Efficient synthesis of isoquinolines and pyridines via copper(I)-catalyzed multi-component reaction†
Kommuri Shekarrao,
Partha Pratim Kaishap,
Sanjib Gogoi* and
Romesh C. Boruah*
Medicinal Chemistry Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, India. E-mail: skgogoi1@gmail.com; rc_boruah@yahoo.co.in; Fax: +913762370011; Tel: +91 3762372948
First published on 31st January 2014
Abstract
A wide range of 3-substituted isoquinolines, steroidal pyridines, 5,6-dihydrobenzo[f]isoquinolines and 1,6-naphthyridine were synthesized in good yield via a ligand-free copper-catalyzed three-component reaction of β-halovinyl/aryl aldehyde, aromatic/aliphatic terminal alkyne and tert-butylamine/benzamidine in DMF under microwave irradiation. A catalyst-free reaction of ortho-alkynyl aryl/vinyl aldehydes with benzamidine under microwave irradiation also provided the 3-substituted isoquinoline and substituted pyridine derivatives in excellent yield.
Introduction
A reaction where one catalyst promotes more than one chemical transformation is always advantageous because this reaction eliminates the isolation of intermediates as well as forming several bonds from readily available starting materials.1 On the other hand, multi-component reactions are recognized as powerful tools for the synthesis of organic compounds, since the products are formed in a single step and the diversity of the products is maintained by simply varying the starting materials.23-Aryl-substituted isoquinolines are of great pharmaceutical importance due to their wide range of biological activities.3 They also constitute the building blocks of numerous biologically important natural products.4 Some of the biologically active 3-arylisoquinoline derivatives are shown in Fig. 1. Synthesis of other pyridine annulated molecules such as steroidal pyridines,5a–f 5,6-dihydrobenzo[f]isoquinolines5g and 1,6-naphthyridine derivatives5h has received considerable attention in the literature as a result of their biological and pharmaceutical importance.
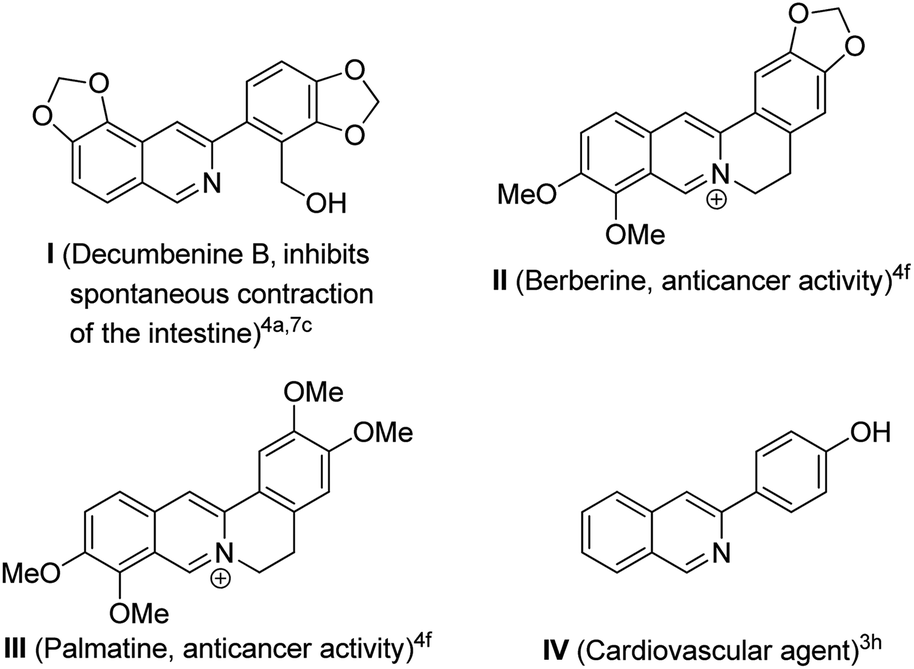 | ||
| Fig. 1 Selected biologically active 3-arylisoquinoline derivatives. | ||
3-Arylisoquinolines are synthesized by Bischler–Napieralski and Pictet–Spengler reactions which require either harsh conditions or tedious reaction procedures.6 Pd-catalyzed annulation processes are widely used to synthesize many nitrogen heterocycles such as isoquinolines7 and 5,6-dihydrobenzo[f]isoquinolines.5g In addition to Pd, other transition metals such as Ni,8a Zr8b and Ag8c are also used to synthesize 3-substituted isoquinoline derivatives. Although these methods are useful for the synthesis of 3-arylisoquinolines and pyridine derivatives, they have some disadvantages such as the stepwise synthesis of tert-butylimines from β-halovinyl aldehydes, followed by coupling reaction of imine intermediates with alkynes and then cyclization of the resultant ortho-alkynyl aryl/vinyl aldehydes in presence of other catalysts (typically CuI),7a–f long reaction time, and low yield of the annulated products with terminal mono-substituted acetylenes.7a Moreover, transition metal complexes used as the catalytic systems are inadequate for large-scale reaction owing to the high cost of these metal complexes. From a synthetic point of view, it is essential for a chemist to develop an improved and facile procedure to synthesize these important heterocycles using a less expensive and more sustainable catalyst. Our interests in the synthesis of biologically important heterocycles have encouraged us to re-investigate a catalytic synthesis version of these important N-heterocycles.9 Herein, we wish to report a microwave-assisted and CuI-catalyzed three-component reaction of β-halovinyl aldehydes, terminal alkynes and tert-butylamine/benzamidine under ligand-free conditions for the synthesis of nitrogen heterocycles. This efficient synthesis of nitrogen heterocycles such as 3-substituted isoquinolines, steroidal pyridines, 5,6-dihydrobenzo[f]isoquinolines and 1,6-naphthyridine eliminates the above-mentioned problems with Pd catalysts.
Results and discussion
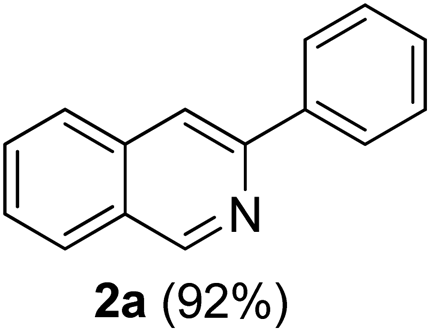
Initial studies were performed by using 2-bromobenzaldehyde 1a, phenylacetylene and tert-butylamine as the model substrates, employing copper(I) iodide as the catalyst (Table 1) and 1,2-dichloroethane (DCE) as the solvent under thermal conditions at 120 °C. Gratifyingly, this reaction proceeded smoothly in DCE to afford regioselectively the annulated product 2a in 27% yield in 12DCEh (entry 1, Table 1). Further investigation revealed that the solvent used played a significant role in this reaction. DMF was the most effective among the tested solvents and the yield of 2a was increased to 61% (entry 2, Table 1), while there was sharp decrease in the yield to 25% (entry 3, Table 1) and 40% (entry 4, Table 1), when toluene and 1,4-dioxane were used respectively. Several other copper sources were also examined (entry 5–8, Table 1) and CuI turned out to be the best among others in view of the yield of the annulated product. The use of Cu(II) salts provided only trace amounts of the product 2a (entry 7–8, Table 1) and no product was formed in the absence of Cu(I) (entry 9,Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Cu salt (mol%) | Solvent | Thermal/MW | Time | Yielda (%) |
| a Isolated yield. | |||||
| 1 | CuI (10) | DCE | 120 °C | 12 h | 27 |
| 2 | CuI (10) | DMF | 120 °C | 12 h | 61 |
| 3 | CuI (10) | Toluene | 120 °C | 12 h | 25 |
| 4 | CuI (10) | 1,4-Dioxane | 120 °C | 12 h | 40 |
| 5 | CuBr (10) | DMF | 120 °C | 12 h | 56 |
| 6 | CuCl (10) | DMF | 120 °C | 12 h | 44 |
| 7 | Cu(OAc)2 (10) | DMF | 120 °C | 12 h | <5 |
| 8 | Cu(OTf)2 (10) | DMF | 120 °C | 12 h | <5 |
| 9 | — | DMF | 120 °C | 12 h | 0 |
| 10 | CuI (10) | DMF | 720 W | 4 min | 92 |
| 11 | CuI (5) | DMF | 720 W | 4 min | 78 |
| 12 | CuI (15) | DMF | 720 W | 4 min | 91 |

At this stage of the investigation, we attempted first to find an efficient system for annulation of the β-bromophenyl aldehyde and terminal alkyne under thermal conditions. Following this we were interested in looking for more attractive reaction conditions in terms of time and reaction yield. Therefore, we examined the influence of microwave (MW) irradiation on the reaction of 1a, phenylacetylene and tert-butylamine in the presence of 10 mol% CuI using the best solvent DMF. A substantial increase in the yield of 2a (92%) along with a significant reduction in the reaction time from 12 h to 4 min was observed (entry 10, Table 1). We also explored the effect of catalyst loading on the yield of the reaction. When the quantity of CuI was reduced to 5 mol%, the yield decreased to 78% (entry 11, Table 1) under microwave irradiation. Increase of catalyst loading to 15 mol% did not affect the yield of the reaction (entry 12, Table 1).
