First total synthesis of fuzanins C, D and their analogues as anticancer agents†
S. Naveen Kumara,
C. H. N. S. Sai Pavan Kumara,
E. Sriharia,
Sravani Kancharlab,
Kolupula Srinivas*b,
Shweta Shrivastavab,
V. G. M. Naidu*b and
V. Jayathirtha Rao*abc
aCrop Protection Chemicals Division, Indian Institute of Chemical Technology, Uppal Road Tarnaka, Hyderabad 500007, India. E-mail: jrao@iict.res.in
bNational Institute of Pharmaceutical Education and Research (NIPER), Balanagar, Hyderabad 500 037, India
cAcSIR-Indian Institute of Chemical Technology, Uppal Road Tarnaka, Hyderabad-500007, India
First published on 14th January 2014
Abstract
The first total synthesis of fuzanins C and D, isolated from the culture supernatant of Kitasatospora sp. IFM10917, is described. Key features of this synthetic strategy involve the use of Sharpless asymmetric epoxidation, dihydroxylation, Mitsunobu reaction and Julia-Kocienski olefination. The total synthesis reported herein also confirmed the absolute configuration of fuzanin D. The optical rotations of synthesized fuzanin D and natural product were opposite in sign, leading to a revision of the reported structure as its enantiomer which is further confirmed by molecular modeling studies. In addition, we also synthesized the analogues of fuzanins C, D containing the quinoline nucleus. All of the synthesized compounds were screened for anticancer activity on four cell lines and found to be potent against HT29 colon cancer cell lines, whereas they were found to be less potent against cervical and breast cancer cell lines.
Introduction
Organic synthesis is the art of building organic molecules through chemical reactions. The synthesis of natural products is one of the most fascinating and challenging areas of research in chemistry. Further, most of the natural products have been at the spearhead of merging the fields of organic chemistry and biology, and the fields will only continue to come together in the future. Nitrogen containing heterocycles such as pyridine, quinoline derivatives are among the most omnipresent azaheterocycles found in many natural products and have been claimed to be the most prevalent heterocycles in pharmaceutically active compounds.1,2 Micrococcin P1, streptonigrin and nemerelline, etoricoxib, rosuvastatin and imatinib are a few examples of commercialized drugs bearing the pyridine motif. The pyridine ring is also ubiquitous in agrochemicals.3 Quinine, chloroquine, pamaquine, tafenoquine, bulaquine are some of the active pharmaceuticals containing quinoline nucleus. Quinoline derivatives exhibit broad range of biological activities such as antimalarial,4 antimicrobial,5 anticancer,6 antifungal,7 antileishmanial,8 anti-inflammatory,9,10 and analgesic activity.10Recently, new carbamate or pyridine containing natural products, fuzanins A (1), B (2), C (3a), and D (4a) were isolated by Ishibashi et al.11 from the culture supernatant of Kitasatospora sp. IFM10917 (active strain from 323 actinomycete strains) obtained from a soil sample collected at Toyama city, Japan (Fig. 1). In addition, they have also evaluated cytotoxicity against human colon carcinoma DLD-1 cells, and inhibition of Wnt signal transcription for the four compounds.
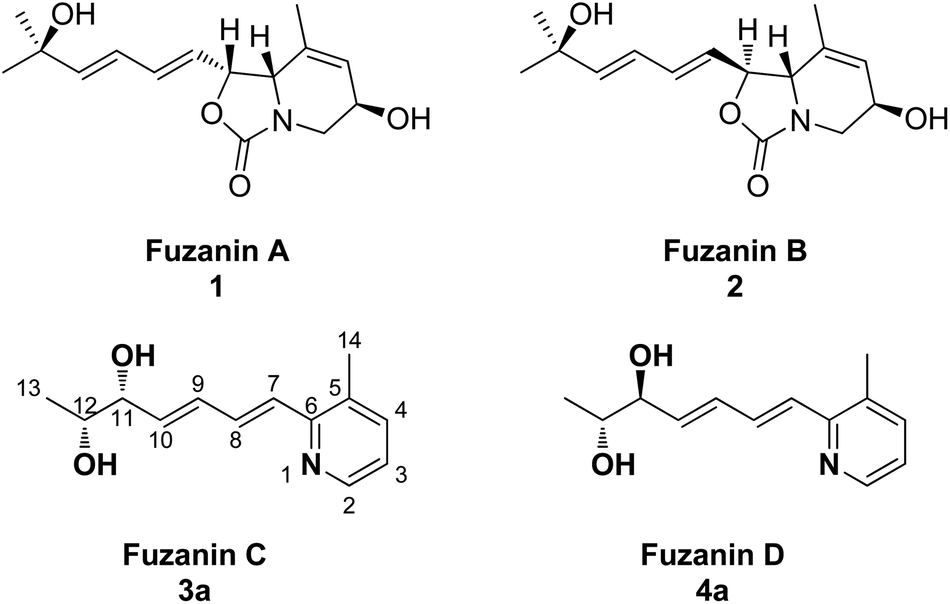 | ||
| Fig. 1 Fuzanins A, B, C and D. | ||
As part of our continuing synthetic efforts towards synthesis of biologically and pharmaceutically favored heterocycles and natural products,12 now we became interested in synthesis of biologically active fuzanin D and its stereo isomer fuzanin C. In this regard, we report the first total synthesis of fuzanin C (3a), D (4a) along with their analogues and also confirmed the absolute configuration of fuzanin D. Some compounds are selectively potent against HT29 colon cancer cell line.
