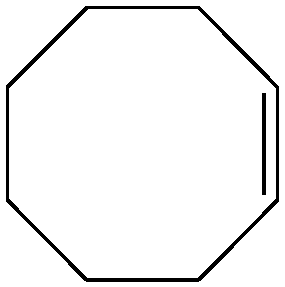
| 3 |
|
100 (95) |
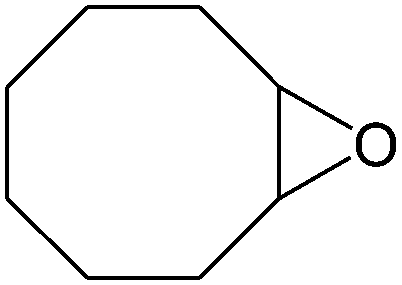
|
10 |
| 4 |

|
100 (93) |

|
22 |
| 5 |

|
10 |

|
24 |
| 6 |
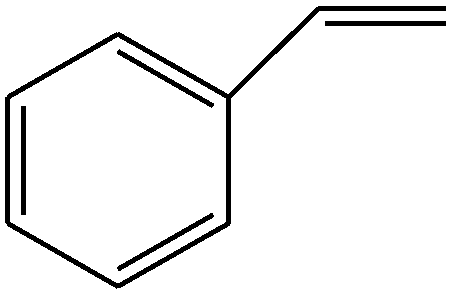
|
50 |

|
24 |
| 90 (85) | 48 | |||
| 7 |
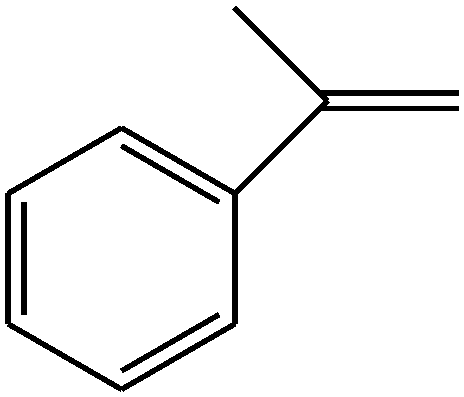
|
100 (96) |
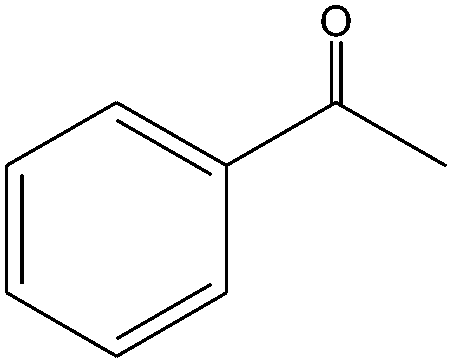
|
20 |
| 8 |
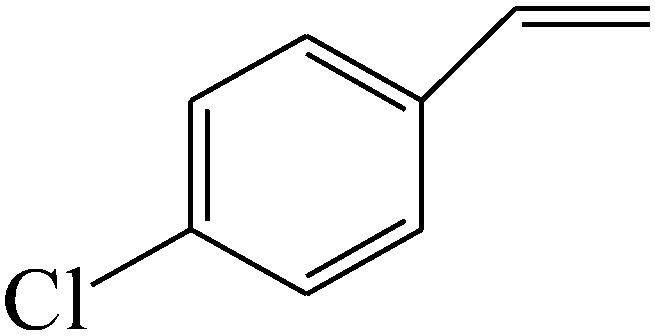
|
73 (65) |
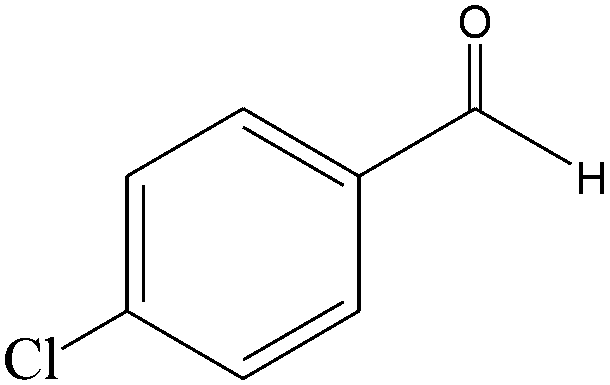
|
48 |
| 9 |