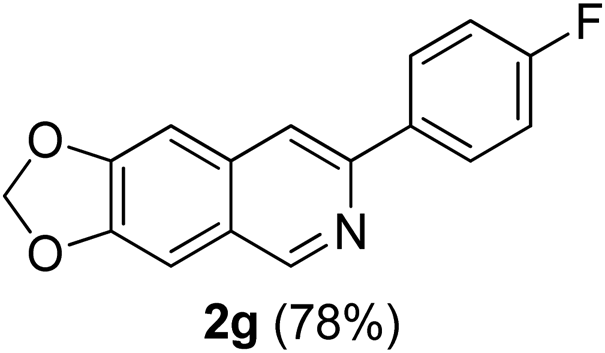
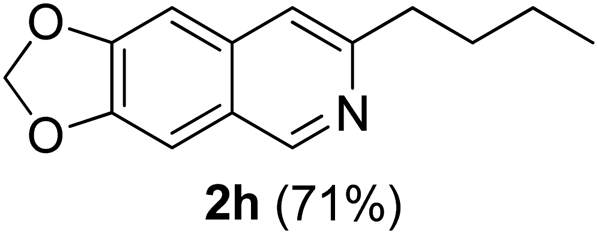
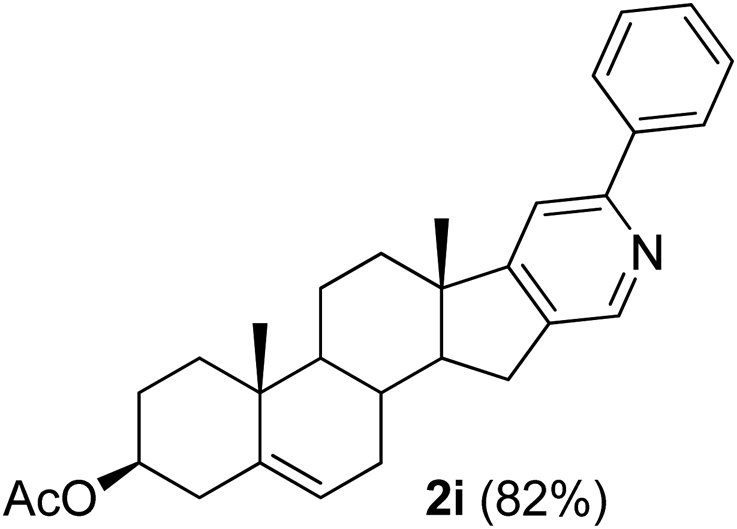
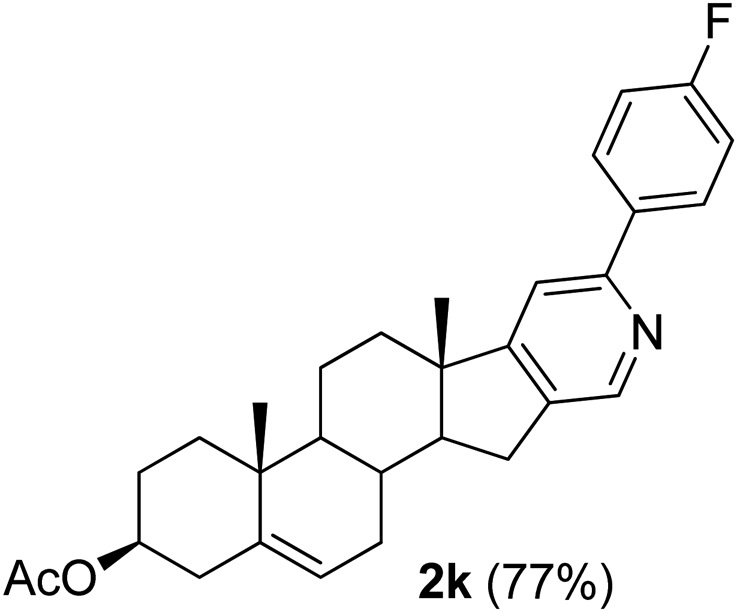
With the optimized reaction conditions in hand (entry 10, Table 1), a series of terminal aliphatic and aromatic alkynes were tested under this condition with 2-bromobenzaldehyde to afford 3-substituted isoquinolines 2b–e in 75–84% yields (entry 2–5, Table 2). In addition to 2-bromobenzaldehyde 1a, we were pleased to observe that 2-chloroaryl aldehydes such as 6-chlorobenzo[1,3]dioxole-5-carbaldehyde (1b) also worked well for this reaction with the same catalyst loading to afford isoquinolines 2f–h in 71–84% yields (entry 6–8, Table 2). It was interesting to note that these optimized reaction conditions were also effective for annulations of β-bromovinyl aldehydes to pyridine derivatives. For example, the annulations of steroidal β-bromovinyl aldehyde 1c9b with different aryl and alkyl group-substituted terminal alkynes under optimized reaction conditions afforded the steroidal pyridines 2i–n in 70–82% yields (entry 9–14, Table 2).
| Entry | β-Halovinyl aldehyde | Alkyne | Productb (% Yield) |
|---|---|---|---|
| a Conditions: 10 mol% CuI, β-halovinyl aldehyde (1 mmol), alkyne (1.1 mmol) and tert-butylamine (2 mmol) in DMF (1 mL) was irradiated in a closed vessel in a Synthos 3000 microwave reactor at 720 W (140 °C and 14 bar) for 4 min.b Refers to the amount of product isolated by chromatography.c The reaction was performed for 8 min. | |||
| 1 |  |
 |
 |
| 2 | 1a | 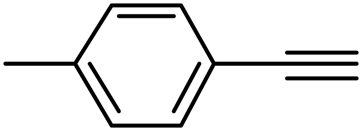 |
 |
| 3 | 1a | 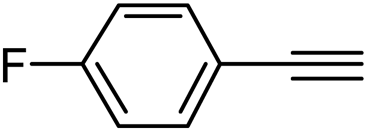 |
 |
| 4 | 1a |  |
 |
| 5 | 1a |  |
 |
| 6 | 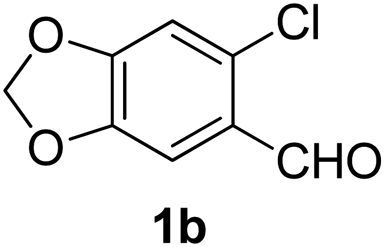 |
 |
 |
| 7 | 1b |  |
 |
| 8 | 1b |  |
 |
| 9 | 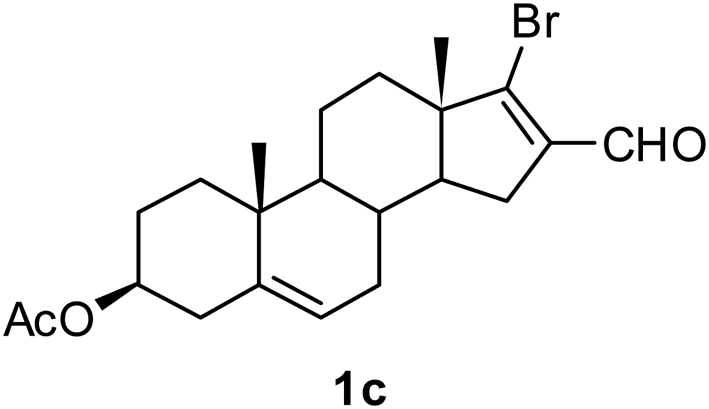 |
 |
 |
| 10 | 1c |  |
 |
| 11 | 1c |  |
 |
| 12 | 1c |  |
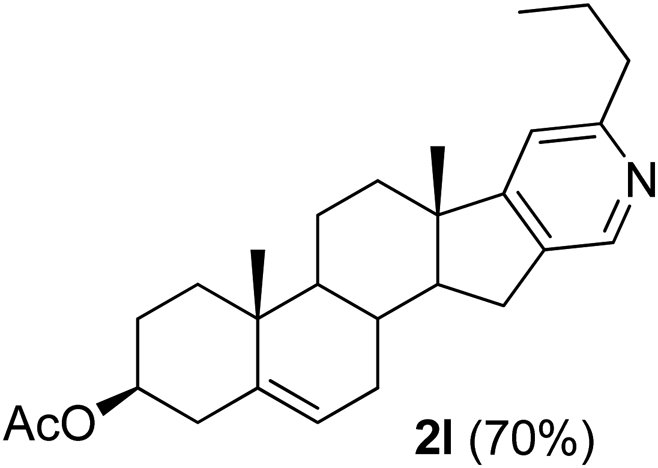 |
| 13 | 1c |  |
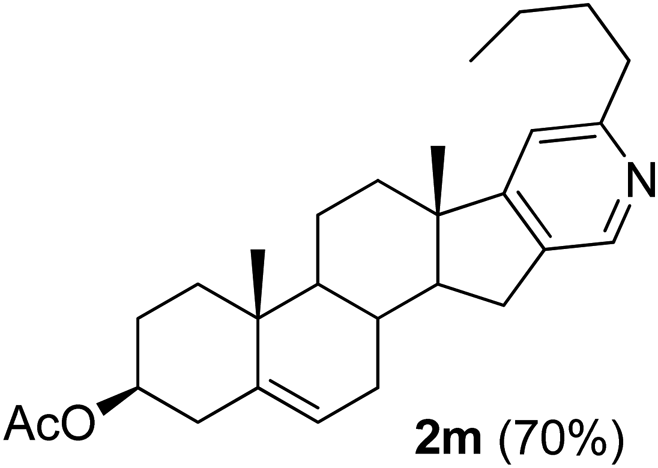 |
| 14 | 1c | 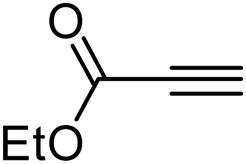 |
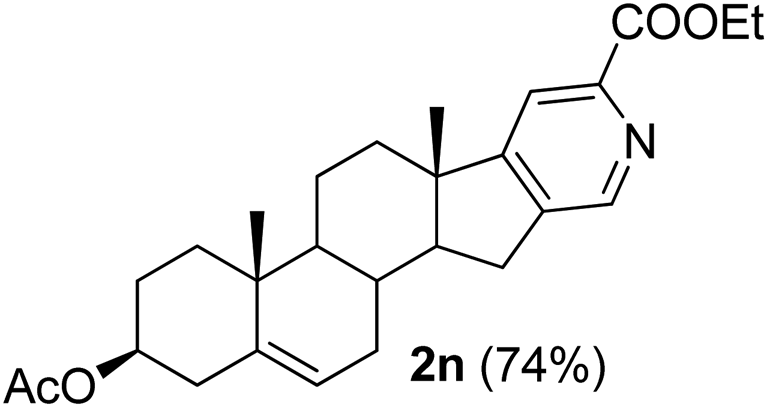 |
| 15 |  |
 |
 |
| 16 | 1d |  |
 |
| 17 | 1d |  |
 |
| 18 |  |
 |
 |
| 19 | 1e | 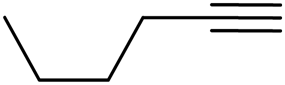 |
 |
| 20 | 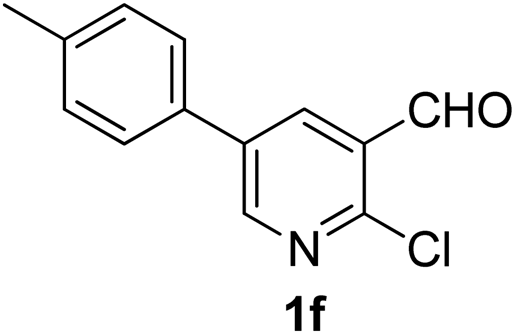 |
 |
 |
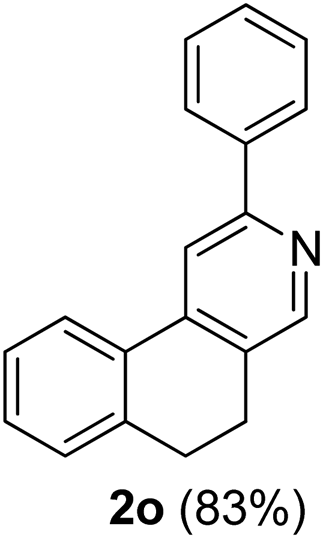
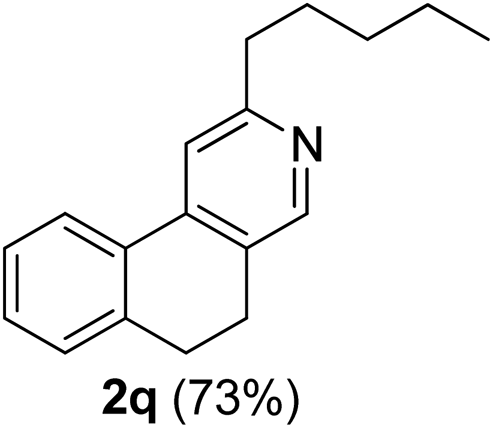
Similarly, β-bromovinyl aldehydes such as 1-bromo-3,4-dihydronaphthalene-2-carbaldehyde (1d)9b and 1-bromo-6-methoxy-3,4-dihydronaphthalene-2-carbaldehyde (1e)9b reacted with a range of aliphatic/aromatic terminal alkynes to afford 9,10-dihydrobenzo[f]isoquinolines 2o–s in 72–83% yields (entry 15–19, Table 2). In order to find out the feasibility of the present methodology for the preparation of the 1,6-naphthyridine derivative, 2-chloro-5-(p-tolyl)pyridine-3-carbaldehyde (1f)9c was treated with phenylacetylene under similar reaction conditions to afford the expected product 2t in 70% yield (entry 20, Table 2). It was observed that aliphatic terminal alkynes afforded relatively low yields of the annulated products in comparison with that of aromatic terminal alkynes. In all these cases, no dimerized by-product of the terminal alkyne was observed. Moreover, good functional-group compatibility was demonstrated with aromatic alkynes substituted with methyl, methoxy and fluoro groups and all proved to be effective annulation partners.