Results and discussion
Fuzanins C (3a) and D (4a) are stereoisomers and differ in stereochemistry at 11th position. The compounds are similar with scaffolds, hence similar synthetic strategies were applied to synthesize them. Retrosynthesis for fuzanins C (3a), D (4a) is depicted in Scheme 1. The analysis revealed that 3a and 4a could be prepared efficiently by a Julia-Kocienski olefination protocol using aldehyde 5a with sulfone 6 and 7 respectively, The intermediate 5a could be prepared from 2,3-dimethyl pyridine and compounds 6, 7 from ethyl sorbate (8).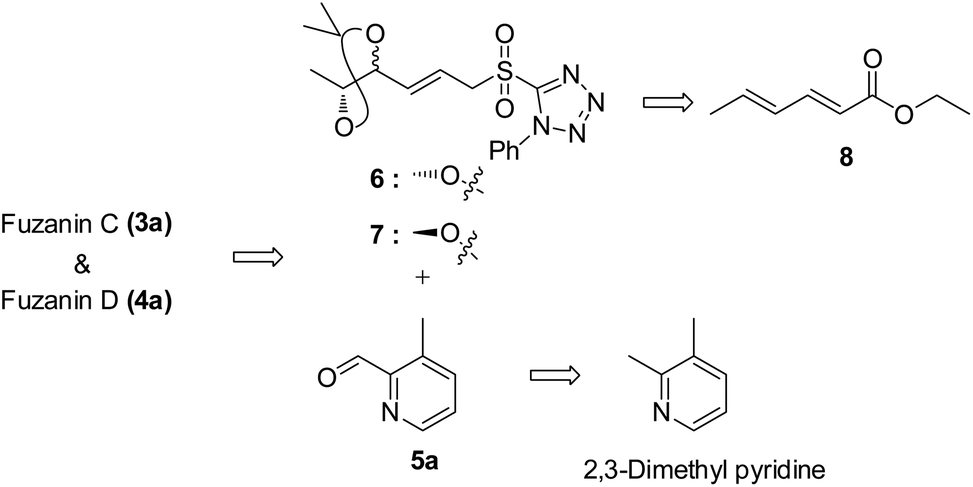 | ||
| Scheme 1 Retrosynthetic analysis of 3a, 4a. | ||
The synthetic strategy for fuzanin C (3a) is described in Scheme 2. The commercially available starting material ethyl sorbate 8, was subjected to Sharpless dihydroxylation conditions using osmium tetraoxide as oxidant and (DHQD)2PHAL as chiral ligand, giving the diol 9.12a,13 Diol 9 was protected as acetonide and the ester functionality was reduced using DIBAL-H to obtain alcohol 10. Mitsunobu reaction of 10 with 1-phenyl-1H-tetrazole-5-thiol to get sulfide, and subsequent oxidation with H2O2 in presence of a molybdenum(VI) catalyst furnished the desired sulfone 6 in 82% yield. The vital coupling reaction of 6 and 5a (5a prepared from SeO2 oxidation of 2,3-dimethyl pyridine14) was performed by the Julia-Kocienski olefination protocol15 using KHMDS as base to afford 11 as mixture of E and Z isomers.
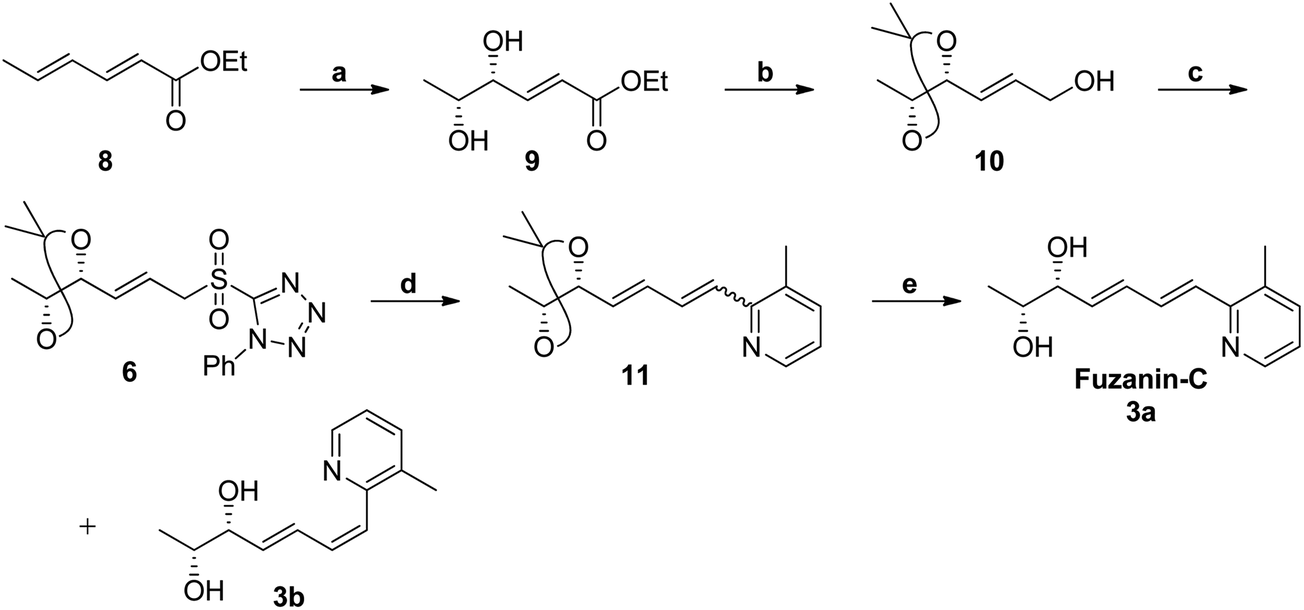 | ||
Scheme 2 Synthesis of fuzanin C (3a). Reagents and conditions: (a) (DHQD)2PHAL, OsO4, CH3SO2NH2, K3Fe(CN)6, K2CO3, t-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0 °C, 12 h, 84%; (b) (i) 2,2-Dimethoxypropane, p-TSA (cat), CH2Cl2, rt, 15 min; (ii) DIBAL-H, CH2Cl2, 0 °C, 1 h, 81% (for two steps); (c) (i) 1-phenyl-1H-tetrazole-5-thiol, PPh3, DIAD, THF, 0 °C, 1 h, 90%; (ii) (NH4)6Mo7O24·4H2O, EtOH, 30% H2O2, rt, 8h, 92%; (d) 5a, KHMDS, THF, ¬78 °C, 2 h, 81%; (e) p-TSA, MeOH, rt, 12 h, 82% (total yield). :1), 0 °C, 12 h, 84%; (b) (i) 2,2-Dimethoxypropane, p-TSA (cat), CH2Cl2, rt, 15 min; (ii) DIBAL-H, CH2Cl2, 0 °C, 1 h, 81% (for two steps); (c) (i) 1-phenyl-1H-tetrazole-5-thiol, PPh3, DIAD, THF, 0 °C, 1 h, 90%; (ii) (NH4)6Mo7O24·4H2O, EtOH, 30% H2O2, rt, 8h, 92%; (d) 5a, KHMDS, THF, ¬78 °C, 2 h, 81%; (e) p-TSA, MeOH, rt, 12 h, 82% (total yield). | ||
The geometrical isomers i.e., E and Z isomers (7:3 ratio by 1H NMR) were inseparable at this stage. However, final acetonide deprotection of 11 with p-TSA afforded fuzanin C (3a) and its Z-isomer 3b respectively (Scheme 2). All characterization data for synthesized fuzanin C (3a) (Fig. S2, ESI†) were in good agreement with those reported by Ishibashi et al.11
Synthetic strategy for fuzanin D (4a) is described in Scheme 3. This synthesis begins with selective protection of C(5)–OH group of 9 with TBS-Cl in presence of triethylamine and DMAP, provided the desired ether 12.16 Mitsunobu inversion of hydroxyl group in 12 with formic acid in the presence of PPh3, DEAD and subsequent hydrolysis of formyl ester using diluted NH4OH–MeOH afforded compound 13 in 55% overall yield.17 TBS deprotection of 13 in presence of tetrabutylammoniumfluoride (TBAF) results trans-1,2-diol 14. trans-1,2-Diol 14 is protected as acetonide, DIBAL-H reduction of ester functionality, Julia-Kocienski olefination protocol, and hydrolysis or deprotection (the same synthetic sequence was followed as in Scheme 2) provided fuzanin D (4a) and its Z-isomer 4b in 6:4 ratio (Scheme 3).