|
30 |
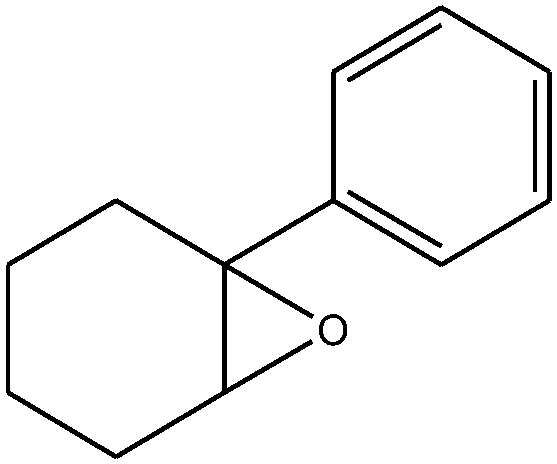
|
48 |
| 10 |

|
50 (43) |
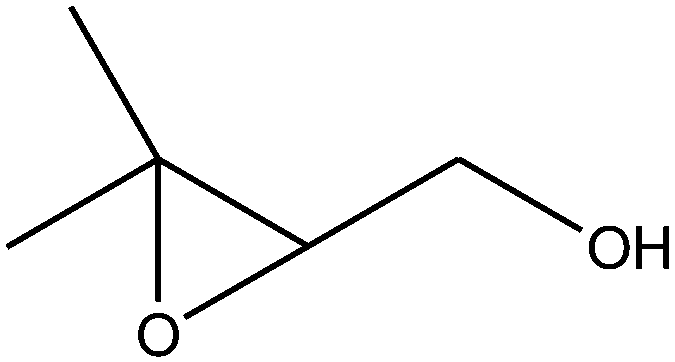
|
22 |
| 11 |

|
45 (38) |

|
22 |
| 12 |

|
100 |
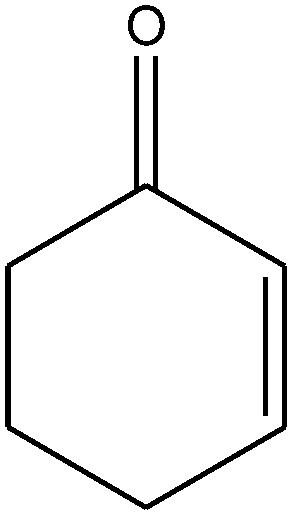
|
12 |
Several useful features of this catalytic method can be seen in Table 1. Different olefins were generally good substrates for this catalyst. It led to complete conversion of cyclooctene, norbornene and cyclohexenes with the formation of the corresponding epoxides as sole products (Table 1, entries 1–4).
The epoxidation of 1-octene as the least reactive terminal olefin proceeded sluggishly (10% yield, entry 5). When the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was conjugated with aromatic ring, the reaction rate was enhanced, however, the epoxide ring was completely opened, producing the related carbonyl compound (Table 1, entries 6–8). It seems that benzaldehyde and acetophenone are favorably formed at high temperature in the oxidation of styrene and α-methylstyrene, respectively, because high temperature will supply enough energy to break the CC bond.33,34 These results were further supported by the oxidation of styrene oxide as substrate under the same conditions. Benzaldehyde was formed quantitatively as the sole product within 48 h. It should be noted that, in all the above mentioned reactions, no phenylacetaldehyde product was detected in the oxidation of either styrene or styrene oxide. Inspection of these results demonstrates that the reaction is significantly affected by electronic factors. While α-methyl styrene was converted completely to acetophenone within 20 h, the oxidation of electron-deficient 4-Cl-styrene proceeded slowly and moderate yield of the related benzaldehyde was obtained (73% after 48 h). It is worth mentioning that a cyclic olefin conjugated with a phenyl ring produced exclusively the pertinent epoxide, albeit with low yield (Table 1, entry 9). The chemoselectivity of the procedure was notable. While primary alcohols containing CC double bonds (allylic and homoallylic alcohols) oxidized completely to the corresponding epoxides (Table 1, entries 10 and 11), a secondary one converted to the related α, β-unsaturated ketone as the only product (entry 12).
