Enantioselective synthesis of pyrazolone derivatives catalysed by a chiral squaramide catalyst†
Jun-Hua Li and
Da-Ming Du*
School of Chemical Engineering and Environment, Beijing Institute of Technology, Beijing 100081, People's Republic of China. E-mail: dudm@bit.edu.cn; Tel: +86 10 68914985
First published on 11th March 2014
Abstract
An efficient chiral squaramide-catalysed enantioselective Michael addition of pyrazolin-5-ones to β,γ-unsaturated α-ketoesters has been developed. The chiral pyrazolone derivatives were obtained in moderate to high yields (up to 99% yield) with high enantioselectivities (up to 96% ee) for most substrates. This catalytic asymmetric reaction provides valuable and easy access to chiral pyrazolone ketoester derivatives.
Introduction
Pyrazoles and pyrazolones, both five-membered heterocyclic compounds containing two adjacent nitrogen atoms, have been widely used in organic synthesis and pharmaceutical chemistry.1 Pyrazolone derivatives were used to synthesize many drugs such as complexes of aminoantipyrine with barbiturate which have different applications under the trade names allonal, veramon, novalgin, phenazone, propylphenazon and dimethylaminophenazone.2 For example, edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one, also known as MCI-186) has been reported to be effective for brain ischemia,3 and myocardial ischemia.4 Owing to the broad biological activities and extensive application of pyrazolone compounds, the development of asymmetric methods for synthesis of chiral pyrazolone derivatives is of considerable interest.There have been an increasing number of reports about the asymmetric synthesis of pyrazole and pyrazolone derivatives in recent years.2,5 We are not aware of any reports about the asymmetric addition of pyrazolones to β,γ-unsaturated α-ketoesters. β,γ-Unsaturated α-ketoesters has been recognized as a useful reaction components in organic synthesis.6 Because of the reported unique feature of β,γ-unsaturated α-ketoesters, which have two adjacent carbonyl groups connected by a single bond, the addition product of 4-hydroxycoumarin to β,γ-unsaturated α-ketoester has more than one forms (Scheme 1).7 This tautomeric form also occurred in the product of pyrazolone to β,γ-unsaturated α-ketoesters. This kind of three structures-tautomeric compounds has few reports ever before.
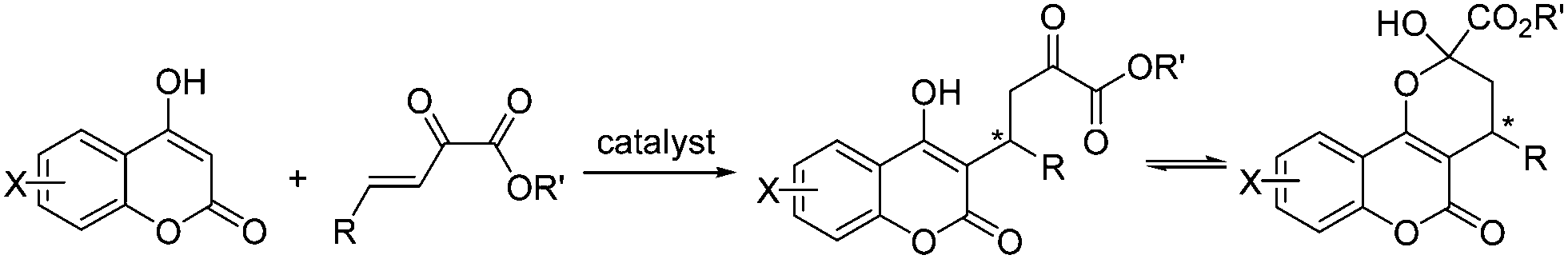 | ||
| Scheme 1 Organocatalyst-promoted Michael addition of coumarin to β,γ-unsaturated α-ketoester. | ||
Herein, we describe the development of a squaramide promoted asymmetric Michael addition of pyrazolones to β,γ-unsaturated α-ketoesters (Scheme 2). β,γ-Unsaturated α-ketoesters, which are electrophiles with two adjacent carbonyl groups connected by a single bond, could be suitably activated and orientated by squaramide, as its two acidic hydrogen atoms are separated by a two-carbon link.7b Satisfactorily, this organocatalytic process is effective, and the corresponding pyrazolone ketoester derivatives were obtained in moderate to high yields with high enantioselectivities (up to 96% ee).
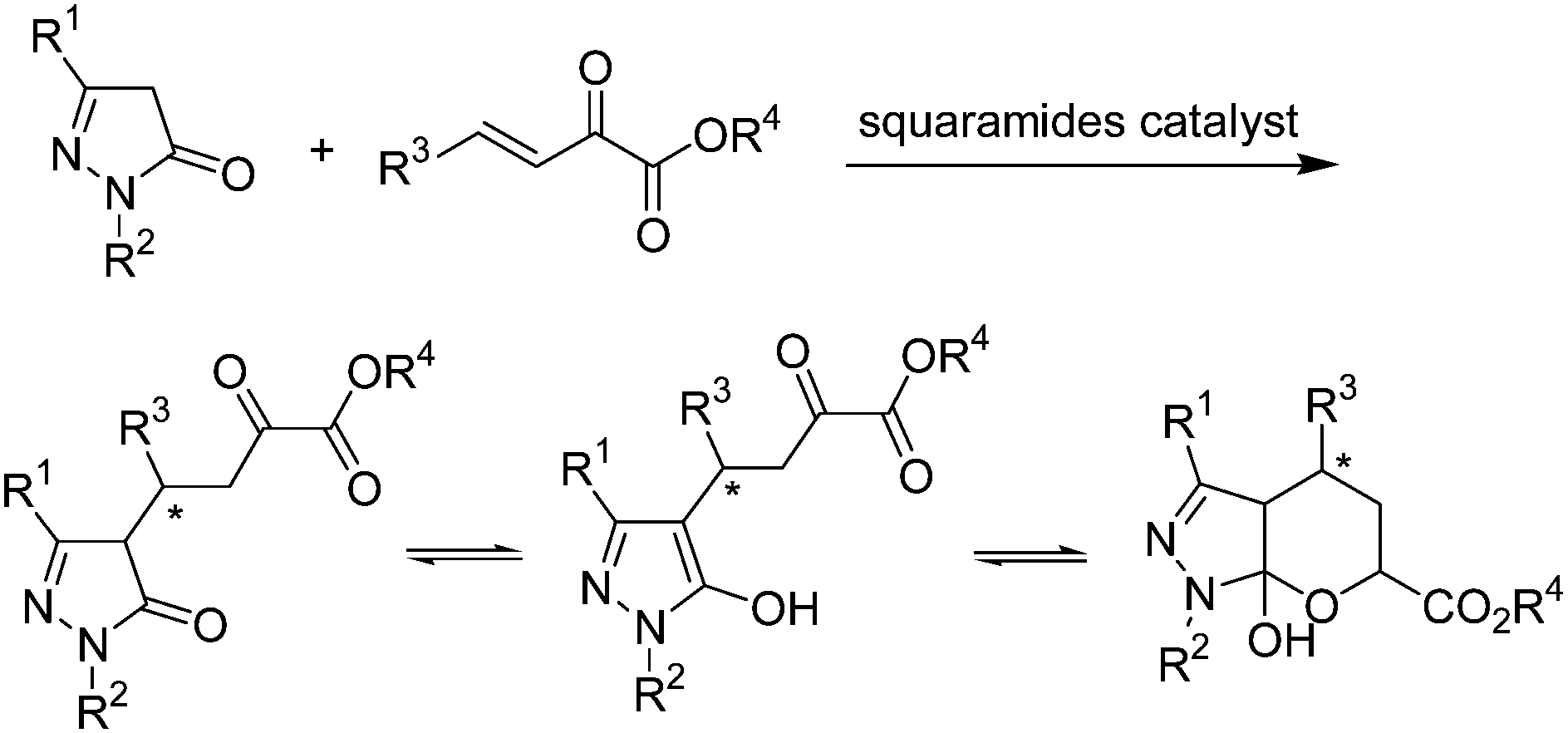 | ||
| Scheme 2 Squaramide-catalysed Michael addition of pyrazolone to β,γ-unsaturated α-ketoester. | ||
Results and discussion
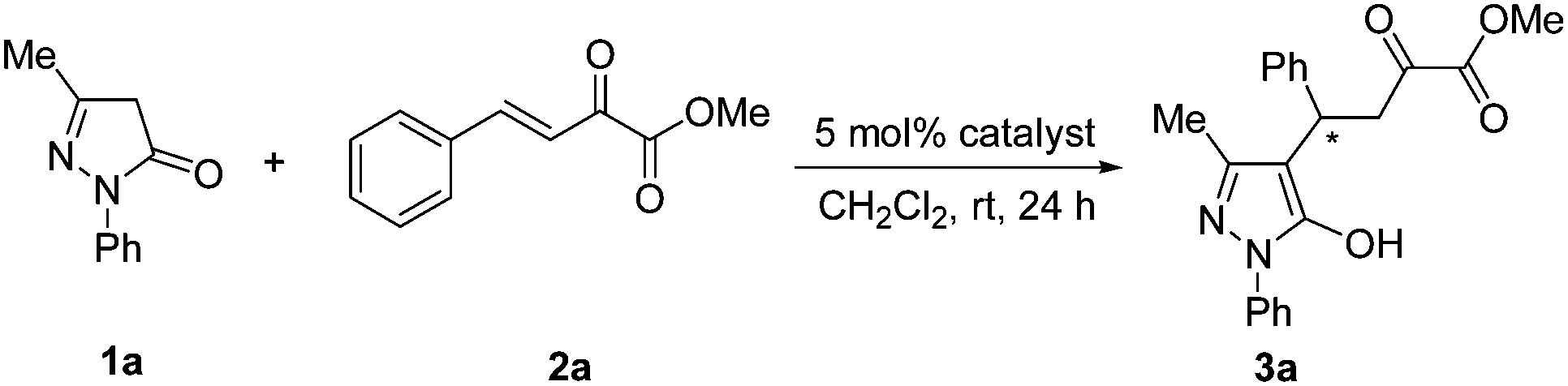