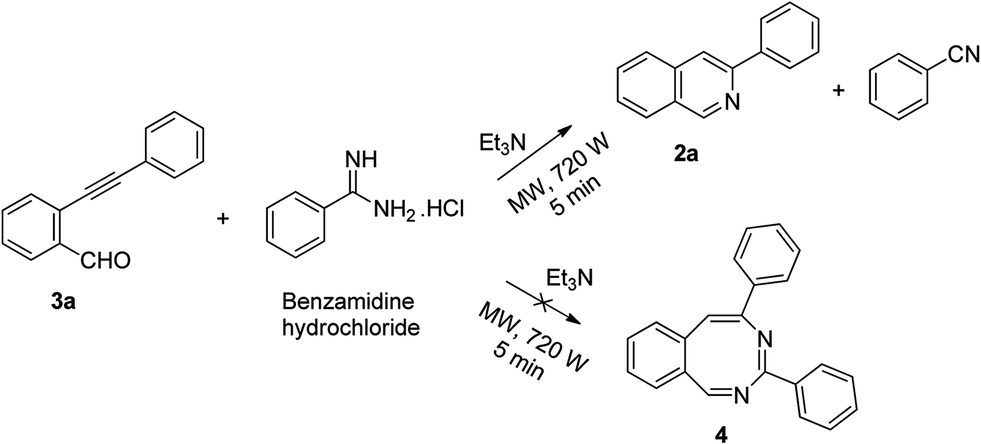
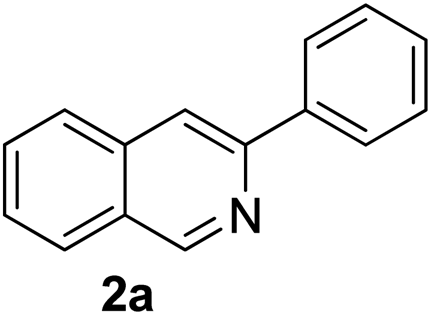
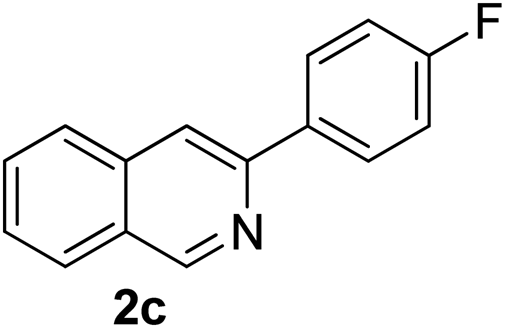
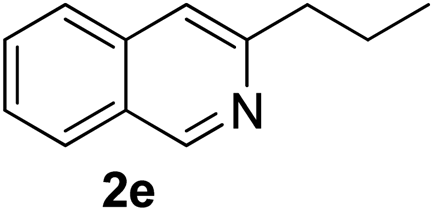
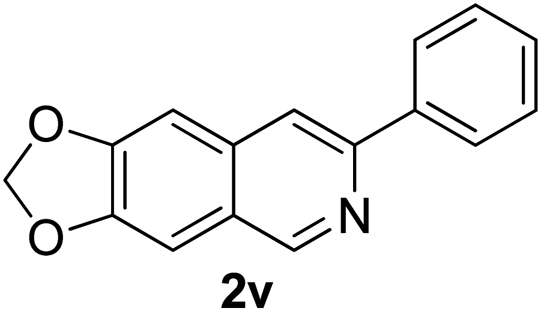
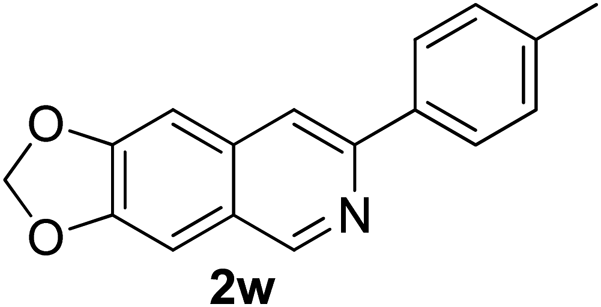
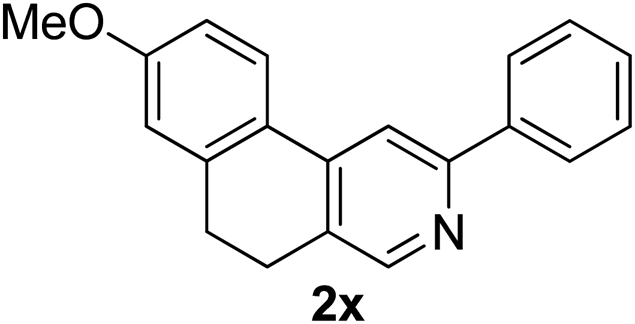
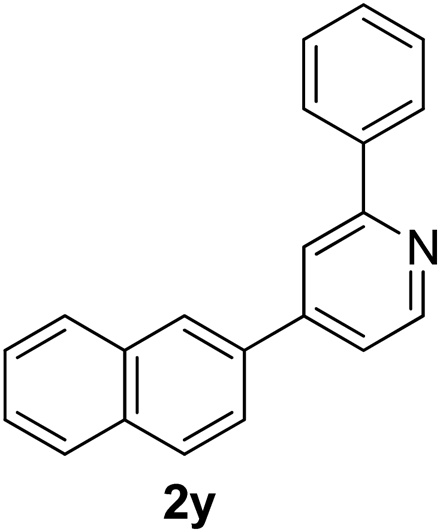
Recently, when we were trying to perform the synthesis of the eight-membered heterocycle 4 (Scheme 1) by the reaction of equimolar amounts of 2-(phenylethynyl)benzaldehyde (3a), benzamidine hydrochloride and triethylamine under microwave irradiation (720 W, 140 °C, 14 bar), we obtained only the isoquinoline derivative 2a (89%) and benzonitrile from the reaction mixture after 5 min of reaction time. A literature search on similar cyclization reactions revealed various methods for the regioselective cyclization reaction of ortho-arylalkynyl aldehyde derivatives to isoquinolines. For example, (i) Gao and Zhang developed an efficient Ag(I)-catalyzed regioselective cyclization reaction of ortho-alkynylaryl aldehyde oxime derivatives to isoquinolines where benzaldehyde was formed as the side product (reaction (1), Scheme 2);10 (ii) Larock et al. reported the synthesis of isoquinolines from N-tert-butyl-2-(1-alkynyl)arylaldimines in the presence of Lewis acid catalyst (Pd, Cu) where isobutene was eliminated (reaction (2), Scheme 2);7a,b (iii) Ghavtadze et al. explored the silver nitrate-assisted cyclization of N-{2-[alkynyl(hetero)aryl]methylene}indolin-1-amines to give the pyridine derivatives where indole was liberated as a leaving group (reaction (3), Scheme 2);11 (iv) Liang et al. studied the synthesis of substituted isoquinolines using 2-alkynylbenzyl azides as the starting material and Ag complex as the catalyst (reaction (4), Scheme 2).8c Our catalyst-free, simple method of cyclization of ortho-arylalkynyl aldehyde (3a) to the isoquinoline derivative 2a using benzamidine (reaction (5), Scheme 2) prompted us to study the cyclization reaction with some more of the known ortho-arylalkynyl aldehydes (3b–h) which were prepared using a known procedure.12 As shown in Table 3, various substituted ortho-alkynyl aryl/vinyl aldehydes (3b–h) were reacted with benzamidine hydrochloride in the presence of triethylamine under microwave irradiation to afford substituted isoquinolines 2a, 2c, 2e, 2u–w in 86–93% yield (entry 1–6, Table 3) and pyridine derivatives 2x,y in 86–93% yield (entry 7–8, Table 3). Encouraged by these results we were interested to see the feasibility of the above copper-catalyzed three-component reaction using benzamidine hydrochloride in place of tert-butylamine as the source of nitrogen under microwave energy. We were delighted to see that this three-component reaction also worked well in the presence of benzamidine hydrochloride even though the yields of the products were less as compared to the above tert-butylamine reaction. Thus, the three-component reaction of β-haloaryl aldehydes 1a, 1b, 1d with phenylacetylene and benzamidine hydrochloride provided isoquinoline derivatives 2a (69%), 2v (74%) and 2o (67%) respectively under microwave irradiation (720 W, 140 °C, 14 bar, 8 min) in the presence of triethylamine (Scheme 3). We also performed the multi-component reactions of 1a and benzamidine hydrochloride with 4-fluorophenylacetylene, n-propylacetylene and 4-methoxyphenylacetylene under the above reaction conditions to afford compounds 2c (64%), 2e (57%) and 2u (66%) respectively (Scheme 3).