C double bond was conjugated with aromatic ring, the reaction rate was enhanced, however, the epoxide ring was completely opened, producing the related carbonyl compound (Table 1, entries 6–8). It seems that benzaldehyde and acetophenone are favorably formed at high temperature in the oxidation of styrene and α-methylstyrene, respectively, because high temperature will supply enough energy to break the CC bond.33,34 These results were further supported by the oxidation of styrene oxide as substrate under the same conditions. Benzaldehyde was formed quantitatively as the sole product within 48 h. It should be noted that, in all the above mentioned reactions, no phenylacetaldehyde product was detected in the oxidation of either styrene or styrene oxide. Inspection of these results demonstrates that the reaction is significantly affected by electronic factors. While α-methyl styrene was converted completely to acetophenone within 20 h, the oxidation of electron-deficient 4-Cl-styrene proceeded slowly and moderate yield of the related benzaldehyde was obtained (73% after 48 h). It is worth mentioning that a cyclic olefin conjugated with a phenyl ring produced exclusively the pertinent epoxide, albeit with low yield (Table 1, entry 9). The chemoselectivity of the procedure was notable. While primary alcohols containing CC double bonds (allylic and homoallylic alcohols) oxidized completely to the corresponding epoxides (Table 1, entries 10 and 11), a secondary one converted to the related α, β-unsaturated ketone as the only product (entry 12).
3.2. Epoxidation of olefins with TBHP
The recent successful results of olefin epoxidation catalyzed by cis-dioxo Mo–Schiff base complexes using TBHP in DCE14–17 encouraged us to evaluate the epoxidation activity of the prepared Mo oxide nanocatalyst under these conditions. Different olefins were oxidized smoothly with 2 equiv. of TBHP by using 1 mol% of nanobelts of α-MoO3 in DCE at 70 °C, albeit with longer reaction times than those in the presence of Schiff base complexes.14–17 Although, α-MoO3 displayed higher epoxidation activity with TBHP than with the H2O2–ethanol system (this work, Table 2), the yields and selectivity remained almost the same, except for 2-cyclohexene-1-ol which produced mainly the pertinent epoxide (85%) along with 15% of the α, β-unsaturated ketone (Table 2, entry 9).| Entry | Olefin | Conversion % (isolated yield) | Productb | Time (h) | |
|---|---|---|---|---|---|
|
a The molar ratio of substrate–TBHP–catalyst was 100:200:1. The reactions were run under air at 70 °C.
b The products were identified by 1H NMR or by by comparison with control sample retention times using GC analysis.35–39 The selectivity of the products was >99%, except for entry 9 which was 85%.
|
|||||
| 1 |

|
100 (94) |

|
6 | |
| 2 |
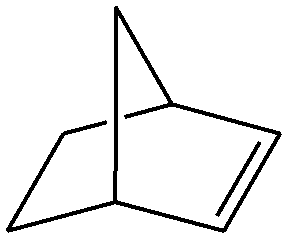
|
100 (93) |

|
6 | |
| 3 |

|
10 |

|
24 | |
| 4 |

|
56 |
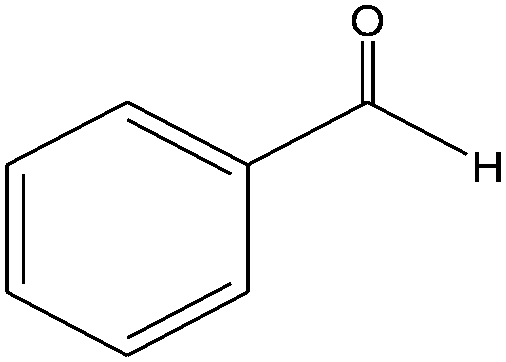
|
12 | |
| 100 (94) | 24 | ||||
| 5 |