A series of chiral squaramide-based organocatalysts I–VI (Fig. 1) were used to catalyze the Michael addition of 3-methyl-1-phenyl-2-pyrazolin-5-one (1a) to β,γ-unsaturated α-ketoester (2a). The reaction was first performed in CH2Cl2 in the presence of 5 mol% catalysts at room temperature, and the screening results are given in Table 1. Squaramides I–III derived from chiral 1,2-diaminocyclohexane afforded the product 3a in good yield with moderate to good enantioselectivity (Table 1, entries 1–3). Squaramides I and II was found to afford a better enantioselectivity than squaramide III, this indicates that the sterically hindered piperidinyl group is beneficial for the enantioselectivity. Compared with other catalysts, the quinine-derived squaramide IV and quinidine-derived squaramide VI provided the best results with good yields and high enantioselectivities (Table 1, entries 4 and 6). Considering both the slightly difference of yields and the market price of quinine and quinidine, catalyst IV was selected as the best catalyst for further optimization of the reaction condition.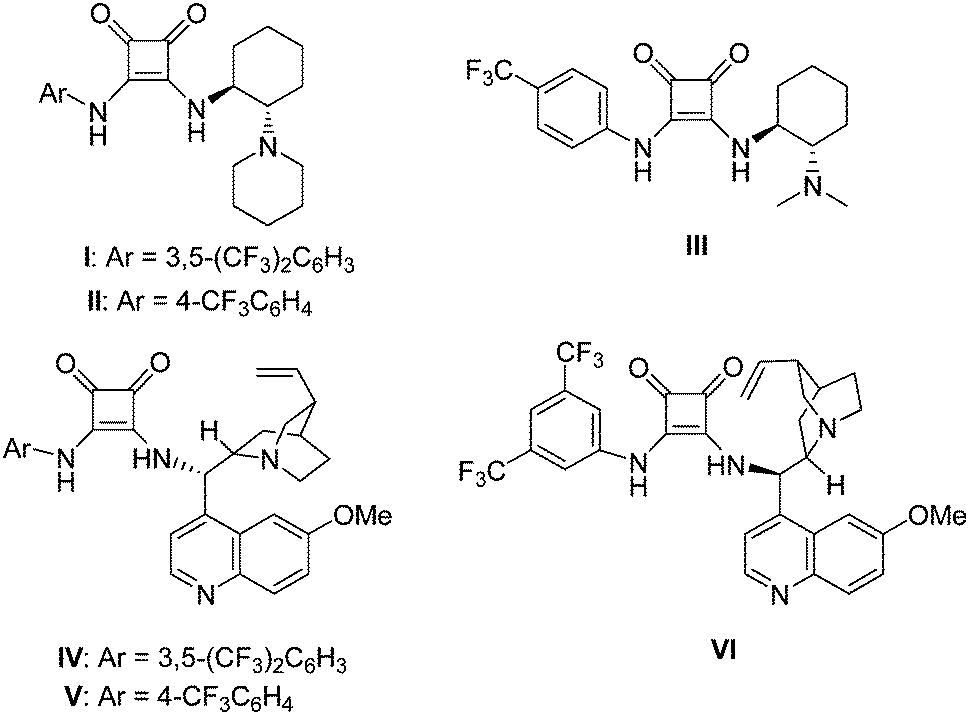 | ||
| Fig. 1 The structures of screened organocatalysts. | ||
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | eec (%) |
| a Reactions were carried out with 3-methyl-1-phenyl-2-pyrazolin-5-one 1a (0.2 mmol) and β,γ-unsaturated α-ketoester 2a (0.25 mmol) in CH2Cl2 (1.0 mL) at room temperature for 24 h.b Isolated yield after column chromatography purification.c Determined by chiral HPLC analysis.d The opposite enantiomer. | |||
| 1 | I | 75 | 88 |
| 2 | II | 83 | 88 |
| 3 | III | 83 | 69 |
| 4 | IV | 75 | 94 |
| 5 | V | 76 | 89 |
| 6 | VI | 73 | 94d |
With squaramide IV as the best catalyst, we further screened the effect of solvents, catalyst loading, temperature and the amounts of solvent for the optimal reaction conditions. The results are summarized in Table 2. Variation of the solvents had a pronounced effect on the yields and enantioselectivities. After CH2Cl2 was used as the solvent, several other common solvents were also been evaluated. Compared with CH2Cl2, the solvents ClCH2CH2Cl, CHCl3, PhCH3, CH3CN, THF and Et2O gave the similar yields but with lower enantioselectives. The proton solvent such as MeOH or H2O is often a poor solvent in hydrogen bond-controlled organocatalysis, but in this reaction even higher enantioselectivities was obtained than in CH2Cl2. Unfortunately, low yield was obtained in MeOH or H2O. After a variety of solvents had been surveyed, the best result was achieved in xylene with good yield and high enantioselectivity (95% ee). Different catalyst loadings were also tried (Table 2, entries 11–12), 10 mol% catalyst IV enhance the enantioselectivity to 96% ee. Thus, 10 mol% catalyst loading was used for further optimization. Next, the effect of temperature and amount of solvent was also investigated (Table 2, entries 13–17), and it was proved that the room temperature and 1 mL of solvent was better than other conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Loading (mol%) | Yieldb (%) | eec (%) |
| a Reactions were carried out with 3-methyl-1-phenyl-2-pyrazolin-5-one 1a (0.20 mmol) and β,γ-unsaturated α-ketoester 2a (0.25 mmol) in solvent (1.0 mL).b Isolated yield after column chromatography purification.c Determined by chiral HPLC analysis.d 0.5 mL solvent was used.e 2.0 mL solvent was used. | |||||
| 1 | CH2Cl2 | rt | 5 | 75 | 94 |
| 2 | ClCH2CH2Cl | rt | 5 | 62 | 78 |
| 3 | CHCl3 | rt | 5 | 75 | 87 |
| 4 | PhCH3 | rt | 5 | 84 | 85 |
| 5 | CH3CN | rt | 5 | 85 | 91 |
| 6 | Xylene | rt | 5 | 75 | 95 |
| 7 | THF | rt | 5 | 74 | 93 |
| 8 | Et2O | rt | 5 | 67 | 88 |
| 9 | MeOH | rt | 5 | 33 | 93 |
| 10 | H2O | rt | 5 | 49 | 96 |
| 11 | Xylene | rt | 2 | 95 | 93 |
| 12 | Xylene | rt | 10 | 72 | 96 |
| 13 | Xylene | −20 | 10 | 85 | 86 |
| 14 | Xylene | 0 | 10 | 83 | 93 |