 | ||
| Scheme 1 Catalyst-free synthesis of isoquinolines. | ||
 | ||
| Scheme 2 Synthesis of isoquinolines. | ||
 | ||
| Scheme 3 Multicomponent synthesis of 3-substituted isoquinolines and substituted pyridines. | ||
| Entry | ortho-Alkynyl aryl/vinyl aldehydes | Product | Yieldb (%) |
|---|---|---|---|
| a Conditions: ortho-alkynyl aryl/vinyl aldehydes (1 mmol), benzamidine hydrochloride (1.1 mmol) and triethylamine (1.1 mmol) was irradiated in a closed vessel in a Synthos 3000 microwave reactor at 720 W (140 °C and 14 bar) for 5 min.b Refers to the amount of product isolated by chromatography. | |||
| 1 |  |
 |
89 |
| 2 |  |
 |
87 |
| 3 | 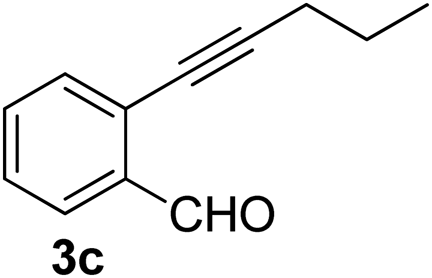 |
 |
86 |
| 4 | 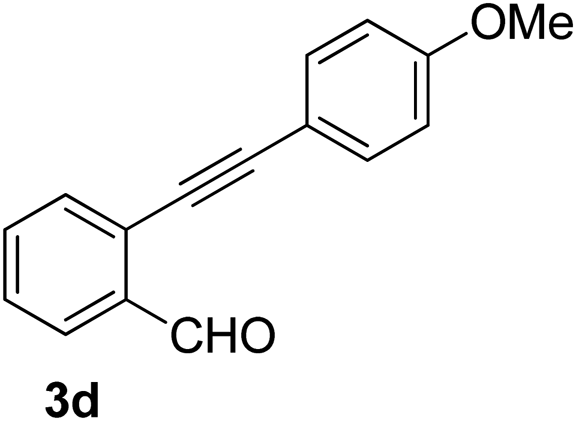 |
 |
93 |
| 5 | 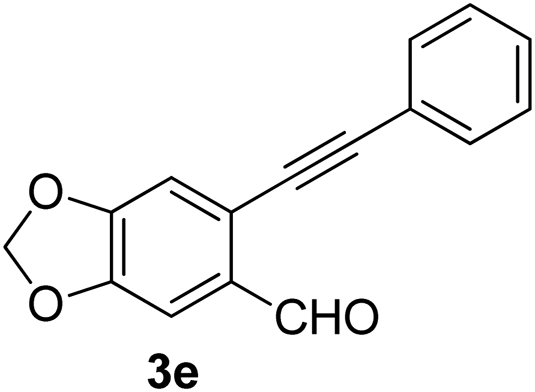 |
 |
90 |
| 6 |  |
 |
89 |
| 7 |  |
 |
86 |
| 8 | 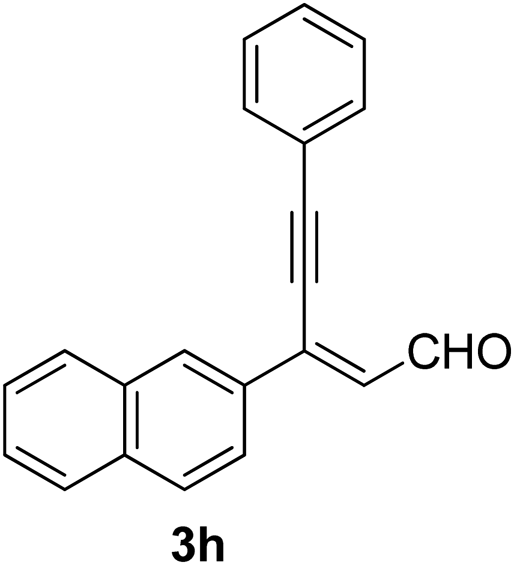 |
 |
93 |
Based on our findings and literature information,13 a plausible mechanism for the tandem reaction is proposed as shown in Scheme 4. The formation of a π-complex between the Cu(I) ion and the triple bond in the presence of the base tert-buylamine activates the triple bond towards deprotonation to form copper(I) acetylide 5a. Subsequently, oxidative addition of this copper(I) acetylide 5a with β-halovinyl aldehyde in the presence of tert-butylamine generates probably copper(III) imine species 5b,13a(Scheme 29),d which on reductive elimination produces copper-coordinated intermediate 5c. The intermediate 5c undergoes 6-endo-dig cyclization to provide regioselectively the iminium intermediate 5d. Fragmentation of the tert-butyl group of 5d to relieve the strain resulting from interaction of the tert-butyl group with the substituent present in the ortho-position, followed by protonation, leads to the formation of compound 2 with the liberation of Cu(I) catalyst.14 Furthermore, a possible mechanism of cyclization of compound 3 in the presence of benzamidine hydrochloride is shown in Scheme 5. Benzamidine hydrochloride forms imine 5e with aldehyde 3 in presence of base triethylamine. The 6-endo-dig-cyclization of 5e and subsequent C–N bond cleavage lead to the formation of compound 2 and benzonitrile. The formation of benzonitrile was proved by taking thin layer chromatography of the crude reaction mixture and confirmed by the presence of the molecular ion peak [M+] at m/z 103 in GC-MS.
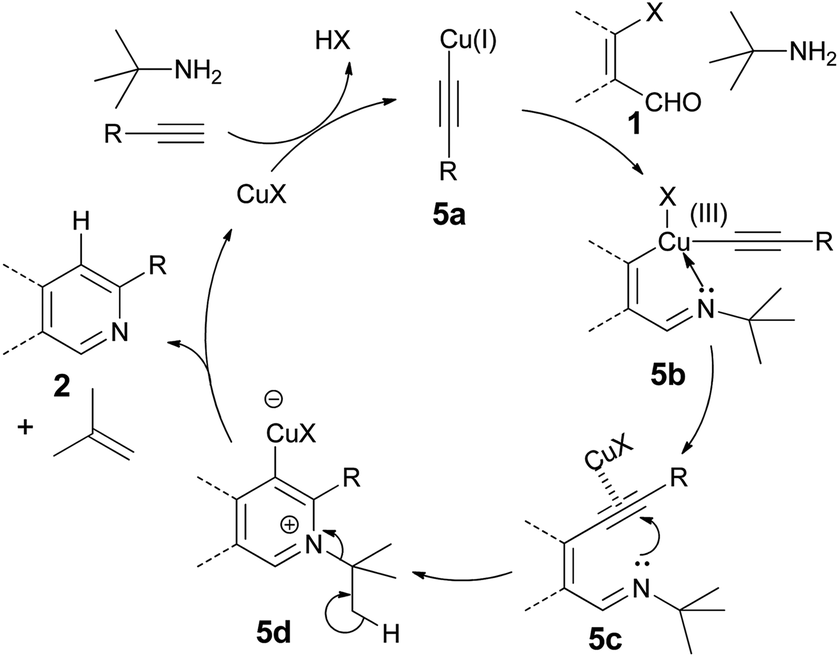 | ||
| Scheme 4 Plausible mechanism of Cu(I)-mediated formation of 2. | ||
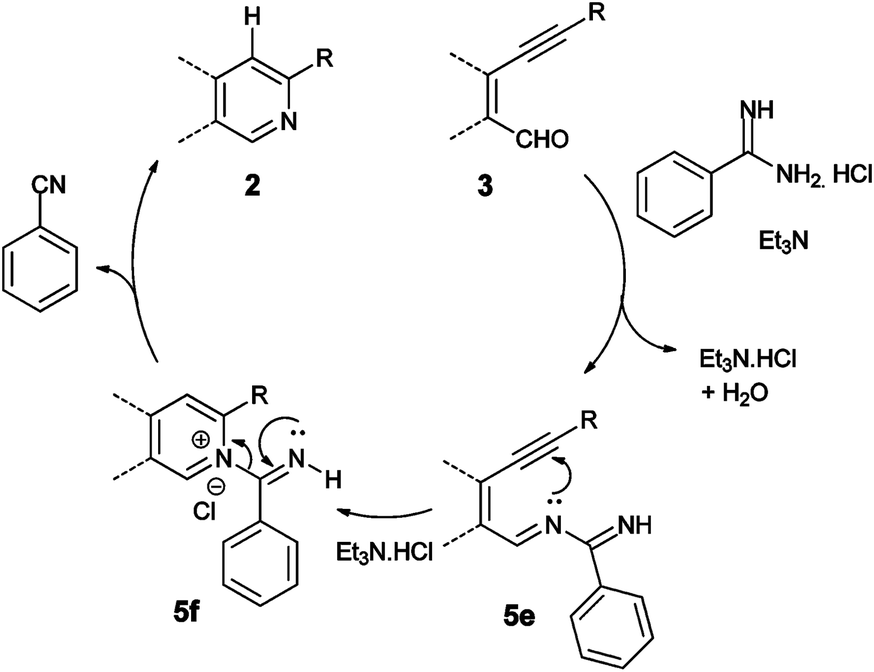 | ||
| Scheme 5 Plausible mechanism of catalyst-free formation of 2. | ||
Because of the anticancer activity shown by different isoquinoline derivatives and heterosteroids, all the compounds were screened in vitro for cytotoxic activities against the cervical HeLa cancer cell line and prostate DU 205 cancer cell line using the MTT-microcultured tetrazolium assay15 and drug doxorubicin as a positive control. Unfortunately, none of the tested compounds could show promising cytotoxic activity against the cervical HeLa cancer cell line (compounds 2a–y, IC50 ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 100 μM, doxorubicin IC50 = 9.76 ± 0.1141 μM) and prostate DU 205 cancer cell line (compounds 2a–y, IC50 100 μM, doxorubicin IC50 = 9.00 ± 0.721 μM).