|
100 (96) |
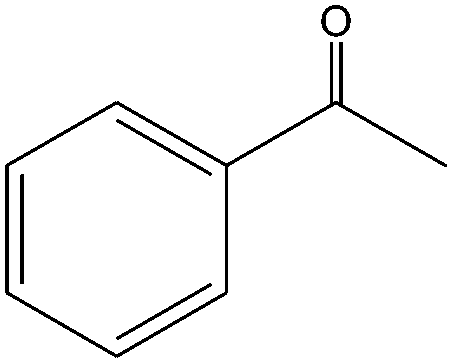
|
14 | |
| 6 |

|
100 (94) |
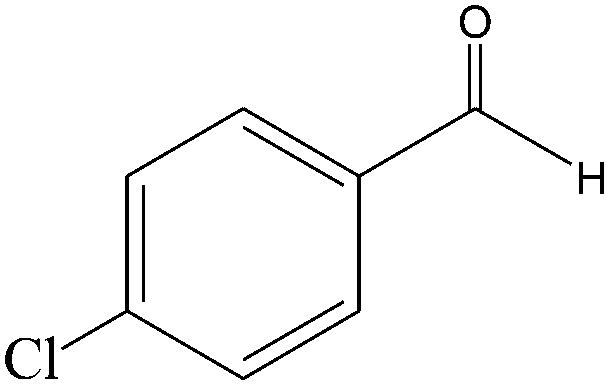
|
24 | |
| 7 |

|
55 (43) |

|
14 | |
| 8 |
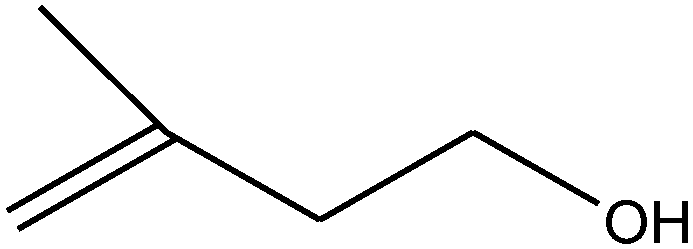
|
50 (45) |

|
14 | |
| 9 |
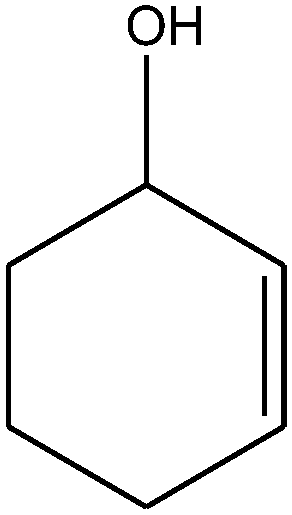
|
100 | 85 |

|
10 |
| 15 |
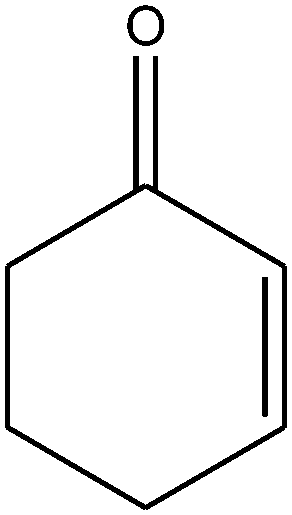
|
||||
3.3. Oxidation of sulfides to sulfoxides
To extend the generality and scope of the presented methodology, oxidation of sulfides was investigated. Oxidation of thioanisole using 2 equivalents of H2O2 in the presence of 1 mol% of nanostructured α-MoO3 in ethanol under mild conditions produced the related sulfoxide as the sole product in excellent yield within 45 min. Accordingly, different sulfides were subjected to this reaction system in the presence of the prepared nanocatalyst (Table 3). The results given in Table 3 illustrate the high efficiency of this protocol for the oxidation of structurally different sulfides. All substrates could be smoothly converted into sulfoxides with excellent conversion rates, and excellent selectivities were obtained under mild conditions.