| 15 | Xylene | 50 | 10 | 66 | 86 |
| 16d | Xylene | rt | 10 | 57 | 96 |
| 17e | Xylene | rt | 10 | 61 | 95 |
Under the optimized conditions with 10 mol% catalyst IV, room temperature, and 1 mL of xylene as the solvent, a diverse array of substituted pyrazolin-5-ones and β,γ-unsaturated α-ketoesters were examined. The results are summarized in Table 3. Various β,γ-unsaturated α-ketoesters bearing either electron-withdrawing or electron-donating groups (F, Cl, Br, Me and OMe) were investigated, and moderate to good yields (64% to 84%) with high enantioselectivities (93% to 96% ee) were obtained (Table 3, entries 2–6). 1-(4-Chlorophenyl)-3-methyl-2-pyrazolin-5-one 1b (Table 3, entry 7), 3-methyl-1-tolyl-2-pyrazolin-5-one 1c (Table 3, entry 8), were also used as the Michael donors, the corresponding products were obtained in good yields with high enantioselectivities (92% and 96% ee). When 3-trifluoromethyl-1-phenyl-2-pyrazolin-5-one 1d was used as Michael donor (Table 3, entry 9 and entry 14), the enantioselectivity was reduced owing to the strong electron-withdrawing ability of the CF3 group. At the same time the occurrence of side reactions was greatly reduced, and excellent yields (99% and 99%) were obtained. In the cases of 1,3-diphenyl-2-pyrazolin-5-one 1e, good yield was still maintained, but with very low enantioselectivity (16% ee and 19% ee) (Table 3, entry 10 and entry 13). This result demonstrated the steric effect in this asymmetric catalytic reaction. 3-Ethyl-1-phenyl-2-pyrazolin-5-one 1f was also used as Michael donor (Table 3, entry 15), compared with 3a, the yield of 3o was slightly increased and similar enantioselectivity was obtained (81% yield and 94% ee). Furthermore, 3k and 3l were also obtained through the Michael addition of 3-methyl-1-phenyl-2-pyrazolin-5-one to the benzyl or allyl α-ketoesters in moderate to good yields (65% and 83%) and high enantioselectivities (90% and 91%) (Table 3, entry 11–12).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | 2 | R3 | R4 | Product | Yieldb (%) | eec (%) |
| a Unless noted otherwise, reactions were carried out with pyrazolin-5-ones 1 (0.2 mmol), β,γ-unsaturated α-ketoesters 2 (0.25 mmol) and catalyst IV (10 mol%) in xylene (1.0 mL) at room temperature.b Isolated yield after column chromatography purification.c Determined by chiral HPLC analysis using a Daicel Chiralpak AD-H or OJ-H column. | |||||||||
| 1 | 1a | Me | Ph | 2a | Ph | Me | 3a | 72 | 96 |
| 2 | 1a | Me | Ph | 2b | 4-ClC6H4 | Me | 3b | 64 | 96 |
| 3 | 1a | Me | Ph | 2c | 4-BrC6H4 | Me | 3c | 84 | 94 |
| 4 | 1a | Me | Ph | 2d | 4-FC6H4 | Me | 3d | 65 | 93 |
| 5 | 1a | Me | Ph | 2e | 4-MeC6H4 | Me | 3e | 71 | 94 |
| 6 | 1a | Me | Ph | 2f | 4-MeOC6H4 | Me | 3f | 64 | 96 |
| 7 | 1b | Me | 4-ClC6H4 | 2a | Ph | Me | 3g | 80 | 92 |
| 8 | 1c | Me | 4-MeC6H4 | 2a | Ph | Me | 3h | 77 | 96 |
| 9 | 1d | CF3 | Ph | 2a | Ph | Me | 3i | 99 | 77 |
| 10 | 1e | Ph | Ph | 2a | Ph | Me | 3j | 75 | 16 |
| 11 | 1a | Me | Ph | 2g | Ph | Bn | 3k | 65 | 90 |
| 12 | 1a | Me | Ph | 2h | Ph | Allyl | 3l | 83 | 91 |
| 13 | 1e | Ph | Ph | 2c | 4-BrC6H4 | Me | 3m | 92 | 19 |
| 14 | 1d | CF3 | Ph | 2c | 4-BrC6H4 | Me | 3n | 99 | 78 |
| 15 | 1f | Et | Ph | 2a | Ph | Me | 3o | 81 | 94 |
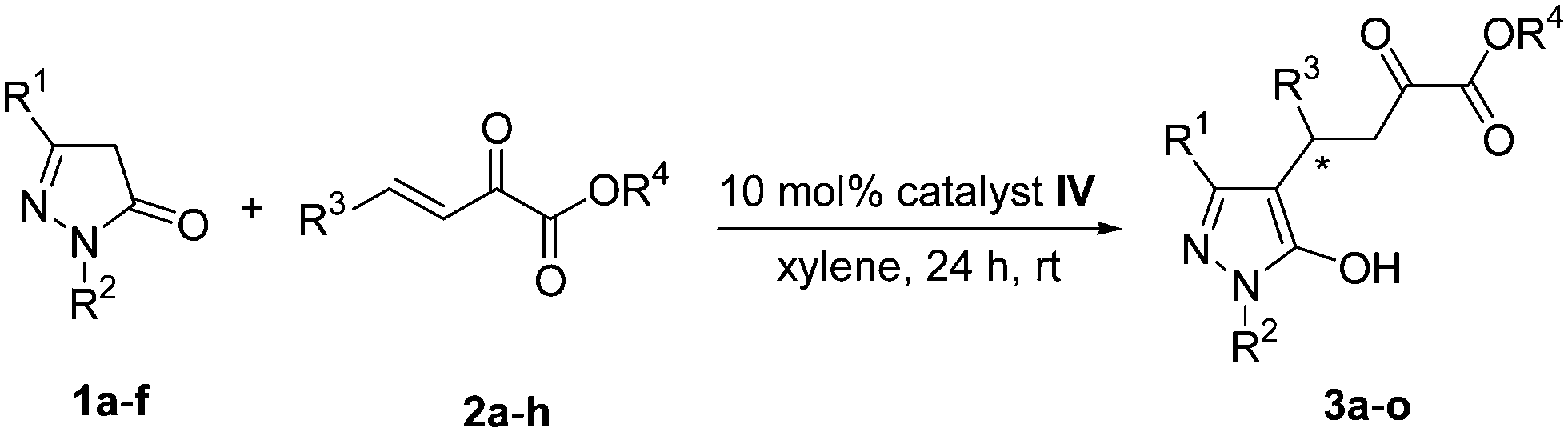
To evaluate the synthetic potential of this catalytic reaction, the synthetic transformation of 3a was performed. As shown in Scheme 3, the reaction of 3a with 3-bromo-1-propyne in the presence of potassium carbonate afforded the propargyloxy derivative 4 in 38% yield with 96% ee. The alkyne group can be used for other valuable transformation.