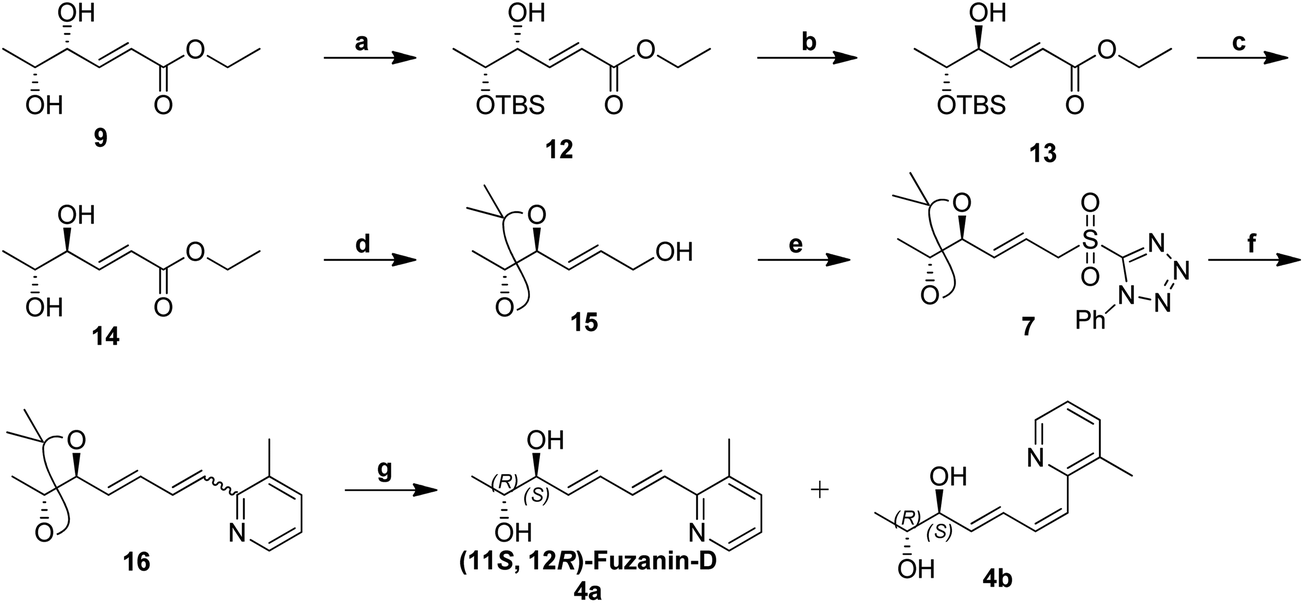 | ||
| Scheme 3 Synthesis of fuzanin D (4a). Reagents and conditions: (a) TBS-Cl, Et3N, DMAP, CH2Cl2, rt, 24 h, 65%; (b) (i) PPh3, DEAD, HCOOH, rt, 2 h; (ii) dil. NH4OH/MeOH, 3 h, 55% (for two steps); (c) TBAF, CH2Cl2, 0 °C, 1 h, 85%; (d) (i) 2,2-Dimethoxypropane, p-TSA (cat), CH2Cl2, rt, 15 min; (ii) DIBAL-H, CH2Cl2, 0 °C, 1 h, 80% (for two steps); (e) (i) 1-phenyl-1H-tetrazole-5-thiol, PPh3, DIAD, THF, 0 °C, 1 h; (ii) (NH4)6Mo7O24·4H2O, EtOH, 30% H2O2, rt, 8 h, 82% (for two steps); (f) 5a, KHMDS, THF, ¬78 °C, 2 h, 81%; (g) p-TSA, MeOH, rt, 12 h, 82% (total yield). | ||
Characterization data for the synthesized fuzanin D (4a) (Fig. S6, ESI†) were in good agreement with those reported by Ishibashi et al. except optical rotation. The [α]D of the natural product is −32.9 (CHCl3), and synthesized fuzanin D (4a) has +29.47. The original work by Ishibashi et al.11 mentioned that fuzanin D as a stereoisomer to fuzanin C and further there was no attempt made to substantiate the stereochemistry of fuzanin D. The fuzanin D synthesized in this work exhibited optical rotation +29.47, whereas the sign of optical rotation is found to be opposite to the natural product reported by Ishibashi et al. In order to investigate the absolute configuration of fuzanin D, we further synthesized the other enantiomer of fuzanin D as given in Scheme 5.