| Entry | Substrate | Conversion % (isolated yield) | Sulfoxide selectivity % | |
|---|---|---|---|---|
| TBHP–DCEb | H2O2–ethanola | |||
|
a The reactions were run at 70 °C and completed within 45 min. The molar ratio of sulfide:H2O2:catalyst was 100:200:1 in 1 mL ethanol.
b The reactions were run at 70 °C and completed within 15 min. The molar ratio of sulfide:TBHP:catalyst was 100:200:1 in 1 mL DCE. The products were identified by comparison with control samples.40–43
|
||||
| 1 |

|
100 (96) | 100 (97) | 96 |
| 2 |

|
95 (90) | 95 (88) | 95 |
| 3 |

|
100 (94) | 100 (95) | 90 |
| 4 |
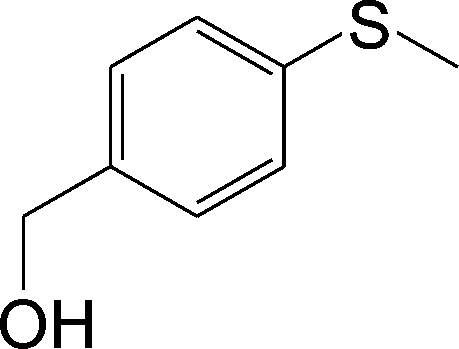
|
100 (96) | 100 (96) | 100 |
| 5 |

|
90 (85) | 90 (87) | 95 |
| 6 |

|
80 (75) | 80 (75) | 90 |
| 7 |

|
100 (94) | 100 (96) | 100 |
| 8 |
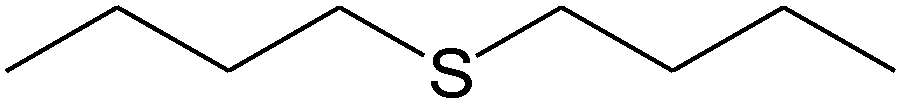
|
95 (89) | 95 (88) | 95 |
| 9 |