 | ||
| Scheme 3 The reaction of 3a with 3-bromo-1-propyne. | ||
Accord the previous reported dual activation model,5e,7b a possible transition state is proposed (Fig. 2). The α-keto ester 2a is assumed to interact with the squaramide moiety of IV through double hydrogen bonds, which activates and fixes the substrate by a bidentate interaction. The 3-methyl-1-phenyl-2-pyrazolin-5-one 1a is deprotonated by the basic nitrogen atom of the tertiary amine via tautomerization. The deprotonated form of pyrazolin-5-one 1a is assumed to interact with the tertiary amine group of quinucidine through the hydrogen bond between the protonated amine and the oxygen atom of 1a. This well-assembled transition state facilitates the nucleophile 1a to attack the α-keto ester 2a from the Re-face and to form the (R) product.
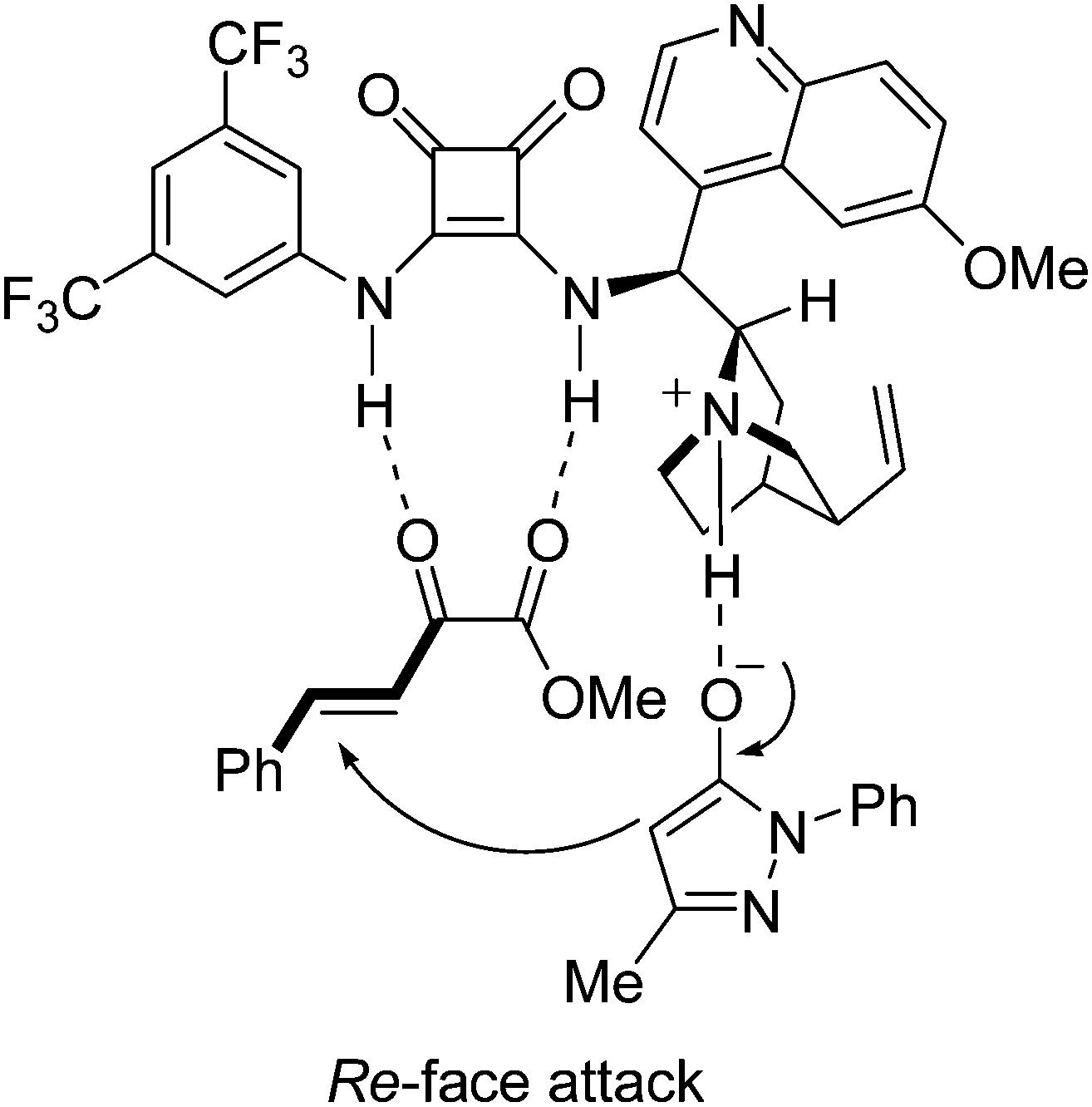 | ||
| Fig. 2 The proposed transition state for enantioselective Michael addition. | ||
Conclusions
In summary, we have successfully developed an organocatalytic enantioselective Michael addition of pyrazolin-5-ones to β,γ-unsaturated α-ketoesters. The corresponding Michael adducts are obtained in moderate to high yields (up to 99%) with high enantioselectivities (up to 96% ee). This catalytic asymmetric reaction provides easy access to chiral pyrazolone ketoester derivatives, which possess potential pharmaceutical activity. Further studies on chiral squaramides are underway in our group to broaden their applications in asymmetric catalysis.Experimental
General information
Commercially available compounds were used without further purification. Column chromatography was carried out using silica gel (200–300 mesh). Melting points were measured with an XT-4 melting point apparatus without correction. The 1H NMR spectra were recorded with a Varian Mercury-plus 400 MHz or a Bruker AVIII 400 MHz spectrometer, while 13C NMR spectra were recorded at 100 MHz. Infrared spectra were obtained with a Perkin Elmer Spectrum One FT-IR spectrometer. The ESI-HRMS spectra were obtained with a Bruker APEX IV FTMS spectrometer. Optical rotations were measured with a WZZ-3 polarimeter at the indicated concentration with unit g per 100 mL. The enantiomeric excesses of the products were determined by chiral HPLC using Agilent 1200 LC instrument using Daicel Chiralpak AD-H or OJ-H columns.Materials
The squaramide organocatalysts were prepared following the reported procedures.8 The racemic compounds 5 were obtained using Et3N as catalyst in Michael addition.General procedure for the enantioselective Michael addition reaction
A mixture of pyrazolone 1 (0.2 mmol), β,γ-unsaturated α-ketoesters 2 (0.25 mmol) and the catalyst IV (0.02 mmol) in xylene (1.0 mL) was stirred at room temperature for 24 h. Next, the mixture was concentrated and purified by silica gel column chromatography (with ethyl acetate–petroleum ether 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 as the eluent) to afford the desired products 3.
:3 as the eluent) to afford the desired products 3.
General procedure for the reaction of products 3 with di-tert-butyl dicarbonate
A mixture of 3 (1 equiv.), di-tert-butyl dicarbonate (2 equiv.) and DMAP (0.2 equiv.) in CH2Cl2 (2 mL) was stirred at room temperature for 2 h. Next, the mixture was concentrated and purified by silica gel column chromatography (with ethyl acetate–petroleum ether 1:8 as the eluent) to afford the desired products 5.