100 μM, doxorubicin IC50 = 9.76 ± 0.1141 μM) and prostate DU 205 cancer cell line (compounds 2a–y, IC50 100 μM, doxorubicin IC50 = 9.00 ± 0.721 μM).
Conclusions
In conclusion, we have demonstrated very efficient multi-component reactions for the synthesis of biologically important 3-substituted isoquinolines, steroid-fused pyridines, 5,6-dihydrobenzo[f]isoquinolines and 1,6-naphthyridine. These copper-catalyzed microwave-assisted annulation reactions are much more efficient than the known transition metal-catalyzed reactions in terms of the rate of reaction, cost of catalyst and steps of the reactions. A wide range of terminal alkynes undergo this reaction with β-halovinyl/aryl aldehydes and tert-butylamine/benzamidine to provide good to excellent yield of products. Moreover, a catalyst-free synthesis of biologically important 3-substituted isoquinoline and substituted pyridine derivatives was developed using ortho-alkynyl aryl/vinyl aldehydes, benzamidine hydrochloride and triethylamine as the starting materials under microwave irradiation.Experimental
General methods
Melting points were measured with a Buchi B-540 melting point apparatus and are uncorrected. IR spectra were recorded using an Elmer FT-IR-2000 spectrometer using KBr pellets or as a thin film using chloroform. NMR spectra were recorded using an Avance DPX 300 MHz FT-NMR spectrometer using tetramethylsilane (TMS) as an internal standard. Mass spectra were recorded on a Trace DSQ GCMS instrument. All the commercially available regents were used as received. All experiments were monitored by thin layer chromatography (TLC). TLC was performed on pre-coated silica gel plates (Merck). Column chromatography was performed on silica gel (100–200 mesh, Merck). All MW reactions were carried out in a Synthos 3000 (Anton Paar) microwave reactor.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) as eluent afforded compounds 2a–t.
:4) as eluent afforded compounds 2a–t.
3-Phenylisoquinoline7c (2a). Yellow gum, yield: 92% (188 mg). 1H NMR (CDCl3, 300 MHz) δ 7.42 (t, J = 7.2 Hz, 1H), 7.52 (t, J = 7.2 Hz, 2H), 7.60 (t, J = 7.9 Hz, 1H), 7.69 (t, J = 7.1 Hz, 1H), 7.89 (d, J = 8.3 Hz, 1H), 8.00 (d, J = 8.1 Hz, 1H), 8.08 (s, 1H), 8.12 (d, J = 7.3 Hz, 2H), 9.35 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 116.6, 126.9, 127.0, 127.1, 127.6, 127.8, 128.5, 128.7, 128.8, 130.2, 130.5, 136.7, 139.6, 151.3, 152.4. IR (CHCl3, cm−1): 3058, 2971, 1790, 1626, 1592, 1492, 1448, 1205, 977, 764. MS (EI, m/z): 205 [M+]. Anal. calcd for C15H11N: C, 87.77; H, 5.40; N, 6.82. Found: C, 87.59; H, 5.49; N, 6.67%.
3-(p-Tolyl)isoquinoline (2b). Brown solid, yield: 84% (184 mg). M.p. 71–72 °C. 1H NMR (CDCl3, 300 MHz) δ 2.44 (s, 3H), 7.33 (d, J = 8.1 Hz, 2H), 7.52 (t, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 8.00 (s, 1H), 8.07 (d, J = 7.8 Hz, 2H), 9.33 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 21.4, 116.0, 126.9, 127.6, 127.63, 129.6, 130.5, 136.7, 136.8, 138.5, 151.2, 152.3. IR (CHCl3, cm−1): 1626, 1589, 1516, 1447, 823, 751. MS (EI, m/z): 219.1 [M+]. Anal. calcd for C16H13N: C, 87.64; H, 5.98; N, 6.39. Found: C, 87.69; H, 5.99; N, 6.57%.
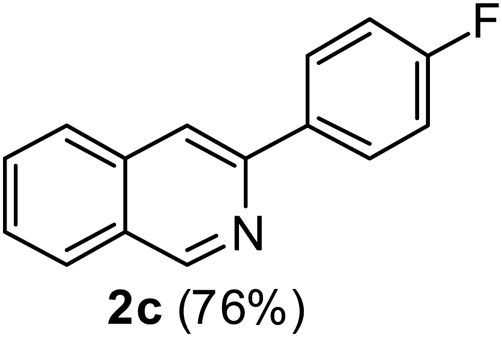
3-(4-Fluorophenyl)isoquinoline (2c). Yellow solid, yield: 76% (170 mg). M.p. 124–125 °C. 1H NMR (CDCl3, 300 MHz) δ 7.13–7.22 (m, 2H), 7.53 (t, J = 6.9 Hz, 1H), 7.65 (t, J = 6.9 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 6.6 Hz, 1H), 7.93 (s, 1H), 8.12–8.02 (m, 2H), 9.28 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 115.5, 115.8, 116.2, 126.8, 127.1, 127.5, 128.7, 128.8, 130.6, 135.7, 136.6, 150.2, 152.4, 164.0 (d, JC–F = 246 Hz). IR (CHCl3, cm−1): 1625, 1600, 1581, 1512, 836. MS (EI, m/z): 223 [M+]. Anal. calcd for C15H10FN: C, 80.70; H, 4.51; N, 6.27. Found: C, 80.82; H, 4.60; N, 6.42%.
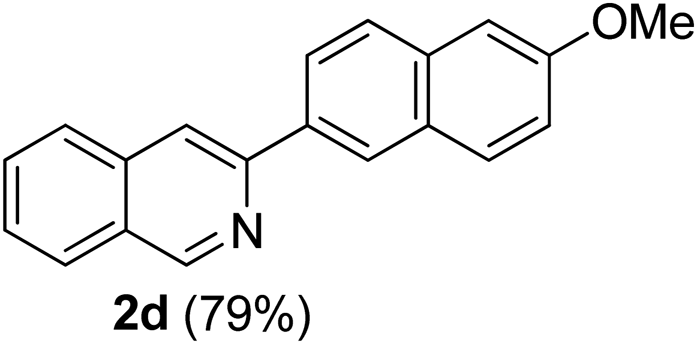
3-(6-Methoxynaphthalen-2-yl)isoquinoline (2d). Light yellow solid, yield: 79% (225 mg). M.p. 171–172 °C. 1H NMR (CDCl3, 300 MHz) δ 3.95 (s, 3H), 7.17–7.26 (m, 2H), 7.58 (t, J = 7.2 Hz, 1H), 7.70 (t, J = 6.9 Hz, 1H), 7.83–7.91 (m, 3H), 8.00 (d, J = 8.1 Hz, 1H), 8.18–8.23 (m, 2H), 8.59 (s, 1H), 9.37 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 55.4, 105.6, 116.4, 119.2, 125.2, 126.1, 126.9, 127.0, 127.3, 127.6, 129.2, 130.3, 130.6, 134.7, 136.8, 151.2, 152.5, 158.1. IR (CHCl3, cm−1): 1621, 1260, 1024, 853, 773, 743. MS (EI, m/z): 285.1 [M+]. Anal. calcd for C20H15NO: C, 84.19; H, 5.30; N, 4.91. Found: C, 84.27; H, 5.36; N 4.76%.
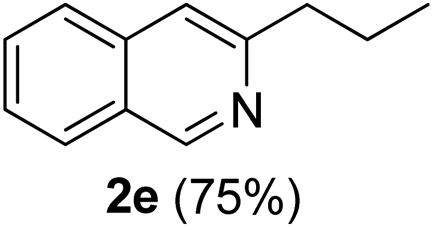
3-Propylisoquinoline (2e). Yellow gum, yield: 75% (128 mg). 1H NMR (CDCl3, 300 MHz) δ 1.00 (t, J = 7.2 Hz, 3H), 1.80 (hextet, J = 7.2 Hz, 2H), 2.91 (t, J = 7.7 Hz, 2H), 7.44 (s, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 9.19 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 13.9, 23.2, 40.1, 118.1, 126.1, 126.3, 127.1, 127.5, 130.2, 136.5, 152.0, 155.5. IR (CHCl3, cm−1): 2960, 2932, 2871, 1630, 1592, 1456, 751. MS (EI, m/z): 171 [M+]. Anal. calcd for C12H13N: C, 84.17; H, 7.65; N, 8.18. Found: C, 84.03; H, 7.55; N, 8.29%.
7-(p-Methoxyphenyl)-[1,3]-dioxolo[4,5-g]isoquinoline (2f). Red solid, yield: 84% (234 mg). M.p. 159–160 °C. 1H NMR (CDCl3, 300 MHz) δ 3.86 (s, 3H), 6.07 (s, 2H), 7.01 (d, J = 7.8 Hz, 2H), 7.06 (s, 1H), 7.16 (s, 1H), 7.79 (s, 1H), 7.99 (d, J = 7.8 Hz, 2H), 9.02 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 55.4, 101.6, 102.6, 103.1, 114.1, 115.5, 124.6, 128.0, 132.2, 135.2, 148.1, 149.9, 150.2, 151.1, 159.9. IR (CHCl3, cm−1): 1598, 1455, 1225, 1026. MS (EI, m/z): 279 [M+]. Anal. calcd for C17H13NO3: C, 73.11; H, 4.69; N, 5.02. Found: C, 73.24; H, 4.80; N, 5.25%.
7-(p-Fluorophenyl)-[1,3]-dioxolo[4,5-g]isoquinoline (2g). Brown solid, yield: 78% (208 mg). M.p. 161–162 °C. 1H NMR (CDCl3, 300 MHz) δ 6.11 (s, 2H), 7.09–7.21 (m, 4H), 7.84 (s, 1H), 8.00–8.07 (m, 2H), 9.05 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 101.7, 102.7, 103.1, 115.5, 115.8, 116.1, 124.9, 128.4, 135.1, 135.8, 148.4, 149.6, 150.1, 151.2, 163.1 (d, JC–F = 246 Hz). IR (CHCl3, cm−1): 1602, 1463, 1233, 956, 843. MS (EI, m/z): 267.0 [M+]. Anal. calcd for C16H10FNO2: C, 71.91; H, 3.77; N, 5.24. Found: C, 71.98; H, 3.83; N, 5.45%.