|
100 (95) | 100 (96) | 100 |
Oxidation of diphenylsulfide (entry 6) under the same conditions produced a lower yield of the desired sulfoxide than other substrates, indicating the influence of steric effects on the reaction rate.
The chemoselectivity of the method is noteworthy, as exemplified by sulfides containing a hydroxyl group (Table 3, entry 4) or CC double bond (Table 3, entry 2, 3, 7). While the sulfide oxidized completely, the alcohol and olefin moieties remained intact. In addition, benzyl phenyl sulfide (Table 3, entry 5) was selectively oxidized to its corresponding sulfoxide without formation of any benzylic oxidation by-products.
As observed in olefin epoxidation, an accelerated oxidation of sulfide occurred when H2O2 was replaced with TBHP (in DCE), albeit with the same yields and selectivity (Table 3).
3.4. Recovery of catalyst
The good/excellent yields of epoxides and sulfoxides obtained using these new catalytic methods demonstrated the high stability and catalytic activity of the prepared nanobelts of α-MoO3. This was further supported by evaluation of the recovery potential of the catalyst in the oxidation of both olefins and sulfides under the different conditions shown in Tables 1–3.Recovery of the nanoMoO3 catalyst was easy and efficient. The catalyst was recovered by centrifuging and decantation of the reaction mixture. It was then washed with ethanol as a safe solvent, dried under vacuum, and used directly for the next round of reaction without further purification. The ease of recovery, combined with the intrinsic stability of the MoO3 nanoparticles, allows the catalyst to be recovered efficiently at least four times in the oxidation of olefins and sulfides under the different conditions used in this study (Fig. 6 and 7).
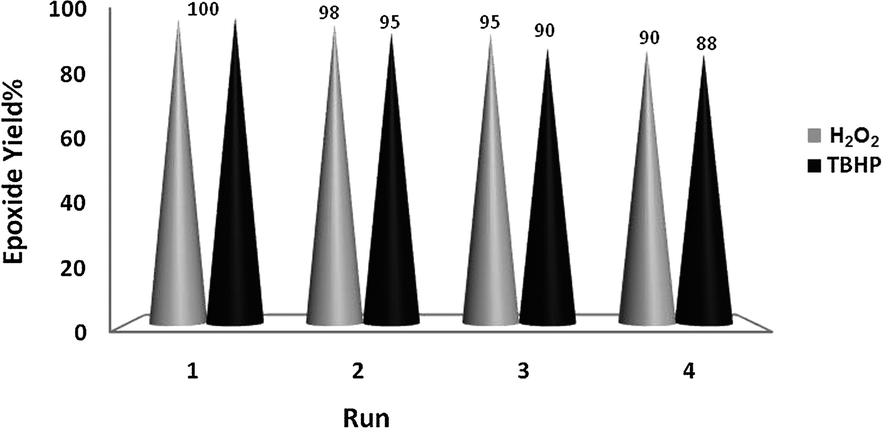 | ||
| Fig. 6 Recycling of the catalytic system for oxidation of cyclooctene (10 mmol) using H2O2 (20 mmol)–EtOH (10 mL) and TBHP (20 mmol)–DCE (10 mL) and catalyzed by nanobelts of α-MoO3 (0.1 mmol) at 70 °C. | ||
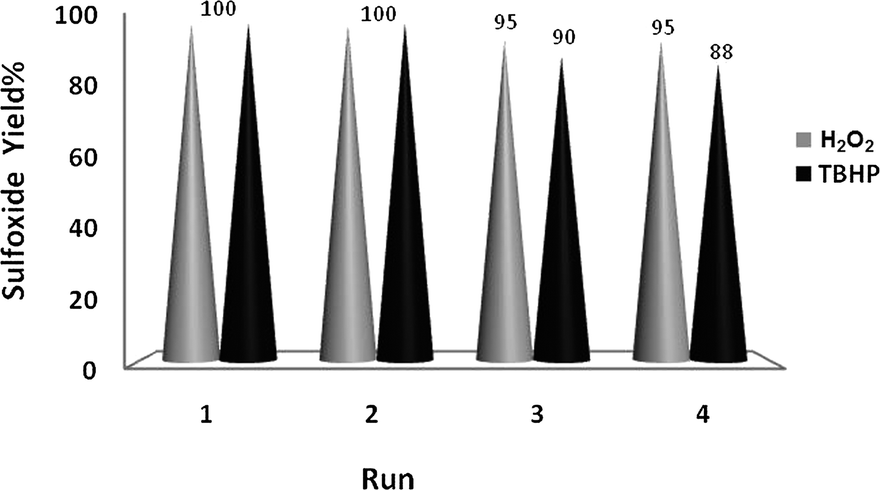 | ||
| Fig. 7 Recycling of the catalytic system for oxidation of thioanisole (10 mmol) using H2O2 (20 mmol)–EtOH (10 mL) and TBHP (20 mmol)–DCE (10 mL) and catalyzed by nanobelts of α-MoO3 (0.1 mmol) at 70 °C. | ||
The comparison of Raman spectra (Fig. 8) and TEM images of used MoO3 nanobelts (Fig. 9) with fresh catalyst showed that the structure, size and morphology of the catalyst remained almost intact after four times recovering.
 | ||
| Fig. 8 Raman spectra of fresh nano α-MoO3 (A) and after being reused 4 times (B) in the oxidation of olefins and sulfides under different conditions. | ||
 | ||
| Fig. 9 TEM images of fresh nano α-MoO3 (A) and after being reused 4 times (B) in the oxidation of olefins and sulfides under different conditions of nanobelts of α-MoO3. | ||