After the products 3 for Michael addition of pyrazolones to β,γ-unsaturated α-ketoesters were obtained, the 1H NMR were measured in DMSO. As shown in Scheme 2, the product was exist with three tautomeric forms in solution, so we can see complex peaks with three tautomeric compounds exit in each NMR spectrum. It is too complicated that can not distinguish between three tautomeric forms. But it was a real compound exist in our hand, so some special method should be performed here to characterize the compounds 3.
As shown in Scheme 4, in order to characterize these compounds 3, Boc protection was performed. In the following characterization part of this paper, the yields, ee values and melting points were measured with compounds 3. The structure confirmation by 1H NMR, 13C NMR, IR and MS were measured with compounds 5. The optical rotations were also measured with compounds 5.
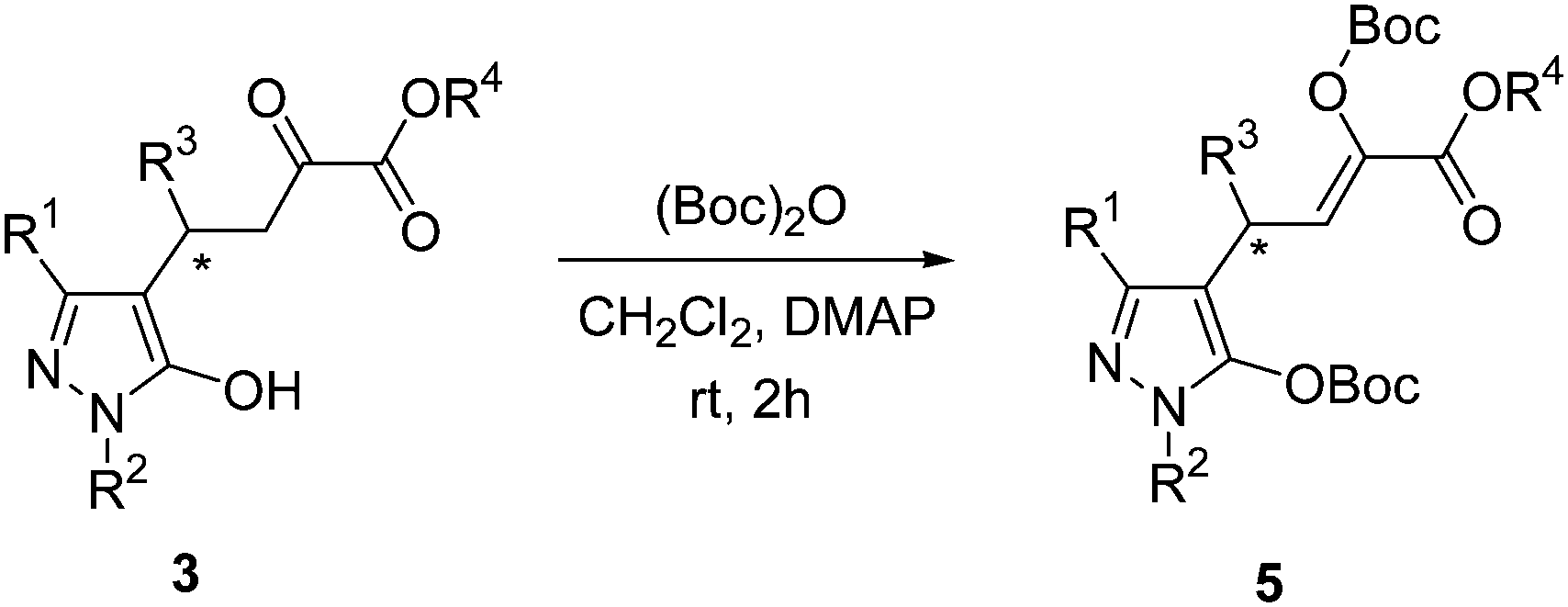 | ||
| Scheme 4 The reaction of compounds 3 with di-tert-butyl dicarbonate. | ||
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 13.9 min, major enantiomer tR = 22.0 min, 96% ee.
Boc-protected compound 5a. [α]25D = +305.77 (c = 1.335, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.54 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 2H, ArH), 7.41 (t, J = 7.8, 2H, ArH), 7.32–7.27 (m, 4H, ArH), 7.25–7.20 (m, 2H, ArH), 6.91 (d, J = 10.4 Hz, 1H, CH), 5.10 (d, J = 10.4, 1H, CH), 3.80 (s, 3H, OCH3), 2.15 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.23 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.3, 150.4, 148.4, 147.4, 142.2, 139.7, 138.1, 137.9, 129.6, 129.1, 128.5, 127.5, 127.1, 126.8, 122.5, 107.9, 85.3, 84.0, 52.3, 36.2, 27.4, 27.0, 13.5 ppm. IR (KBr): ν 3063, 2979, 2934, 1760, 1734, 1598, 1505, 1437, 1371, 1286, 1247, 1149, 1119, 1079, 761, 697 cm−1. HRMS (ESI): m/z calcd for C31H37N2O8 [M + H]+ 565.25444, found 565.25547.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 16.5 min, major enantiomer tR = 33.4 min, 96% ee.
Boc-protected compound 5b. [α]25D = −21.5 (c = 0.4, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.52 (d, J = 8.4 Hz, 2H, ArH), 7.41 (t, J = 8.0 Hz, 2H, ArH), 7.30–7.20 (m, 5H, ArH), 6.85 (d, J = 10.4 Hz, 1H, CH), 5.06 (d, J = 10.4 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 2.16 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.24 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.1, 150.3, 148.4, 147.2, 142.1, 138.3, 138.2, 137.8, 132.6, 129.1, 129.0, 128.9, 128.6, 127.2, 122.5, 107.6, 85.4, 84.2, 52.4, 35.7, 27.4, 27.0, 13.4 ppm. IR (KBr): ν 2982, 2934, 1778, 1764, 1737, 1599, 1507, 1491, 1437, 1387, 1372, 1272, 1246, 1148, 1120, 879, 764, 695 cm−1. HRMS (ESI): m/z calcd for C31H36ClN2O8 [M + H]+ 599.21547, found 599.21541.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 19.0 min, major enantiomer tR = 34.8 min, 94% ee.
Boc-protected compound 5c. [α]25D = −100.9 (c = 0.56, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.52 (d, J = 7.6 Hz, 2H, ArH), 7.43–7.39 (m, 4H, ArH), 7.32–7.27 (m, 1H, ArH), 7.15 (d, J = 8.0 Hz, 2H, ArH), 6.84 (d, J = 10.4, 1H, CH), 5.03 (d, J = 10.0 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 2.16 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.24 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.1, 150.3, 148.4, 147.3, 142.2, 138.8, 138.4, 137.8, 131.6, 129.4, 129.1, 128.8, 127.2, 122.6, 120.8, 107.5, 85.5, 84.2, 52.5, 35.8, 27.4, 27.0, 13.4 ppm. IR (KBr): ν 2982, 2934, 1764, 1736, 1598, 1507, 1488, 1437, 1371, 1272, 1246, 1148, 1120 1072, 1011, 878, 763, 694 cm−1. HRMS (ESI): m/z calcd for C31H36BrN2O8 [M + H]+ 643.16496, found 643.16637.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 16.9 min, major enantiomer tR = 42.3 min, 93% ee.