The retrosynthesis of other enantiomer fuzanin D i.e., (11R,12S)-fuzanin D [(11R,12S)-4a] is depicted in Scheme 4. We envisioned the two stereocenters in (11R,12S)-4a and this could be constructed by the Sharpless asymmetric epoxidation. In this regard, the synthesis was started from 2-buten-1-ol, which was subjected to Sharpless epoxidation to afford the epoxy alcohol 18 (Scheme 5).18
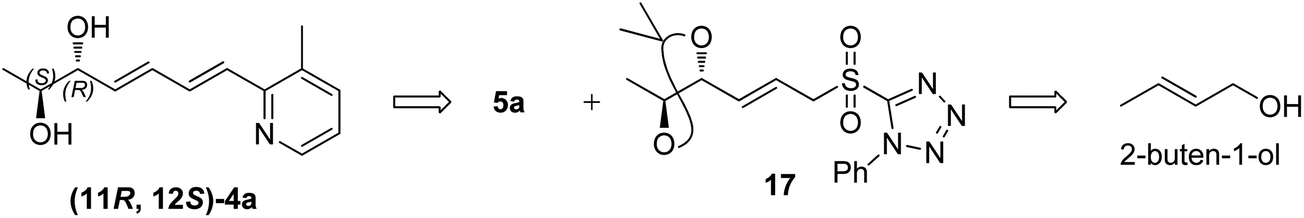 | ||
| Scheme 4 Retrosynthetic analysis of (11R,12S)-4a. | ||
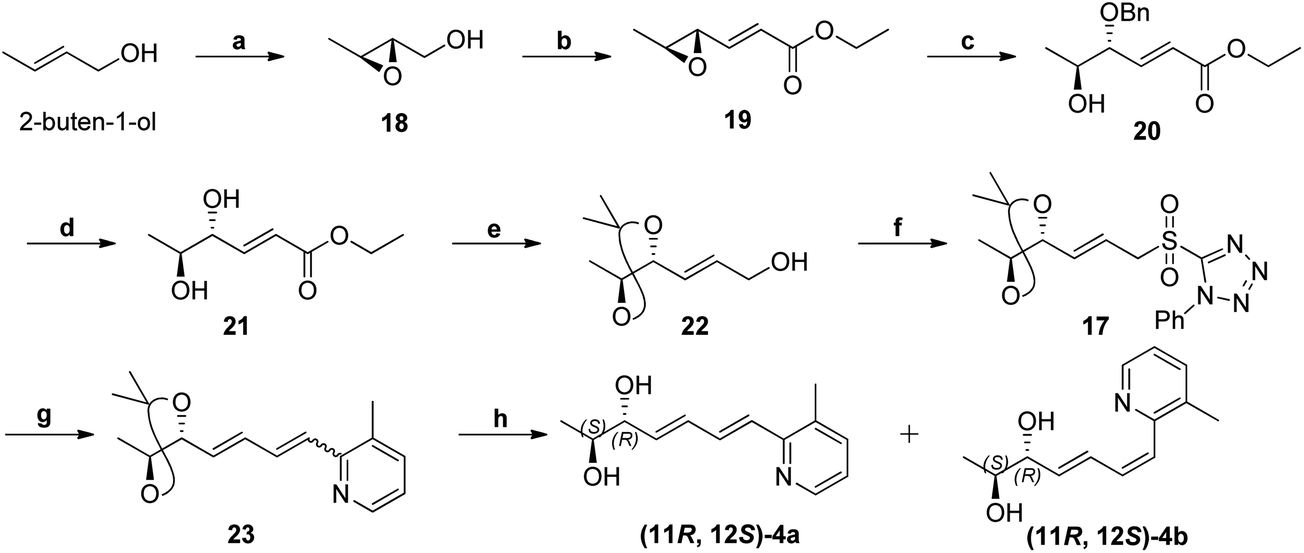 | ||
Scheme 5 Synthesis of (11R,12S)-4a. Reagents and conditions: (a) Ti(OiPr)4, (+)-DIPT, anhydrous TBHP, CH2Cl2, ¬20 °C, 2 h; (b) (i) DMSO, (COCl)2, Et3N, CH2Cl2, ¬78 °C, 30 min. (ii) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOEt, CH2Cl2, rt, 24 h, 56% (for two steps); (c) BnOH, BF3·OEt2, DCM, ¬20 °C, 1 h, 60%; (d) AlCl3, m-xylene, 0 °C, 15 min, 76%; (e) (i) 2,2-Dimethoxypropane, p-TSA, CH2Cl2, rt, 15 min. (ii) DIBAL-H, CH2Cl2, 0 °C, 1 h, 86% (for two steps); (f) (i) 1-phenyl-1H-tetrazole-5-thiol, PPh3, DIAD, THF, 0 °C, 1 h. (ii) (NH4)6Mo7O24·4H2O, EtOH, 30% H2O2, rt, 81% (for two steps); (g) 5a, KHMDS, THF, ¬78 °C, 2 h, 80%; (h) p-TSA, MeOH, rt, 12 h, 82% (total yield). CHCOOEt, CH2Cl2, rt, 24 h, 56% (for two steps); (c) BnOH, BF3·OEt2, DCM, ¬20 °C, 1 h, 60%; (d) AlCl3, m-xylene, 0 °C, 15 min, 76%; (e) (i) 2,2-Dimethoxypropane, p-TSA, CH2Cl2, rt, 15 min. (ii) DIBAL-H, CH2Cl2, 0 °C, 1 h, 86% (for two steps); (f) (i) 1-phenyl-1H-tetrazole-5-thiol, PPh3, DIAD, THF, 0 °C, 1 h. (ii) (NH4)6Mo7O24·4H2O, EtOH, 30% H2O2, rt, 81% (for two steps); (g) 5a, KHMDS, THF, ¬78 °C, 2 h, 80%; (h) p-TSA, MeOH, rt, 12 h, 82% (total yield). | ||
Swern oxidation of epoxy alcohol 18 and C2-Wittig homologation with [(ethoxycarbonyl)methylene]triphenyl-phosphorane, afforded requisite epoxy ester 19.19 The trans regioselective opening of epoxide of 19 was accomplished with benzyl alcohol in presence of BF3·OEt2 as lewis acid catalyst to get benzyl ether 20,20 which was deprotected using AlCl3 to get diol 21.20,21 Acetonide protection of 21, DIBAL-H reduction, Julia-Kocienski olefination protocol, and hydrolysis or deprotection (the same synthetic sequence was followed as in Scheme 2) provided (11R,12S)-4a and its Z-isomer (11R,12S)-4b in 6:4 ratio (Scheme 5) (Fig. S11, ESI†).
The optical rotation of (11R,12S)-4a −26.9, which is in good accordance with isolated fuzanin D. This lead unambiguously to the conclusion that the natural product isolated has opposite configuration to that of reported fuzanin D (Fig. 2).