The presented methodologies are therefore cost effective and industrially important, as they allow recycling of the catalyst and use of H2O2 and TBHP as environmentally-friendly oxidants, particularly in ethanol as a safe reaction media. In addition to these advantages, the high yielding oxidation methods also offered ready scalability. For example, the use of a semi scale-up procedure (10 mmol) for epoxidation of cyclooctene and oxidation of thioanisole in the presence of nanobelts of α-MoO3 led to isolation of the related epoxide and sulfoxide in 93 and 95% yield, respectively.
4. Conclusions
In conclusion, nanobelts of α-MoO3 prepared by a new and safe sol–gel method, with sizes ranging from 20–70 nm in width and 200–400 nm in length, efficiently catalyzed epoxidation of olefins and oxygenation of sulfides to sulfoxides using H2O2 and TBHP in good/excellent yields and excellent selectivity. It was observed that the catalytic activity of the prepared heterogeneous nanocatalyst was strikingly different from the bulk one. Our results demonstrated clearly the efficiency, selectivity and oxidative stability of this nanocatalyst, given its effective reusability and strength in removing by-products. By using ethanol as a “green” solvent, these conditions are cost effective, environmentally benign and possess high generality, which make the presented methodologies suitable for future industrial use.Acknowledgements
Support for this work by Research Council of University of Birjand is highly appreciated.References
- J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004 Search PubMed.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 1998 Search PubMed.
- M. R. dos Santos, J. R. Diniz, A. M. Arouca and A. F. Gomes, ChemSusChem, 2012, 5, 716–726 CrossRef CAS PubMed.
- K. A. Srinivas, A. Kumar and S. M. S. Chauhan, Chem. Commun., 2002, 2456–2457 RSC.
- U. Arnold, W. Habicht and M. Döring, Adv. Synth. Catal., 2006, 348, 142–150 CrossRef CAS.
- H. H. Kung, Transition Metal Oxides: Surface Chemistry and Catalysis, Stud. Surf. Sci. Catal., Elsevier, Amsterdam, 1989, vol. 45, pp. 1–277 Search PubMed.
- V. E. Henrichand and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, Cambridge, UK, 1994 Search PubMed.
- C. Noguera, Physics and Chemistry at Oxide Surface, Cambridge University Press, Cambridge, UK, 1996 Search PubMed.
- S. U. Sonavane, M. B. Gawande, S. S. Deshpande and R. V. Jayaram, Catal. Commun., 2007, 8, 1803–1806 CrossRef CAS PubMed.
- Molybdenum and Tungsten: Their Roles in Biological Processes, Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 2002, vol. 39 Search PubMed.
- J. H. Enemark and J. J. A. Cooney, Chem. Rev., 2004, 104, 1175–1200 CrossRef CAS PubMed.
- K. C. Gupta, A. K. Sutar and C.-C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS PubMed.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS PubMed.
- A. Rezaeifard, I. Sheikhshoaie, N. Monadi and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2010, 799–806 CrossRef CAS.
- I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733–738 CrossRef CAS PubMed.
- A. Rezaeifard, I. Sheikhshoaie, N. Monadi and M. Alipour, Polyhedron, 2010, 29, 2703–2709 CrossRef CAS PubMed.