7-Butyl-[1,3]-dioxolo[4,5-g]isoquinoline (2h). Brown solid, yield: 71% (163 mg). M.p. 68–69 °C. 1H NMR (CDCl3, 300 MHz) δ 0.91 (t, J = 7.2 Hz, 3H), 1.25–1.41 (m, 2H), 1.72 (pentate, J = 7.8 Hz, 2H), 2.83 (t, J = 6.6 Hz, 2H), 6.03 (s, 2H), 6.95 (s, 1H), 7.11 (s, 1H), 7.27 (s, 1H), 8.88 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 14.0, 22.5, 32.2, 37.5, 101.4, 102.0, 103.0, 117.9, 124.1, 135.0, 147.8, 149.6, 151.0, 154.7. IR (CHCl3, cm−1): 1605, 1483, 1457, 1237, 1037, 940. MS (EI, m/z): 229.1 [M+]. Anal. calcd for C14H15NO2: C, 73.34; H, 6.59; N, 6.11. Found: C, 73.37; H, 6.71; N, 6.39%.
3β-Acetoxy-6′-phenyl-5,16-androstadieno[16,17-c]pyridine (2i). Yellow solid, yield: 82% (362 mg). M.p. 185–186 °C. 1H NMR (CDCl3, 300 MHz) δ 0.94 (s, 3H), 1.04 (s, 3H), 0.79–2.4 (m, 14H), 1.97 (s, 3H), 2.40–2.58 (m, 1H), 2.64–2.82 (m, 1H), 4.47–4.64 (m, 1H), 5.36 (d, J = 4.6 Hz, 1H), 7.22 (s, 1H), 7.25–7.50 (m, 4H), 7.88 (d, J = 7.3 Hz, 2H), 8.46 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 18.7, 19.4, 20.6, 21.5, 27.7, 29.8, 30.8, 31.6, 34.2, 36.9, 38.1, 45.7, 50.4, 57.2, 73.8, 113.8, 122.1, 127.0, 128.7, 137.4, 139.6, 140.0, 145.2, 155.8, 164.5, 170.6. IR (CHCl3, cm−1) 2926, 2854, 1731, 1657, 1243, 1031. MS (EI, m/z): 381.2 [M+ − 60]. Anal. calcd for C30H35NO2: C, 81.59; H, 7.99; N, 3.17. Found: C, 81.49; H, 7.96; N, 3.24%.
3β-Acetoxy-6′-(p-tolyl)-5,16-androstadieno[16,17-c]pyridine (2j). Black solid, yield: 79% (360 mg). M.p. 206–208 °C. 1H NMR (CDCl3, 300 MHz) δ 0.97 (s, 3H), 1.09 (s, 3H), 2.05 (s, 3H), 0.90–2.42 (m, 15H), 2.40 (s, 3H), 2.55 (t, J = 11.4 Hz, 1H), 2.80 (dd, J = 14.7 Hz and 5.1 Hz, 1H), 4.49–4.78 (m, 1H), 5.42 (d, J = 4.6 Hz, 1H), 7.26 (d, J = 7.8 Hz, 2H), 7.45 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 8.50 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 19.4, 20.6, 21.3, 21.5, 22.7, 27.7, 29.7, 30.8, 31.6, 34.1, 36.85, 36.9, 38.1, 45.5, 50.4, 57.2, 73.8, 113.4, 122.1, 126.8, 129.4, 137.0, 137.2, 138.4, 140.0, 145.4, 155.9, 164.0, 170.6. IR (CHCl3, cm−1): 1729, 1374, 1254, 1034, 823, 753. MS (EI, m/z): 395.2 [M+ − 60]. Anal. calcd for C31H37NO2: C, 81.72; H, 8.19; N, 3.07. Found: C, 81.75; H, 8.28; N, 3.27%.
3β-Acetoxy-6′-[p-fluorophenyl]-5,16-androstadieno[16,17-c]pyridine (2k). Red solid, yield: 77% (354 mg). M.p. 176–177 °C. 1H NMR (CDCl3, 300 MHz) δ 1.0 (s, 3H), 1.11 (s, 3H), 2.05 (s, 3H), 0.89–2.36 (m, 15H), 2.59 (t, J = 11.2 Hz, 1H), 2.78 (dd, J = 14.6 Hz and 5.0 Hz, 1H), 4.49–4.77 (m, 1H), 5.44 (d, J = 7.5 Hz, 1H), 7.14 (t, J = 8.4 Hz, 2H), 7.43 (s, 1H), 7.93 (t, J = 6.0 Hz, 2H), 8.50 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 18.6, 19.4, 20.6, 21.5, 27.7, 29.6, 29.7, 29.8, 30.8, 31.6, 34.1, 36.85, 36.9, 38.1, 45.7, 50.3, 57.2, 73.8, 115.5, 115.7, 122.1, 128.8, 128.9, 140.0, 163.1 (d, JC–F = 223 Hz), 170.6. IR (CHCl3, cm−1): 2927, 1722, 1245, 1033. MS (EI, m/z): 399.2 [M+ − 60]. Anal. calcd for C30H34FNO2: C, 78.40; H, 7.46; N, 3.05. Found: C, 78.51; H, 7.29; N, 3.13%.
3β-Acetoxy-6′-propyl-5,16-androstadieno[16,17-c]pyridine (2l). Yellow solid, yield: 70% (285 mg). M.p. 161–162 °C. 1H NMR (CDCl3, 300 MHz) δ 0.95 (s, 3H), 1.10 (s, 3H), 2.04 (s, 3H), 0.90–2.60 (m, 20H), 2.71–2.80 (m, 4H) 4.55–4.68 (m, 1H), 5.42 (d, J = 5.3 Hz, 1H), 6.93 (s, 1H), 8.35 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 13.9, 18.6, 19.3, 20.6, 21.4, 23.4, 27.7, 29.7, 30.8, 31.6, 34.1, 36.8, 36.9, 38.1, 39.9, 45.5, 50.4, 57.1, 73.7, 115.8, 122.0, 136.1, 140.0, 143.9, 159.8, 164.6, 170.5. IR (CHCl3, cm−1) 2928, 2853, 1733, 1244, 1033. MS (EI, m/z): 347.3 [M+ − 60]. Anal. calcd for C27H37NO2: C, 79.56; H, 9.15; N, 3.44. Found: C, 79.58; H, 9.20; N, 3.30%.
3β-Acetoxy-6′-butyl-5,16-androstadieno[16,17-c]pyridine (2m). Yellow solid, yield: 69% (291 mg). M.p. 168 °C. 1H NMR (CDCl3, 300 MHz) δ 0.94 (s, 3H), 1.10 (s, 3H), 2.04 (s, 3H), 0.90–2.62 (m, 22H), 2.71–2.80 (m, 4H) 4.55–4.68 (m, 1H), 5.43 (d, J = 4.5 Hz, 1H), 6.91 (s, 1H), 8.34 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 14.0, 18.6, 19.3, 20.6, 21.4, 22.7, 28.8, 29.7, 30.8, 31.6, 32.4, 34.1, 36.8, 38.1, 45.4, 50.4, 57.2, 73.8, 115.5, 122.1, 135.8, 140.0, 144.8, 160.5, 163.7, 170.5. IR (CHCl3, cm−1): 2955, 2934, 2855, 1733, 1245, 1033, 755. MS (EI, m/z): 361.2 [M+ − 60]. Anal. calcd for C28H39NO2: C, 79.76; H, 9.32; N, 3.32. Found: C, 79.78; H, 9.37; N, 3.42%.
3β-Acetoxy-6′-ethoxycarbonyl-5,16-androstadieno[16,17-c]pyridine (2n). Yellow solid, yield: 74% (324 mg). M.p. 129 °C. 1H NMR (CDCl3, 300 MHz) δ 0.98 (s, 3H), 1.10 (s, 3H), 1.45 (t, J = 7.4 Hz, 3H), 2.05 (s, 3H), 1.11–3.00 (m, 17H), 4.48 (q, J = 7.1 Hz, 2H), 4.50–4.65 (m, 1H), 5.43 (d, J = 4.6 Hz, 1H), 7.90 (s, 1H), 8.59 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 14.4, 18.5, 19.3, 20.5, 21.4, 27.7, 30.2, 30.8, 31.5, 34.0, 36.9, 38.1, 45.7, 50.3, 57.1, 61.9, 73.7, 118.2, 121.9, 140.0, 142.8, 145.9, 146.9, 164.4, 170.6. IR (CHCl3, cm−1) 2850, 1728, 1374, 1246, 1031. MS (EI, m/z): 377.2 [M+ − 60]. Anal. calcd for C27H35NO4: C, 74.11; H, 8.06; N, 3.20. Found: C, 74.25; H, 8.14; N, 3.39%.
3-Phenyl-9,10-dihydrobenzo[f]isoquinoline (2o). Yellow gum, yield: 83% (213 mg). 1H NMR (CDCl3, 300 MHz) δ 2.86–3.05 (m, 4H), 7.02–7.52 (m, 6H), 7.82–8.04 (m, 4H), 8.56 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 25.2, 28.5, 114.8, 124.3, 126.9, 127.3, 128.7, 128.8, 129.6, 130.5, 132.1, 138.3, 139.7, 142.7, 148.8, 156.6. IR (CHCl3, cm−1): 2926, 2850, 1599, 1475, 1448, 771, 743. MS (EI, m/z): 257.1 [M+]. Anal. calcd for C19H15N: C, 88.68; H, 5.88; N, 5.44. Found: C, 88.76; H, 5.64; N, 5.59%.
3-Propyl-9,10-dihydrobenzo[f]isoquinoline (2p). Yellow gum, yield: 75% (167 mg). 1H NMR (CDCl3, 300 MHz) δ 0.93 (t, J = 7.4 Hz, 3H), 1.74 (hextet, J = 7.3 Hz, 2H), 2.71–2.90 (m, 6H), 7.10–7.32 (m, 3H), 7.40 (s, 1H), 7.72 (t, J = 3.6 Hz, 1H), 8.32 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 13.9, 23.3, 25.1, 28.5, 40.0, 116.7, 124.3, 127.2, 128.6, 129.2, 129.5, 132.0, 138.4, 142.6, 147.7, 160.6. IR (CHCl3, cm−1): 2959, 2933, 2871, 1603, 1544, 1483, 772. MS (EI, m/z): 223 [M+]. Anal. calcd for C16H17N: C, 86.05; H, 7.67; N, 6.27. Found: C, 85.94; H, 7.60; N, 6.19%.