Boc-protected compound 5d. [α]25D = +17.5 (c = 0.72, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.0 Hz, 2H, ArH), 7.41 (t, J = 7.8 Hz, 2H, ArH), 7.31–7.22 (m, 3H, ArH), 6.99 (t, J = 8.6 Hz, 2H, ArH), 6.86 (d, J = 10.0 Hz, 1H, CH), 5.06 (d, J = 10.0 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 2.16 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.24 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.2, 161.7 (d, 1JC–F = −244.0 Hz), 150.3, 148.4, 147.3, 142.1, 138.2, 137.8, 135.4 (d, 3JC–F = 3.1 Hz), 129.3, 129.2, 129.1, 127.2, 122.6, 115.3 (d, 2JC–F = 21.2 Hz), 107.8, 85.4, 84.1, 52.4, 35.5, 27.4, 27.0, 13.4 ppm. IR (KBr): ν 2984, 1776, 1764, 1736, 1599, 1508, 1438, 1372, 1265, 1246, 1147, 1119, 879, 842, 763, 748, 703 cm−1. HRMS (ESI): m/z calcd for C31H36FN2O8 [M + H]+ 583.24502, found 583.24430.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 10.3 min, major enantiomer tR = 17.0 min, 94% ee.
Boc-protected compound 5e. [α]25D = −7.4 (c = 1.81, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.4 Hz, 2H, ArH), 7.41 (t, J = 8.0 Hz, 2H, ArH), 7.30–7.27 (m, 1H, ArH), 7.16–7.04 (m, 4H, ArH), 6.90 (d, J = 10.4 Hz, 1H, CH), 5.05 (d, J = 10.4 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 2.30 (s, 3H, CH3), 2.15 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.23 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.3, 150.4, 148.4, 147.5, 142.2, 138.0, 137.9, 136.7, 136.3, 129.9, 129.2, 129.1, 127.4, 127.1, 122.6, 108.1, 85.2, 84.0, 52.3, 35.9, 27.5, 27.0, 20.9, 13.5 ppm. IR (KBr): ν 2983, 1776, 1764, 1736, 1598, 1507, 1435, 1371, 1270, 1246, 1147, 1117, 878, 763 cm−1. HRMS (ESI): m/z calcd for C32H39N2O8 [M + H]+ 579.27009, found 579.26889.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 22.3 min, major enantiomer tR = 41.4 min, 96% ee.
Boc-protected compound 5f. [α]25D = −8.79 (c = 2.275, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.54–7.52 (m, 2H, ArH), 7.41 (t, J = 7.8 Hz, ArH), 7.30 (d, J = 7.6 Hz, 1H, ArH), 7.17 (d, J = 8.4 Hz, 2H, ArH), 6.88 (d, J = 10.4 Hz, 1H, CH), 6.83 (d, J = 8.8 Hz, 2H, ArH), 5.04 (d, J = 10.4 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 2.15 (s, 3H, CH3), 1.49 (s, 9H, tBu), 1.24 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.3, 158.4, 150.4, 148.5, 147.4, 142.2, 138.0, 137.8, 131.8, 129.9, 129.1, 128.6, 127.1, 122.6, 113.9, 108.1, 85.2, 84.0, 55.2, 52.3, 35.5, 27.5, 27.0, 13.5 ppm. IR (KBr): ν 2983, 2956, 2930, 1775, 1763, 1735, 1598, 1508, 1437, 1371, 1272, 1248, 1148, 1121, 1032, 764, 694 cm−1. HRMS (ESI): m/z calcd for C32H39N2O9 [M + H]+ 595.26501, found 595.26485.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 12.0 min, major enantiomer tR = 9.5 min, 92% ee.
Boc-protected compound 5g. [α]25D = +16.9 (c = 0.675, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.49 (d, J = 8.8 Hz, 2H, ArH), 7.38 (d, J = 8.8 Hz, 2H, ArH), 7.32–7.20 (m, 5H, ArH), 6.88 (d, J = 10.4 Hz, 1H, CH), 5.08 (d, J = 10.4 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 2.13 (s, 3H, CH3), 1.48 (s, 9H, tBu), 1.28 (s, 9H, tBu) ppm; 13C NMR (400 MHz, CDCl3): δ = 162.3, 150.4, 148.5, 147.9, 142.2, 139.6, 138.2, 136.6, 132.7, 129.4, 129.2, 128.6, 127.6, 126.9, 123.6, 108.4, 85.6, 84.1, 52.4, 36.2, 27.5, 27.1, 13.5 ppm. IR (KBr): ν 2983, 2924, 2854, 1778, 1764, 1737, 1599, 1503, 1438, 1371, 1274, 1244, 1147, 1119, 1053, 878, 834, 768, 700 cm−1. HRMS (ESI): m/z calcd for C31H36ClN2O8 [M + H]+ 599.21547, found 599.21504.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 13.6 min, major enantiomer tR = 10.7 min, 96% ee.
Boc-protected compound 5h. [α]25D = +17.0 (c = 2.015, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.40 (d, J = 8.0 Hz, 2H, ArH), 7.29–7.25 (m, 5H, ArH), 7.20 (d, J = 8.0 Hz, 2H, ArH), 6.90 (d, J = 10.4 Hz, 1H, CH), 5.09 (d, J = 10.4 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 2.35 (s, 3H, CH3), 2.14 (s, 3H), 1.48 (s, 9H, tBu), 1.24 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.3, 150.4, 148.5, 147.1, 142.1, 139.8, 138.0, 137.0, 135.5, 129.7, 129.6, 128.5, 127.6, 126.8, 122.6, 107.7, 85.2, 84.0, 52.4, 36.2, 27.5, 27.1, 21.0, 13.5 ppm. IR (KBr): ν 2983, 2926, 1776, 1764, 1736, 1600, 1519, 1440, 1371, 1270, 1246, 1148, 1119, 1084, 880, 821, 767, 700 cm−1. HRMS (ESI): m/z calcd for C32H39N2O8 [M + H]+ 579.27009, found 579.26950.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 5.2 min, major enantiomer tR = 7.3 min, 77% ee.
Boc-protected compound 5i. [α]25D = +13.3 (c = 2.065, CH2Cl2). 1H NMR (400 MHz, CDCl3): 7.56 (d, J = 8.4 Hz, 2H, ArH), 7.46 (t, J = 7.2 Hz, 2H, ArH), 7.40 (d, J = 8.0 Hz, 1H, ArH), 7.31–7.21 (m, 5H, ArH), 6.96 (d, J = 10.4 Hz, 1H, CH), 5.33 (d, J = 10.0 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 1.48 (s, 9H, tBu), 1.16 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.2, 150.2, 147.4, 143.2, 139.5 (q, 2JC–F = 37.2 Hz), 139.3, 138.7, 137.1, 129.3, 128.63, 128.58, 128.51, 127.4, 127.1, 123.4, 121.0 (q, 1JC–F = 69.1 Hz), 109.2, 86.2, 84.2, 52.5, 35.5, 27.4, 26.9 ppm. IR (KBr): ν 2984, 2934, 1784, 1765, 1739, 1599, 1497, 1451, 1392, 1373, 1282, 1254, 1229, 1155, 1136, 1012, 876, 765, 749, 698 cm−1. HRMS (ESI): m/z calcd for C31H33F3N2NaO8 [M + Na]+ 641.20812, found 641.20956.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 18.1 min, major enantiomer tR = 15.2 min, 16% ee.