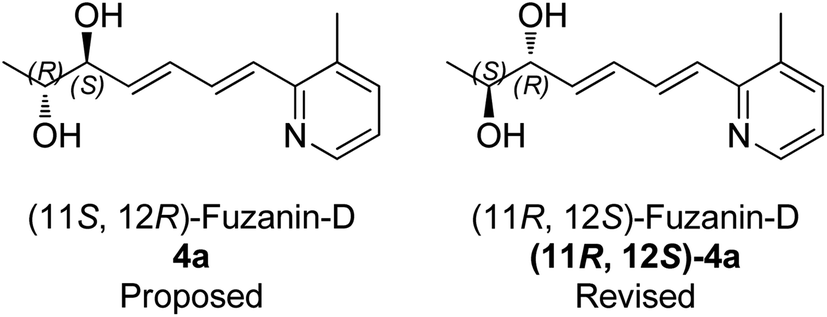 | ||
| Fig. 2 Revised structure of fuzanin D. | ||
This observation was further corroborated by computational study. Optical rotations (at 589.3 nm) of fuzanin C (3a), fuzanin D (4a) and (11R,12S)-fuzanin D [(11R,12S)-4a] were computed using B3LYP/Aug-CC-pVDZ method for geometries obtained at B3LYP/6-31G(d) basis set (Fig. 3).22 Calculated [α]D for (3a), (4a) and (11R,12S)-4a are +66.5, +22.4 and −8.5 degrees[dm(g cm−3)]−1 respectively (Table 1). Optical rotations of computationally calculated fuzanins and synthesized fuzanins were having same sign (Table 1). The comparison of experimental and calculated optical rotations allows us to define the stereochemistry fuzanin D reported by Ishibashi et al. as (11R,12S)-fuzanin D (Fig. 2).
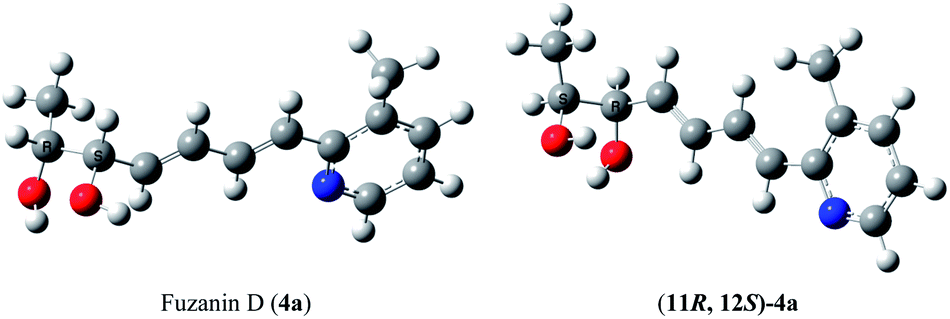 | ||
| Fig. 3 Energy minimized geometries of fuzanin D (4a) & (11R,12S)-4a. | ||
| Compounds | [α]D (in ° dm−1 g−1 cm3) | ||
|---|---|---|---|
| Isolation papera | Synthesized compounds | Calculatedb | |
| a Ref. 11.b Optical rotations were computed using B3LYP/aug-cc-pVDZ//B3LYP/6-31G(d) basis set in gas phase. | |||
| Fuzanin C (3a) | [α]15D = +34.5 | [α]24D = +39.89 | +66.5 |
| Fuzanin D (4a) | [α]15D = −32.9 | [α]24D = +29.47 | +22.4 |
| (11R,12S)-4a | — | [α]24D = −26.9 | −8.5 |
Intrigued by this, we also synthesized different analogues (3c, 3d, 4c and 4d) using substituted 2-chloroquinoline-3-carboxaldehydes (5b and 5c)23 by same synthetic strategy of Julia-Kocienski olefination protocol, and hydrolysis (Scheme 6) (Fig. S4, S5, S8 and S9, ESI†).
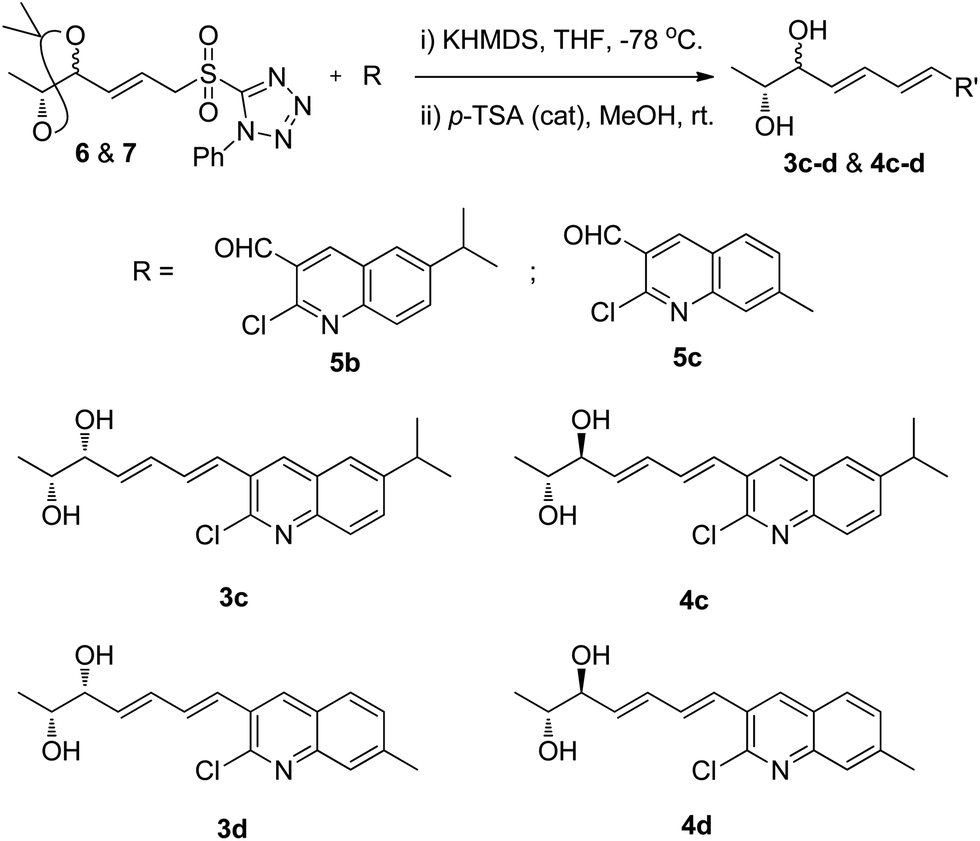 | ||
| Scheme 6 Analogues of fuzanins C, D. | ||
Furthermore, all the synthesized compounds were subjected to anticancer activity on various cell lines such as HT29 (Colon cancer), ME-180 (Cervical cancer), MCF-7 and MDA-MB-453 (Breast cancer) by employing MTT assay (details of bio assay are presented in Experimental section). For comparison purpose, the cytotoxicity of salinomycin was evaluated under the same experimental conditions. The compounds that exhibiting ≥50% cell inhibition at 100 μM were considered for determination of IC50 value, which was calculated from the % cell viability (from control) versus concentration curves obtained after 24 h drug treatment from MTT assay, are shown in Table 2.