3-Pentyl-9,10-dihydrobenzo[f]isoquinoline (2q). Yellow gum, yield: 73% (183 mg). 1H NMR (CDCl3, 300 MHz) δ 0.91 (t, J = 6.9 Hz, 3H), 1.25–1.52 (m, 4H), 1.71–1.81 (m, 2H), 2.79–2.94 (m, 6H), 7.15–7.39 (m, 3H), 7.47 (s, 1H), 7.77–7.83 (m, 1H), 8.39 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 14.1, 22.6, 25.1, 28.5, 29.8, 31.6, 38.0, 116.7, 124.3, 127.2, 128.6, 129.2, 129.5, 132.0, 138.4, 142.6, 147.6, 160.9. IR (CHCl3, cm−1): 2955, 2930, 2857, 1603, 1545, 1451, 754. MS (EI, m/z): 251.1 [M+]. Anal. calcd for C18H21N: C, 86.01; H, 8.42; N, 5.57. Found: C, 86.29; H, 8.53; N, 5.78%.
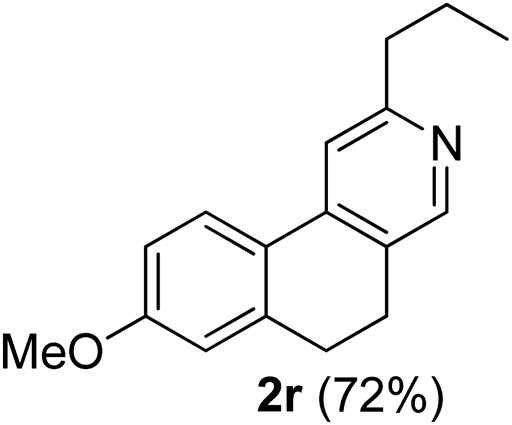
7-Methoxy-3-propyl-9,10-dihydrobenzo[f]isoquinoline (2r). Yellow gum, yield: 72% (182 mg). 1H NMR (CDCl3, 300 MHz) δ 1.01 (t, J = 7.4 Hz, 3H), 1.72–1.89 (m, 2H), 2.72–2.97 (m, 6H), 3.87 (s, 3H), 6.82 (d, J = 2.1 Hz, 1H), 6.89 (dd, J = 8.5 Hz and 2.3 Hz, 1H), 7.43 (s, 1H), 7.75 (d, J = 8.6 Hz, 1H), 8.36 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 13.9, 23.2, 25.1, 28.8, 39.3, 55.4, 113.0, 113.8, 116.4, 124.5, 126.1, 128.3, 140.5, 161.0. IR (CHCl3, cm−1): 2960, 2932, 1602, 1464, 1259, 1045. MS (EI, m/z): 253 [M+]. Anal. calcd for C17H19NO: C, 80.60; H, 7.56; N, 5.53. Found: C, 80.69; H, 7.55; N, 5.75%.
3-Butyl-7-methoxy-9,10-dihydrobenzo[f]isoquinoline (2s). Yellow gum, yield: 72% (192 mg). 1H NMR (CDCl3, 300 MHz) δ 0.89 (t, J = 7.3 Hz, 3H), 1.29–1.40 (m, 2H), 1.62–1.72 (m, 2H), 2.72–2.81 (m, 6H), 3.81 (s, 3H), 6.82 (dd, J = 8.3 Hz and 2.6 Hz, 1H), 7.25 (d, J = 2.7 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 7.35 (s, 1H), 8.32 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 14.0, 22.6, 25.4, 27.7, 32.3, 37.9, 55.5, 109.9, 114.8, 116.6, 119.2, 129.3, 129.4, 130.6, 133.1, 142.3, 148.1, 158.8, 161.0. IR (CHCl3, cm−1) 2962, 2930, 1601, 1463, 1257, 1042. MS (EI, m/z): 267 [M+]. Anal. calcd for C18H21NO: C, 80.86; H, 7.92; N, 5.24. Found: C, 80.96; H, 7.87; N, 5.56%.
7-Phenyl-3-(p-tolyl)-1,6-naphthyridine (2t). Yellow solid, yield: 70% (207 mg). M.p. 190–191 °C. 1H NMR (CDCl3, 300 MHz) δ 2.38 (s, 3H), 7.02–7.60 (m, 7H), 8.12 (d, J = 7.3 Hz, 2H), 8.18–8.34 (m, 2H), 9.28 (s, 1H), 9.33 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 21.3, 117.6, 122.6, 127.2, 129.0, 129.2, 130.1, 131.9, 134.1, 135.0, 138.7, 138.9, 150.2, 152.8, 154.6, 154.9. IR (KBr, cm−1) 2923, 2850, 1352, 810, 767. MS (EI, m/z): 296 [M+]. Anal. calcd for C21H16N2: C, 85.11; H, 5.44; N, 9.45. Found: C, 85.13; H, 5.44; N, 9.58%.
:4) as eluent afforded compounds 2a, 2c, 2e, 2u–y. The spectral data of compounds 2a (89%, 182 mg), 2c (87%, 194 mg) and 2e (86%, 147 mg) are similar to those mentioned above.
3-(4-Methoxyphenyl)isoquinoline (2u). Red solid, yield: 93% (219 mg). M.p. 85–87 °C. 1H NMR (CDCl3, 300 MHz) δ 3.87 (s, 3H). 7.04 (d, J = 8.7 Hz, 2H), 7.54 (t, J = 7.8 Hz, 1H), 7.65 (t, J = 6.9 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 11.7 Hz, 1H), 7.98 (s, 1H), 8.09 (d, J = 9.3 Hz, 2H), 9.30 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 55.4, 114.2, 115.4, 126.7, 126.8, 127.4, 127.6, 128.2, 130.5, 132.2, 136.8, 151.0, 152.3, 160.1. IR (CHCl3, cm−1): 1608, 1515, 1249, 834. MS (EI, m/z): 235 [M+]. Anal. calcd for C16H13NO: C, 81.68; H, 5.57; N, 5.95. Found: C, 81.78; H, 5.63; N, 5.79%.
7-Phenyl-[1,3]-dioxolo[4,5-g]isoquinoline (2v). Brown solid, yield: 90% (224 mg). M.p. 111–112 °C. 1H NMR (CDCl3, 300 MHz) δ 5.96 (s, 2H), 6.97 (d, J = 3.3 Hz, 1H), 7.08 (d, J = 2.4 Hz, 1H), 7.35–7.51 (m, 3H), 7.79 (s, 1H), 8.05 (d, J = 6.9 Hz, 2H), 9.00 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 101.6, 102.7, 103.0, 116.3, 124.9, 126.8, 127.2, 128.3, 128.8, 129.2, 135.0, 139.6, 148.3, 150.0, 150.3, 151.0. IR (CHCl3, cm−1): 1600, 1481, 1459, 1232, 1038. MS (EI, m/z): 249 [M+]. Anal. calcd for C16H11NO2: C, 77.10; H, 4.45; N, 5.62. Found: C, 77.31; H, 4.37; N, 5.68%.
7-(p-Tolyl)-[1,3]-dioxolo[4,5-g]isoquinoline (2w). Brown solid, yield: 89% (234 mg). M.p. 152–153 °C. 1H NMR (CDCl3, 300 MHz) δ 2.41 (s, 3H), 6.01 (s, 2H), 7.01 (s, 1H), 7.13 (s, 1H), 7.29 (d, J = 8.1 Hz, 2H), 7.80 (s, 1H), 7.95 (d, J = 8.1 Hz, 2H), 9.01 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 21.3, 101.6, 102.7, 103.0, 116.0, 124.8, 126.6, 129.5, 135.1, 136.6, 138.2, 148.2, 149.9, 150.3, 151.1. IR (CHCl3, cm−1): 1598, 1480, 1456, 1224, 1035. MS (EI, m/z): 263 [M+]. Anal. calcd for C17H13NO2: C, 77.55; H, 4.98; N, 5.32. Found: C, 77.49; H, 4.78; N, 5.53%.
7-Methoxy-3-phenyl-9,10-dihydrobenzo[f]isoquinoline (2x). Yellow gum, yield: 86% (247 mg). 1H NMR (CDCl3, 300 MHz) δ 2.86–2.96 (m, 4H), 3.90 (s, 3H), 6.93 (dd, J = 8.3 Hz and 2.5 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.30–7.57 (m, 5H), 7.98 (s, 1H), 8.02 (d, J = 7.3 Hz, 1H), 8.59 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ 25.5, 27.6, 55.6, 110.0, 114.9, 127.0, 128.5, 128.8, 129.0, 129.5, 130.6, 130.8, 132.9, 139.2, 143.0, 148.4, 158.9. IR (CHCl3, cm−1): 2926, 2851, 1601, 1542, 1481, 1220, 771. MS (EI, m/z): 287.1 [M+]. Anal. calcd for C20H17NO: C, 83.59; H, 5.96; N, 4.87. Found: C, 83.57; H, 6.09; N, 4.78%.
4-(Naphthalen-2-yl)-2-phenylpyridine (2y). Yellow gum, yield: 93% (261 mg). 1H NMR (CDCl3, 300 MHz) δ 7.35–7.54 (m, 6H), 7.74 (dd, J = 8.5 Hz and 1.6 Hz, 1H), 7.82–7.93 (m, 1H), 7.91 (t, J = 8.3 Hz, 2H), 8.00 (d, J = 0.5 Hz, 1H), 8.05–8.13 (m, 3H), 8.74 (d, J = 5.1 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ 119.0, 120.5, 124.5, 126.5, 126.8, 126.9, 127.2, 127.8, 128.5, 128.9, 129.0, 129.2, 133.5, 133.51, 135.7, 139.4, 149.3, 150.1, 158.1. IR (CHCl3, cm−1): 2924, 1621, 1222, 776. MS (EI, m/z): 281.1 [M+]. Anal. calcd for C21H15N: C, 89.65; H, 5.37; N, 4.98. Found: C, 89.49; H, 5.21; N, 4.76%.