Boc-protected compound 5j. [α]25D = +2.18 (c = 2.645, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.65 (d, J = 7.6 Hz, 2H, ArH), 7.58–7.56 (m, 2H, ArH), 7.44 (t, J = 7.6 Hz, 2H, ArH), 7.40–7.32 (m, 4H, ArH), 7.27–7.26 (m, 4H, ArH), 7.22–7.16 (m, 1H, ArH), 6.99 (d, J = 10.0 Hz, 1H, CH), 5.30 (d, J = 10.0 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 1.40 (s, 9H, tBu), 1.18 (s, 9H, tBu) ppm; 13C NMR (400 MHz, CDCl3): δ = 162.3, 150.6, 150.3, 147.9, 142.8, 140.3, 138.04, 137.96, 132.9, 130.1, 129.1, 128.5, 128.4, 128.1, 127.53, 127.45, 126.8, 122.8, 107.8, 85.5, 83.9, 52.3, 36.4, 27.4, 26.9 ppm. IR (KBr): ν 3063, 2982, 2934, 1781, 1763, 1736, 1597, 1571, 1503, 1455, 1372, 1273, 1244, 1149, 1136, 1115, 877, 763, 698 cm−1. HRMS (ESI): m/z calcd for C36H39N2O8 [M + H]+ 627.27009, found 627.27162.
:15, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 12.6 min, major enantiomer tR = 15.2 min, 90% ee.
Boc-protected compound 5k. [α]25D = +13.3 (c = 1.46, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.4 Hz, 2H, ArH), 7.43–7.31 (m, 6H, ArH), 7.29–7.20 (m, 7H, ArH), 6.96 (d, J = 10.4 Hz, 1H, CH), 5.24 (s, 2H, CH2), 5.10 (d, J = 10.4 Hz, 1H, CH), 2.15 (s, 3H, CH3), 1.41 (s, 9H, tBu), 1.20 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 161.8, 150.3, 147.4, 139.7, 138.2, 137.9, 135.2, 129.9, 129.1, 128.7, 128.55, 128.47, 128.3, 128.2, 127.6, 127.1, 126.8, 122.6, 109.6, 107.9, 85.3, 84.0, 67.2, 36.2, 27.4, 27.0, 13.5 ppm. IR (KBr): ν 2981, 2930, 1776, 1762, 1731, 1598, 1505, 1454, 1371, 1270, 1246, 1221, 1147, 1117, 1081, 799, 759, 697 cm−1. HRMS (ESI): m/z calcd for C37H41N2O8 [M + H]+ 641.28574, found 641.28647.
:15, flow rate 1.0 mL min−1, detection at 254 nm): major enantiomer tR = 9.6 min, minor enantiomer tR = 13.0 min, 91% ee.
Boc-protected compound 5l. [α]25D = −6.8 (c = 0.97, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.0 Hz, 2H, ArH), 7.41 (t, J = 7.6 Hz, 2H, ArH), 7.32–7.21 (m, 6H, ArH), 6.94 (d, J = 9.6 Hz, 1H, CH), 5.99–5.89 (m, 1H, CH), 5.36 (d, J = 17.2 Hz, 1H, CH), 5.25 (d, J = 10.4 Hz, 1H, CH), 5.11 (d, J = 10.4 Hz, 1H, CH), 4.71 (d, J = 6.8 Hz, 2H, CH2), 2.16 (s, 3H, CH3), 1.48 (s, 9H, tBu), 1.23 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 161.5, 150.4, 148.4, 147.4, 142.2, 139.7, 138.2, 138.0, 131.5, 129.7, 129.1, 128.6, 127.6, 127.1, 126.8, 122.6, 118.6, 107.9, 85.3, 84.0, 66.1, 36.2, 27.5, 27.0, 13.5 ppm. IR (KBr): ν 2982, 2934, 1776, 1762, 1733, 1598, 1506, 1454, 1371, 1270, 1246, 1225, 1148, 1119, 1081, 878, 762, 698 cm−1. HRMS (ESI): m/z calcd for C33H39N2O8 [M + H]+ 591.27009, found 591.27083.
:15, flow rate 1.0 mL min−1, detection at 254 nm): major enantiomer tR = 22.7 min, minor enantiomer tR = 15.7 min, 19% ee.
Boc-protected compound 5m. [α]25D = −3.89 (c = 3.14, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.64 (d, J = 8.0 Hz, 2H, ArH), 7.56 (d, J = 7.2 Hz, 2H, ArH), 7.44 (t, J = 7.8 Hz, 2H, ArH), 7.40–7.30 (m, 6H, ArH), 7.13 (d, 2H, J = 8.4 Hz, 2H, ArH), 6.92 (d, J = 10.0 Hz, 1H, CH), 5.23 (d, J = 10.0 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 1.40 (s, 9H, tBu), 1.19 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.1, 150.4, 150.2, 147.9, 142.7, 139.3, 138.4, 137.8, 132.7, 131.5, 129.40, 129.36, 129.2, 128.5, 128.4, 128.3, 127.6, 122.8, 120.8, 107.6, 85.7, 84.0, 52.4, 36.0, 27.4, 26.9 ppm. IR (KBr): ν 2980, 2934, 1780, 1764, 1737, 1596, 1572, 1503, 1486, 1455, 1396, 1371, 1274, 1244, 1149, 1136, 1121, 1073, 1010, 876, 763, 696 cm−1. HRMS (ESI): m/z calcd for C36H38BrN2O8 [M + H]+ 705.18061, found 705.18110.
:15, flow rate 1.0 mL min−1, detection at 254 nm): major enantiomer tR = 10.7 min, minor enantiomer tR = 6.2 min, 78% ee.
Boc-protected compound 5n. [α]25D = −6.0 (c = 4.56, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.55 (d, J = 6.4 Hz, 2H, ArH), 7.46 (t, J = 6.8 Hz, 3H, ArH), 7.41 (d, J = 8.0 Hz, 2H, ArH), 7.14 (d, J = 6.8 Hz, 2H, ArH), 6.88 (d, J = 10.0 Hz, 1H, CH), 5.28 (d, J = 9.6 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 1.48 (s, 9H, tBu), 1.18 (s, 9H, tBu) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.0, 150.1, 147.5, 143.2, 139.4 (q, 2JC–F = 37.3 Hz), 139.0, 138.4, 137.0, 131.6, 129.3, 129.2, 128.7, 127.7, 123.3, 121.1, 121.0 (q, 1JC–F = −268.5 Hz), 108.7, 86.5, 84.3, 52.5, 35.1, 27.4, 26.9 ppm. IR (KBr): ν 2983, 2956, 2934, 1783, 1765, 1738, 1599, 1550, 1489, 1451, 1394, 1372, 1278, 1253, 1228, 1155, 1138, 1075, 1011, 875, 765, 698 cm−1. HRMS (ESI): m/z calcd for C31H32BrF3N2NaO8 [M + Na]+ 719.11863, found 719.11938.
:5, flow rate 1.0 mL min−1, detection at 254 nm): major enantiomer tR = 9.3 min, minor enantiomer tR = 8.4 min, 91% ee.
Boc-protected compound 5o. [α]25D = +2.67 (c = 3.15, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.56–7.54 (m, 2H, ArH), 7.41 (t, J = 7.8 Hz, 2H, ArH), 7.30–7.26 (m, 5H, ArH), 7.23–7.19 (m, 1H, ArH), 6.93 (d, J = 10.4 Hz, 1H, CH), 5.12 (d, J = 10.4 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 2.54 (q, J = 7.6 Hz, 2H, CH2), 1.48 (s, 9H, tBu), 1.21–1.18 (m, 12H, tBu + CH3) ppm. 13C NMR (400 MHz, CDCl3): δ = 162.3, 152.4, 150.3, 148.4, 142.1, 140.0, 138.1, 137.9, 129.8, 129.0, 128.5, 127.5, 127.0, 126.8, 122.6, 107.3, 85.2, 83.9, 52.3, 36.1, 27.4, 27.0, 20.9, 12.9 ppm. IR (KBr): ν 2980, 2937, 1778, 1764, 1737, 1598, 1506, 1449, 1437, 1394, 1372, 1270, 1244, 1147, 1119, 1087, 878, 765, 698 cm−1. HRMS (ESI): m/z calcd for C32H39N2O8 [M + H]+ 579.27009, found 579.27103.