| Compound | IC50/μg mL−1 |
|---|---|
| HT-29 (colon cancer) | |
| 3a | 96.2 ± 2.65 |
| 3b | 85.3 ± 4.72 |
| 3c | 48.5 ± 2.56 |
| 3d | 76.2 ± 2.45 |
| 4a | 77.9 ± 2.95 |
| 4b | 98.5 ± 1.88 |
| 4c | 35.3 ± 0.83 |
| 4d | 27.4 ± 0.12 |
| (11R,12S)-4a | >100 |
| (11R,12S)-4b | >100 |
| Salinomycin | 20.5 ± 1.26 |
All compounds were found to be relatively potent against colon cancer cell line and less potent against cervical and breast cancer cell lines. Among all the compounds, quinoline derivatives of fuzanin D (4c, 4d) were found to be potent with an IC50 value of 35.3 ± 0.83, 27.4 ± 0.12 μM respectively. Compound 3c has exhibited moderate activity. Remaining compounds were found to be less potent against HT-29 cell lines. The reference compound salinomycin has IC50 value of 20.5 ± 1.26 μM (Fig. 4).
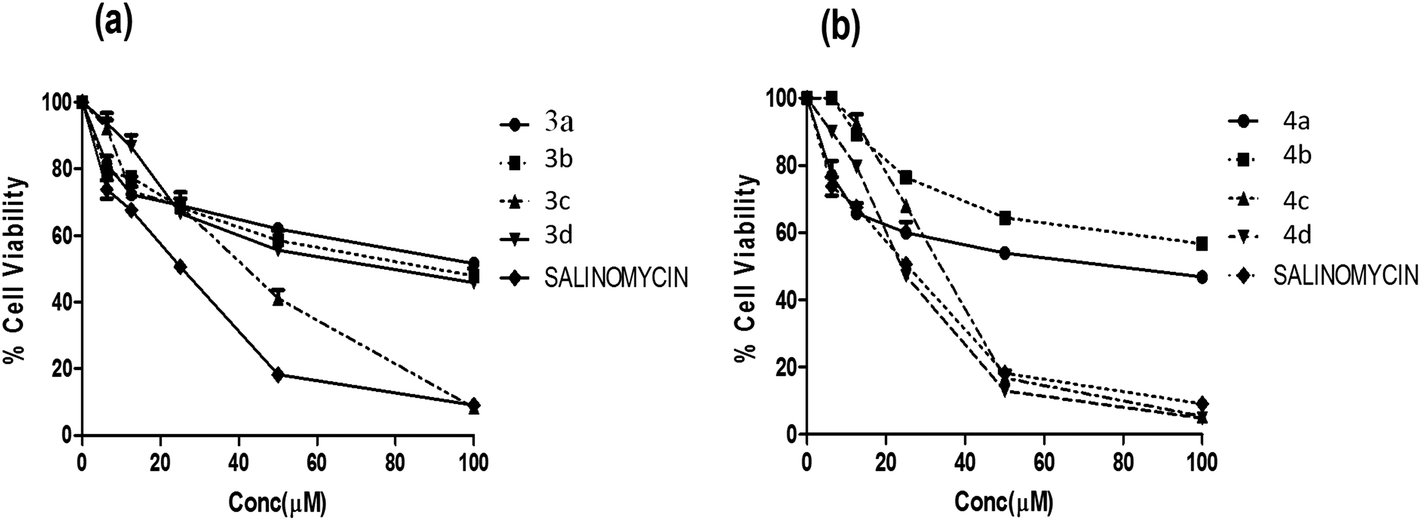 | ||
| Fig. 4 Dose response of fuzanin compounds against HT-29 cancer cell line. | ||
Conclusion
In conclusion, we have unveiled the first stereoselective total synthesis of fuzanins C, D and their analogues. Synthesis of fuzanin D described here also serves to establish its absolute configuration. Specific optical rotations of synthesized compound and reported fuzanin D indicated opposite signs. This was confirmed by total synthesis of its enantiomer (11R,12S)-4a. Stereochemistry of reported fuzanin D (4a) should be 11R,12S instead of 11S,12R configuration. Molecular modeling studies also supported this observation. Further, fuzanins C, D and their analogues were screened for anticancer activity in four cancer cell lines. The compounds were found to exert cytotoxicity selectively on HT29 cancer cell lines. Quinoline nucleus containing analogues 3c, 4c and 4d are relatively more potent.Experimental section
All reactions were carried out under an inert atmosphere unless mentioned otherwise, and standard syringe-septa techniques were followed. Solvents were freshly dried and purified by conventional methods prior to use. The progress of all reactions were monitored by TLC, using TLC aluminium backed sheets precoated with silicagel 60 F254 to a thickness of 0.25 mm (Merck). Column chromatography was performed on silica gel (60–120 mesh and 100–200 mesh), and EtoAc, hexane were used as eluents. Optical rotation values were measured with a Perkin-Elmer P241 polarimeter and a JASCO DIP-360 digital polarimeter, and IR spectra were recorded with a Perkin-Elmer FT-IR spectrophotometer. 1H and 13C NMR spectra were recorded with Varian Gemini 200 MHz, Bruker Avance 300 MHz, Varian Unity 400 MHz or Varian Inova 500 MHz spectrophotometers. TMS was used as an internal standard in CDCl3. Mass spectra were recorded with a VG micromass 7070H (EI), QSTAR XL high-resolution mass spectrophotometer, a Thermo Finnigan ESI Ion trap Mass spectrophotometer.(4R,5R,E)-Ethyl-4,5-dihydroxyhex-2-enoate (9)
To a mixture of K3Fe(CN)6 (28.22 g, 85.71 mmol), K2CO3 (11.80 g, 85.71 mmol), and (DHQD)2PHAL (0.41 g, 0.57 mmol) in 45 mL of t-BuOH–H2O (1:1), was added OsO4 (70.56 mg, 0.28 mmol) followed by methane sulfonamide (2.71 g, 28.57 mmol) at 0 °C. After stirring for 15 min at 0 °C, ethyl sorbate 8 (4 g, 28.57 mmol) was added and stirred vigorously at 0 °C for 12 h, and then quenched with sat. sodium sulfite (35 mL). The reaction mixture was extracted with ethyl acetate (3 × 40 mL). The organic layer washed with 2 N KOH (20 mL), brine solution and dried over Na2SO4. Solvent removed under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 7:3) to afford diol 9 (3.97 g, 84%) as light yellow oil; [α]25D + 50.21 (c 0.1, EtOH); IR (Neat): 3396, 2924, 1704, 1371, 1281, 1036 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.89 (dd, 1H, J = 15.5, 5.2 Hz), 6.10 (dd, 1H, J = 15.5, 1.5 Hz), 4.17 (q, 2H, J = 7.5 Hz), 4.02 (brt, 1H, J = 5.2 Hz), 3.75–3.63 (m, 1H), 1.27 (t, 3H, J = 7.5 Hz), 1.20 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 75 MHz): δ 166.5, 146.6, 122.4, 75.6, 70.2, 60.7, 19.0, 14.1; anal. calcd for C8H14O4 (174.19): C, 55.16; H, 8.10. Found: C, 55.15; H, 8.17%.