:4) as eluent afforded compounds 2a, 2c, 2e, 2o, 2u,v whose spectral data are similar to those mentioned above.Acknowledgements
We thank the Department of Science & Technology, New Delhi for financial support. K.S. thanks CSIR, New Delhi for the award of Senior research fellowship (CSIR-SRF). We are grateful to Director, CSIR-NEIST for his keen interests. We gratefully acknowledge financial support from CSIR-ORIGIN (CSC 0108), CSIR-ACT (CSC0301) and CSIR-OSDD (HCP-0001) projects.References
- (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed; (b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed.
- (a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef; (b) Multicomponent Reactions, ed. J. Zhu and H. Bienayme, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) W.-J. Cho, E.-K. Kim, M.-J. Park, S.-U. Choi, C.-O. Lee, S. H. Cheon, B.-G. Choi and B.-H. Chung, Bioorg. Med. Chem., 1998, 6, 2449 CrossRef CAS; (b) W.-J. Cho, S. Y. Min, T. N. Le and T. S. Kim, Bioorg. Med. Chem. Lett., 2003, 13, 4451 CrossRef CAS PubMed; (c) W.-J. Cho, M.-J. Park, B.-H. Chung and C.-O. Lee, Bioorg. Med. Chem. Lett., 1998, 8, 41 CrossRef CAS; (d) W.-J. Cho, S.-J. Yoo, M.-J. Park, B.-H. Chung and C.-O. Lee, Arch. Pharmacal Res., 1997, 20, 264 CrossRef CAS PubMed; (e) W.-J. Cho, E.-K. Kim, I. Y. Park, E. Y. Jeong, T. S. Kim, T. N. Le, D.-D. Kim and E.-S. Lee, Bioorg. Med. Chem., 2002, 10, 2953 CrossRef CAS; (f) W.-J. Cho, S.-J. Yoo, B.-H. Chung, B.-G. Choi, S.-H. Cheon, S.-H. Whang, S.-K. Kim, B.-H. Kang and C.-O. Lee, Arch. Pharmacal Res., 1996, 19, 321 CrossRef CAS; (g) D. L. C. McCall, J. Alexander, J. Barber, R. G. Jaouhari, A. Satoskar and R. D. Waigh, Bioorg. Med. Chem. Lett., 1994, 4, 1663 CrossRef CAS; (h) N. C. W. Wong, J. E. L. Tucker, H. C. Hansen, F. S. Chiacchia and D. McCaffrey, US Pat., Appl. US, 2008188467, 2008, Chem. Abstr., 2008, 149, 246409 Search PubMed.
- (a) Y. Wada, N. Nishida, N. Kurono, T. Ohkuma and K. Orito, Eur. J. Org. Chem., 2007, 4320 CrossRef CAS; (b) G. Nonaka and I. Nishioka, Chem. Pharm. Bull., 1973, 21, 1410 CrossRef CAS; (c) G.-L. Zhang, G. Rücker, E. Breitmaier and R. Mayer, Phytochemistry, 1995, 40, 1813 CrossRef CAS; (d) G. Nonaka, Y. Kodera and I. Nishioka, Chem. Pharm. Bull., 1973, 21, 1020 CrossRef CAS; (e) N. Takao and K. Iwasa, Chem. Pharm. Bull., 1973, 21, 1587 CrossRef CAS; (f) Y. Sun, K. Xun, Y. Wang and X. Chen, Anti-Cancer Drugs, 2009, 20, 757 CrossRef CAS PubMed; (g) C. Moulis, J. Gleye and E. Stanislas, Phytochemistry, 1977, 16, 1283 CrossRef CAS; (h) S. Aoki, Y. Watanabe, M. Sanagawa, A. Setiawan, N. Kotoku and M. Kobayashi, J. Am. Chem. Soc., 2006, 128, 3148 CrossRef CAS PubMed; (i) K. W. Bentley, Nat. Prod. Rep., 1992, 9, 365 RSC; (j) Y.-H. Li, P. Yang, W.-J. Kong, Y.-X. Wang, C.-Q. Hu, Z.-Y. Zuo, Y.-M. Wang, H. Gao, L.-M. Gao, Y.-C. Feng, N.-N. Du, Y. Liu, D.-Q. Song and J.-D. Jiang, J. Med. Chem., 2009, 52, 492 CrossRef CAS PubMed.
- (a) J.-Z. Yan, J. Li and G.-W. Rao, Steroids, 2007, 72, 736 CrossRef CAS PubMed; (b) G. A. Potter, S. E. Barrie, M. Jarman and M. G. Rowlands, J. Med. Chem., 1995, 38, 2463 CrossRef CAS; (c) M. G. Barthakur, M. Borthakur and R. C. Boruah, Steroids, 2008, 73, 1137 CrossRef CAS PubMed; (d) G. H. Rasmusson, G. F. Reynolds, T. Utne, R. B. Jobson, R. L. Primka, C. Berman and J. R. Brooks, J. Med. Chem., 1984, 27, 1690 CrossRef CAS; (e) A.-G. E. Amr and M. M. Abdulla, Bioorg. Med. Chem., 2006, 14, 4341 CrossRef CAS PubMed; (f) G. Abbiati, A. Arcadi, G. Bianchi, S. Di Giuseppe, F. Marinelli and E. Rossi, J. Org. Chem., 2003, 68, 6959 CrossRef CAS PubMed; (g) S. Hesse and G. Kirsch, Synthesis, 2003, 717 CAS; (h) C. Mukhopadhyay, P. Das and R. J. Butcher, Org. Lett., 2011, 13, 4664 CrossRef CAS PubMed.
- Selected articles for the preparation of 3-arylisoquinoline derivatives by Bischler–Napieralski reaction, see: (a) N. Sotomayor, T. Vicente, E. Domínguez, E. Lete and M.-J. Villa, Tetrahedron, 1994, 50, 2207 CrossRef CAS; (b) E. Dominguez, E. Lete, M. D. Badia, M. J. Villa, L. Castedo and D. Dominguez, Tetrahedron, 1987, 43, 1943 CrossRef CAS; (c) N. Sotomayor, E. Domínguez and E. Lete, Tetrahedron Lett., 1994, 35, 2973 CrossRef CAS; (d) I. Tellitu, D. Badía, E. Domínguez and F. J. García, Tetrahedron: Asymmetry, 1994, 5, 1567 CrossRef CAS , By Pictet–Spengler reaction: ; (e) S. F. Dyke, D. W. Brown, M. Sainsbury and G. Hardy, Tetrahedron, 1971, 27, 3495 CrossRef CAS; (f) D. Badia, E. Dominguez, C. Iriondo and E. Martinez de Marigorta, Heterocycles, 1986, 24, 1867 CrossRef CAS; (g) E. Dominguez, E. Lete, D. Badia, M. J. Villa, L. Castedo and D. Dominguez, Tetrahedron, 1987, 43, 1943 CrossRef CAS; (h) J. L. Vicario, D. Badia, E. Dominguez and L. Carrillo, J. Org. Chem., 1999, 64, 4610 CrossRef CAS PubMed.
- (a) K. R. Roesch and R. C. Larock, Org. Lett., 1999, 1, 553 CrossRef CAS; (b) H. Zhang and R. C. Larock, J. Org. Chem., 2002, 67, 7048 CrossRef CAS PubMed; (c) S. Roy, B. Neuenswander, D. Hill and R. C. Larock, J. Comb. Chem., 2009, 11, 1061 CrossRef CAS PubMed; (d) S. V. Aeken, S. Verbeeck, J. Deblander, B. U. W. Maes and K. A. Tehrani, Tetrahedron, 2011, 67, 2269 CrossRef PubMed; (e) M. Dell'Acqua, G. Abbiati and E. Rossi, Synlett, 2010, 2672 CAS; (f) L. Lin, Q. Wu, S. Huang and G. Yang, Chin. J. Chem., 2012, 30, 1075, DOI:10.1002/cjoc.201100560 , CAN 157:133619; (g) G. Dai and R. C. Larock, J. Org. Chem., 2002, 67, 7042 CrossRef CAS PubMed; (h) M. Alfonsi, M. Dell'Acqua, D. Facoetti, A. Arcadi, G. Abbiati and E. Rossi, Eur. J. Org. Chem., 2009, 2852 CrossRef CAS; (i) E.-i. Negishi, C. Copéret and S.-Y. Liou, Chem. Rev., 1996, 96, 365 CrossRef CAS PubMed.
- (a) R. P. Korivi and C.-H. Cheng, Org. Lett., 2005, 7, 5179 CrossRef CAS PubMed; (b) T. V. V. Ramakrishna and P. R. Sharp, Org. Lett., 2003, 5, 877 CrossRef CAS PubMed; (c) Y.-N. Niu, Z.-Y. Yan, G.-L. Gao, H.-L. Wang, X.-Z. Shu, K.-G. Ji and Y.-M. Liang, J. Org. Chem., 2009, 74, 2893 CrossRef CAS PubMed.
- (a) U. Bora, A. Saikia and R. C. Boruah, Org. Lett., 2003, 5, 435 CrossRef CAS PubMed; (b) S. Gogoi, K. Shekarrao, A. Duarah, T. C. Bora, S. Gogoi and R. C. Boruah, Steroids, 2012, 77, 1438 CrossRef CAS PubMed , and refs cited therein; (c) S. Gogoi, S. Gogoi and R. C. Boruah, Synthesis, 2013, 45, 219 CrossRef CAS PubMed.
- H. Gao and J. Zhang, Adv. Synth. Catal., 2009, 351, 85 CrossRef CAS.
- N. Ghavtadze, R. Fröhlich and E.-U. Würthwein, Eur. J. Org. Chem., 2010, 1787 CrossRef CAS.
- V. Rustagi, R. Tiwari and A. K. Verma, Eur. J. Org. Chem., 2012, 4590 CrossRef CAS.
- (a) P. Siemsen, R. C. Livingston and F. Deiderich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS; (b) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC; (c) N. T. Patil, A. Nijamudheen and A. Datta, J. Org. Chem., 2012, 77, 6179 CrossRef CAS PubMed; (d) P. Cadiot and W. Chodkiewicz, in Chemistry of acetylenes, ed. H. G. Viehe, Dekker, New York, 1969, ch. 9, pp. 597–647 Search PubMed.
- G. Wu, A. L. Rheingold, S. J. Geib and R. F. Heck, Organometallics, 1987, 6, 1941 CrossRef CAS.
- For the MTT assay protocol see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR spectra of 2a–y and MTT assay protocol. See DOI: 10.1039/c3ra46722h |
| This journal is © The Royal Society of Chemistry 2014 |