The reaction of 3a with 3-bromo-1-propyne
A mixture of 3a (44.3 mg, 0.122 mmol), propargyl bromide (29 mg 0.224 mmol) and anhydrous potassium carbonate (50.6 mg, 0.366 mmol) in CH3COCH3 (2 mL) was stirred at room temperature for 3 h. Next, the mixture was concentrated and purified by silica gel column chromatography (with ethyl acetate–petroleum ether 1:2 as the eluent) to afford the desired products 4 as colorless oil (18.7 mg, 38% yield). Enantiomeric excess was determined by HPLC (Daicel Chiralpak OJ-H column, n-hexane–isopropanol 70:30, flow rate 1.0 mL min−1, detection at 254 nm): minor enantiomer tR = 36.3 min, major enantiomer tR = 74.6 min, 96% ee; [α]25D = −23.3 (c = 0.935, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ = 7.48–7.38 (m, 6H, ArH), 7.30–7.18 (m, 4H, ArH), 4.37 (dd, J1 = 4.0 Hz, J2 = 9.6 Hz, 1H), 4.32–4.25 (m, 1H, CH), 4.10 (d, J = 2.0 Hz, 2H, CH2), 3.82 (s, 3H, OCH3), 3.45 (dd, J1 = 4.0 Hz, J2 = 18.8 Hz, 1H, CH), 2.24 (s, 3H, CH3), 2.13 (br s, 1H, CH) ppm. 13C NMR (400 MHz, CDCl3): δ = 192.1, 165.4, 160.9, 154.0, 142.6, 134.5, 130.9, 129.1, 128.5, 127.7, 126.6, 126.4, 123.4, 114.9, 74.3, 74.1, 52.9, 42.9, 38.7, 35.7, 11.1 ppm. IR (KBr): ν 2953, 2924, 2854, 1730, 1660, 1594, 1495, 1456, 1276, 1267, 1263, 1134, 1125, 1087, 1071, 763, 751, 698 cm−1. HRMS (ESI): m/z calcd for C24H23N2O4 [M + H]+ 403.16523, found 403.16552.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (grant no. 21272024).Notes and references
- For selected examples, see: (a) R. K. Boeckman, Jr, J. E. Reed and P. Ge, Org. Lett., 2001, 3, 3651 CrossRef; (b) M. Abass and B. B. Mostafa, Bioorg. Med. Chem., 2005, 13, 6133 CrossRef CAS PubMed; (c) G. Mariappan, B. P. Saha, N. R. Bhuyan, P. R. Bharti and D. Kumar, J. Adv. Pharm. Technol. Res., 2010, 1, 260 CAS; (d) R. Ma, J. Zhu, J. Liu, L. Chen, X. Shen, H. Jiang and J. Li, Molecules, 2010, 15, 3593 CrossRef CAS PubMed; (e) S. Gogoi and C.-G. Zhao, Tetrahedron Lett., 2009, 50, 2252 CrossRef CAS PubMed; (f) M. Radi, V. Bernardo, B. Bechi, D. Castagnolo, M. Pagano and M. Botta, Tetrahedron Lett., 2009, 50, 6572 CrossRef CAS; (g) S. Gogoi, C.-G. Zhao and D. Ding, Org. Lett., 2009, 11, 2249 CrossRef CAS PubMed; (h) Z. G. Yang, Z. Wang, S. Bai, X. Liu, L. Lin and X. Feng, Org. Lett., 2011, 13, 596 CrossRef CAS PubMed; (i) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928 CrossRef CAS PubMed.
- A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Org. Biomol. Chem., 2011, 9, 6519 CAS.
- (a) H. Kawai, H. Nakai, M. Suga, S. Yuki, T. Watanabe and K. I. Saito, J. Pharmacol. Exp. Ther., 1997, 281, 921 CAS; (b) T. Watanabe, S. Yuki, M. Egawa and H. Nishi, J. Pharmacol. Exp. Ther., 1994, 268, 1597 CAS.
- T. W. Wu, L. H. Zeng, J. Wu and K. P. Fung, Life Sci., 2002, 71, 2249 CrossRef CAS PubMed.
- For selected examples, see: (a) F. Li, L. Sun, Y. Teng, P. Yu, J. C.-G. Zhao and J.-A. Ma, Chem. – Eur. J., 2012, 18, 14255 CrossRef CAS PubMed; (b) Z.-C. Geng, X. Chen, J.-X. Zhang, N. Li, J. Chen, X.-F. Huang, S.-Y. Zhang, J.-C. Tao and X.-W. Wang, Eur. J. Org. Chem., 2013, 4738 CrossRef CAS; (c) H.-X. Wang, Y.-M. Wang, H.-B. Song, Z.-H. Zhou and C.-C. Tang, Eur. J. Org. Chem., 2013, 4844 CrossRef CAS; (d) Y.-H. Liao, W.-B. Chen, Z.-J. Wu, X.-L. Du, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Adv. Synth. Catal., 2010, 352, 827 CrossRef CAS; (e) J.-H. Li and D.-M. Du, Org. Biomol. Chem., 2013, 11, 6215 RSC; (f) Z. Wang, Z.-L. Chen, S. Bai, W. Li, X.-H. Liu, L.-L. Lin and X.-M. Feng, Angew. Chem., Int. Ed., 2012, 51, 2776 CrossRef CAS PubMed.
- For selected examples utilizing β,γ-unsaturated α-ketoesters as electrophiles, see: (a) J. Zhou and Y. Tang, Org. Biomol. Chem., 2004, 2, 429 RSC; (b) S.-L. Zhao, C.-W. Zheng and G. Zhao, Tetrahedron: Asymmetry, 2008, 19, 2608 CrossRef; (c) Y.-F. Wang, K. Wang, W. Zhang, B.-B. Zhang, C.-X. Zhang and D.-Q. Xu, Eur. J. Org. Chem., 2012, 3691 CrossRef; (d) D. T. Cohen, C.-D. Benoit and K.-A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 1678 CrossRef CAS PubMed; (e) L. Liu, H.-L. Ma, Y.-M. Xiao, F.-P. Du, Z.-H. Qin, N. Li and B. Fu, Chem. Commun., 2012, 48, 9281 RSC; (f) Z.-H. Dong, J.-H. Feng, W.-D. Cao, X.-H. Liu, L.-L. Lin and X.-M. Feng, Tetrahedron Lett., 2011, 52, 3433 CrossRef CAS.
- For selected examples, see: (a) Y.-J. Gao, Q. Ren, L. Wang and J. Wang, Chem. – Eur. J., 2010, 16, 13068 CrossRef CAS PubMed; (b) D.-Q. Xu, Y.-F. Wang, W. Zhang, S.-P. Luo, A.-G. Zhong, A.-B. Xia and Z.-Y. Xu, Chem. – Eur. J., 2010, 16, 4177 CrossRef CAS PubMed; (c) X.-K. Chen, C.-W. Zheng, S.-L. Zhao, Z. Chai, Y.-Q. Yang, G. Zhao and W.-G. Cao, Adv. Synth. Catal., 2010, 352, 1648 CrossRef CAS.
- (a) W. Yang and D.-M. Du, Org. Lett., 2010, 12, 5450 CrossRef CAS PubMed; (b) W. Yang and D.-M. Du, Adv. Synth. Catal., 2011, 353, 1241 CrossRef CAS; (c) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of new compounds, and HPLC chromatograms. See DOI: 10.1039/c3ra45974h |
| This journal is © The Royal Society of Chemistry 2014 |