(E)-3-((4R,5R)-2,2,5-Trimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (10)
To the solution of 9 (3 g, 17.24 mmol) in 30 mL CH2Cl2, 2,2-dimethoxypropane (5.37 g, 51.72 mmol), catalytic amount of p-TSA were added and stirred for 15 min at rt. The reaction mixture was washed with saturated aqueous NaHCO3 solution (10 mL), dried over Na2SO4, concentrated under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 1:9) to afford cyclic acetonide (3.46 g, 94%) as colour less oil. To the solution cyclic acetonide (3.2 g, 14.9 mmol) in 30 mL of dry CH2Cl2 at 0 °C, DIBAL-H (21.01 mL, 37.25 mmol, 25% solution in THF) was added, and stirred for 1 h at rt. The reaction mixture was quenched by slow addition of aq. sodium potassium tartate and stirred for 2 h. Organic layer was separated and aqueous layer was extracted by chloroform (20 mL), combined organic layer was dried over Na2SO4, solvent removed under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 6:4) to afford 10 (2.4 g, 81%) as light yellow, viscous oil; [α]24D + 46.01 (c 0.1, CHCl3); IR (Neat): 3395, 2923, 2851, 1449, 1259, 998 cm−1; 1H NMR (CDCl3, 300 MHz): δ 5.99 (dtd, 1H, J = 15.5, 5.1, 0.7 Hz), 5.70 (tdd, 1H, J = 15.5, 7.5, 1.5 Hz), 4.19 (d, 2H, J = 5.1 Hz), 3.9 (t, 1H, J = 7.5 Hz), 3.84–3.75 (m, 1H), 1.42 (s, 3H), 1.41 (s, 3H), 1.26 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 50 MHz): δ 134.2, 127.0, 108.2, 83.1, 76.5, 62.3, 27.2, 26.8, 16.3; anal. calcd for C9H16O3 (172.10): C, 62.77; H, 9.36. Found: C, 62.58; H, 9.57%.
1-Phenyl-5-(((E)-3-((4R,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl)allyl)sulfonyl)-1H-tetrazole (6)
To a solution of alcohol 10 (0.6 g, 3.48 mmol) and 1-phenyl-1H-tetrazole-5-thiol (0.74 g, 4.15 mmol) in 20 mL THF at 0 °C, DIAD (0.82 mL, 4.16 mmol) and triphenyl phosphine (0.84 g, 4.15 mmol) were added, and stirred for 1 h at same temperature. The reaction was quenched by addition of saturated aqueous NH4Cl solution, extracted with ethyl acetate (2 × 20 mL), dried over Na2SO4, concentrated under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 3:7) to afford sulphide (1.0 g, 90%) as colour less, viscous oil; [α]24D − 24.80 (c 0.1, CHCl3); IR (Neat): 2923, 1679, 1618, 1185, 1001, 741 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.61–7.55 (m, 5H), 6.05–5.95 (m, 1H), 5.81 (dd, 1H, J = 15.1, 6.7 Hz), 4.13–3.97 (m, 2H), 3.90 (t, 1H, J = 8.3 Hz), 3.79–3.70 (m, 1H), 1.40 (s, 3H), 1.39 (s, 3H), 1.22 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 50 MHz): δ 153.3, 133.4, 132.2, 130.0, 129.6, 127.5, 123.6, 108.4, 82.5, 76.4, 34.4, 27.1, 26.7, 16.2; MS (ESI): m/z 355 (M + Na)+; HRMS (ESI): m/z 355.1200 (M + Na)+ (calcd for C16H21N4O2S 355.1199).
To a solution of sulphide (0.50 g, 1.50 mmol) and ammonium heptamolybdate tetrahydrate (0.55 g, 0.45 mmol) in 12 mL of ethanol at 0 °C, H2O2 (30% solution in water, 2.04 mL, 18.07 mmol) was added and stirred for 8 h at rt. The reaction mixture was poured in to saturated aqueous NaHCO3 solution (40 mL) and extracted with ethyl acetate (3 × 30 mL), dried over Na2SO4, concentrated under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 3:7) to afford sulphone 4 (0.5 g, 92%) as colour less, viscous oil; [α]24D − 28.70 (c 0.1, CHCl3); IR (Neat): 2922, 1679, 1618, 1222, 741 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.68–7.59 (m, 5H), 5.98 (dd, 1H, J = 15.8, 6.0 Hz), 5.93–5.83 (m, 1H), 4.45 (m, 2H), 3.93 (dd, 1H, J = 8.3, 6.0 Hz), 3.77–3.68 (m, 1H), 1.41 (s, 3H), 1.37 (s, 3H), 1.22 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 50 MHz): δ 152.8, 140.0, 132.8, 131.4, 129.5, 125.0, 116.6, 108.9, 82.1, 76.4, 59.0, 27.1, 26.6, 16.3; MS (ESI): m/z 365 (M + H)+; HRMS (ESI): m/z 387.1093 (M + Na)+ (calcd for C16H20O4N4NaS 387.1097).