- A. Rezaeifard, M. Jafarpour, H. Raissi, M. Alipour and H. Stoeckli-Evans, Z. Anorg. Allg. Chem., 2012, 638, 1023–1030 CrossRef CAS.
- A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, J. Am. Chem. Soc., 2013, 135, 10036–10039 CrossRef CAS PubMed.
- J. A. Rodríguez and M. Fernández-García, Synthesis, properties, and applications of oxide nanomaterials, JohnWiley& Sons, Inc, 2007 Search PubMed.
- F. Wang and W. Ueda, Chem. Commun., 2008, 3196–3198 RSC.
- B. Zhu, C. R. Xia and X. G. Luo, Thin Solid Films, 2001, 385, 209–214 CrossRef CAS.
- E. d. P. Carreiro and A. J. Burke, J. Mol. Catal. A: Chem., 2006, 249, 123–128 CrossRef CAS PubMed.
- H. Tian, C. A. Roberts and I. E. Wachs, J. Phys. Chem. C, 2010, 114, 14110–14120 CAS.
- V. R. Choudhary, R. Jha and P. Jana, Catal. Commun., 2008, 10, 205–207 CrossRef CAS PubMed.
- M. Bowker, A. F. Carley and M. House, Catal. Lett., 2007, 120, 34–39 CrossRef.
- T. Ressler, J. Wienold, R. E. Jentoft and F. Girgsdies, Eur. J. Inorg. Chem., 2003, 301–312 CrossRef CAS.
- M. Abrantes, T. R. Amarante, M. M. Antunes, S. Gago, F. A. Almeida Paz, I. Margiolaki, A. E. Rodrigues, M. Pillinger, A. A. Valente and I. S. Gonçalves, Inorg. Chem., 2010, 49, 6865–6873 CrossRef CAS PubMed.
- E. da Palma Carreiro and A. J. Burke, J. Mol. Catal. A: Chem., 2006, 249, 123–128 CrossRef PubMed.
- E. da Palma Carreiro, C. Monteiro, G. Yong-en, A. J. Burke and A. I. Rodrigues, J. Mol. Catal. A: Chem., 2006, 260, 295–298 CrossRef PubMed.
- A. Corma and H. García, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS PubMed.
- F. Wang and W. Ueda, Chem.–Eur. J., 2009, 15, 742–753 CrossRef CAS PubMed.
- M. Jafarpour, A. Rezaeifard, M. Ghahramaninezhad and T. Tabibi, New J. Chem., 2013, 37, 2087–2095 RSC.
- B. Tyagi, U. Sharma and R. V. Jasra, Appl. Catal., A, 2011, 408, 171–177 CrossRef CAS PubMed.
- X. Meng, K. Lin, J. Sun, M. Yang, D. Jiang and F.-S. Xiao, Catal. Lett., 2001, 76, 105–109 CrossRef CAS.
- A. Rezaeifard, M. Jafarpour, A. Naeimi and S. Kaafi, Catal. Commun., 2011, 12, 761–765 CrossRef CAS PubMed.
- A. Rezaeifard, M. Jafarpour, A. Naeimi and K. Mohammadi, J. Mol. Catal. A: Chem., 2012, 357, 141–147 CrossRef CAS PubMed.
- Y. Ding, W. Zhao, H. Hua and B. Ma, Green Chem., 2008, 10, 910–913 RSC.
- E. G. Ankudey, H. F. Olivo and T. L. Peeples, Green Chem., 2006, 8, 923–926 RSC.
- F. Bruyneel, C. Letondor, B. Bastürk, A. Gualandi, A. Pordea, H. Stoeckli-Evans and R. Neier, Adv. Synth. Catal., 2012, 354, 428–440 CrossRef CAS.
- F. Rajabi, S. Naserian, A. Primo and R. Luque, Adv. Synth. Catal., 2011, 353, 2060–2066 CrossRef CAS.
- L. Xu, J. Cheng and M. L. Trudel, J. Org. Chem., 2003, 68, 5388–5391 CrossRef CAS PubMed.
- J. M. Samanen and E. Brandeis, J. Org. Chem., 1988, 53, 561–569 CrossRef CAS.
- M. H. Ali and G. J. Bohnert, Synth. Commun., 1998, 28, 2983–2998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the nanobelts of α-MoO3. See DOI: 10.1039/c3ra44404j |
| This journal is © The Royal Society of Chemistry 2014 |