3-Methyl-2-((3E)-4-((4R,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl)buta-1,3-dien-1-yl)pyridine (11)
To a solution of sulphone 6 (0.40 g, 1.09 mmol) in 10 mL THF at −78 °C, potassium bis(trimethylsilyl)amide (0.5 M in THF solution, 2.85 mL, 1.42 mmol) was added. After 30 min of stirring at same temperature, the solution of aldehyde 5a (0.14 g, 1.19 mmol) in 2 mL THF was added. The reaction mixture was stirred for further 1.5 h at −78 °C, the reaction allowed to warm to rt and was stirred for additional 2 h. Brine solution was added to the reaction mixture and extracted with ethyl acetate (3 × 10 mL), dried over Na2SO4, concentrated under vacuo. The crude residue was purified by silica gel column chromatography (EtOAc–hexane, 1:4) to afford 11 (0.23 g, 81%) (inseparable cis, trans mixture, 3:7) as viscous, light yellow oil; IR (Neat): 2984, 2932, 1581, 1498, 1380, 1028, 858 cm−1; 1H NMR (CDCl3, 300 MHz): δ 8.45 (d, 0.3H, J = 4.7 Hz), 8.40 (d, 0.7H, J = 4.1 Hz), 7.45–7.35 (m, 2H), 7.07–7.01 (m, 1H), 6.79 (d, 0.7H, J = 15.1 Hz), 6.60–6.65 (m, 1.5H), 5.92–5.76 (m, 1H), 4.02 (brt, 1H, J = 7.5 Hz), 3.84–3.75 (m, 1.5H), 2.36 (s, 2.1H), 2.31 (s, 0.9H), 1.44 (s, 4.2H), 1.41 (s, 1.8H), 1.29–1.26 (m, 3H); 13C NMR (CDCl3, 50 MHz): δ 155.0, 153.4, 147.3, 146.8, 138.3, 137.8, 133.7, 132.8, 132.3, 131.7, 130.9, 129.1, 127.0, 122.3, 122.0, 116.8, 108.7, 108.5, 83.7, 77.7, 77.0, 27.5, 27.1, 19.3, 18.9, 16.8, 16.7; MS (ESI): m/z 260 (M + H)+; HRMS (ESI): m/z 260.1641 (M + H)+ (calcd for C16H22O2N 260.1645).
(2R,3R,4E,6E)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (3a)
To a solution of 11 (0.10 g, 0.38 mmol) in 10 mL methanol, was added p-toluenesulphonic acid (1.32 g, 0.77 mmol) and the reaction mixture stirred for 12 h at rt, and solvent removed under vacuo. The crude residue was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3 solution and the solvent removed under vacuo. The crude compound was purified by silica gel column chromatography (EtOAc–hexane, 1:1) to afford fuzanin C (3a) (0.05 g) as viscous, colour less oil, and cis isomer 3b (0.02 g) as viscous, colour less oil, (total yield 82%); [α]24D + 39.89 (c 0.055, CHCl3); IR (Neat): 3395, 2923, 1679, 1618, 772 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.39 (d, 1H, J = 4.7 Hz), 7.42 (d, 1H, J = 7.7 Hz), 7.37 (dd, 1H, J = 15.0, 11.0 Hz), 7.04 (dd, 1H, J = 7.7, 4.7 Hz), 6.78 (d, 1H, J = 15.0 Hz), 6.54 (dd, 1H, J = 15.3, 11.0 Hz), 5.94 (dd, 1H, J = 15.3, 6.9 Hz), 3.95 (t, 1H, J = 6.9 Hz), 3.68 (q, 1H, J = 6.6 Hz), 2.34 (s, 3H), 1.18 (d, 3H, J = 6.6 Hz); 13C NMR (CDCl3, 100 MHz): δ 153.2, 146.8, 138.2, 135.7, 133.3, 132.1, 130.8, 128.0, 122.0, 77.1, 70.7, 18.8, 18.6; MS (ESI): m/z 220 (M + H)+; HRMS (ESI): m/z 242.1148 (M + Na)+ (calcd for C13H17O2NNa 242.1151).
(2R,3R,4E,6Z)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (3b)
Yield: 0.02 g; [α]24D + 82.23 (c 0.051, CHCl3); IR (Neat): 3421, 2924, 1619, 1449, 772 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.42 (d, 1H, J = 4.7 Hz), 7.45 (d, 1H, J = 7.3 Hz), 7.29 (dd, 1H, J = 15.2, 11.5 Hz), 7.06 (dd, 1H, J = 7.3, 4.7 Hz), 6.46 (d, 1H, J = 11.5 Hz), 6.38 (t, 1H, J = 11.5 Hz), 5.87 (dd, 1H, J = 15.2, 6.6 Hz), 3.93 (t, 1H, J = 6.6 Hz), 3.66 (quin, 1H, J = 6.2 Hz), 2.31 (s, 3H), 1.17 (d, 3H, J = 6.2 Hz); 13C NMR (CDCl3, 100 MHz): δ 154.5, 146.2, 137.8, 136.9, 132.9, 131.9, 129.2, 126.0, 121.8, 76.7, 70.4, 19.0, 18.8; MS (ESI): m/z 220 (M + H)+; HRMS (ESI): m/z 220.1329 (M + H)+ (calcd for C13H18O2N 220.1332).(2R,3R,4E,6E)-7-(2-Chloro-6-isopropylquinolin-3-yl)hepta-4,6-diene-2,3-diol (3c)
According to the procedure 3a, the sulphone 6 (0.10 g, 0.27 mmol) and aldehyde 5b (0.07 g, 0.32 mmol), gave cyclic acetonide (inseparable cis, trans mixture, 6:94 by NMR) as light yellow oil. Cyclic acetonide dissolved in 10 mL of methanol, catalytic amount of p-TSA was added and stirred for 30 min at rt, and solvent removed under vacuo. The crude residue was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3 solution and the solvent removed under vacuo. The crude compound was purified by silica gel column chromatography (EtOAc–hexane, 1:1) to afford compound 3c (0.06 g, 70%, for two steps) as white solid; mp 120–122 °C; [α]24D − 89.56 (c 0.022, CHCl3); IR (KBr): 3409, 2963, 2926, 1584, 1047, 768 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.19 (s, 1H), 7.89 (d, 1H, J = 9.0 Hz), 7.60–7.56 (m, 2H), 6.98 (d, 1H, J = 15.9 Hz), 6.83 (dd, 1H, J = 15.9, 10.9 Hz), 6.59 (dd, 1H, J = 14.9, 10.9 Hz), 5.91 (dd, 1H, J = 14.9, 5.9 Hz), 4.01 (t, 1H, J = 5.9 Hz), 3.73 (quin, 1H, J = 5.9 Hz), 3.07 (sep, 1H, J = 6.9 Hz), 1.33 (d, 6H, J = 6.9 Hz), 1.24 (d, 3H, J = 5.9 Hz); 13C NMR (CDCl3, 100 MHz): δ 149.0, 148.0, 145.5, 134.7, 133.3, 132.3, 131.9, 130.3, 129.5, 127.8, 127.6, 127.3, 123.5, 77.0, 70.8, 34.0, 23.6, 19.0; MS (ESI): m/z 332 [M + H]+; HRMS (ESI): m/z 332.1410 (M + H)+ (calcd for C19H23O2NCl 332.1411).
(2R,3R,4E,6E)-7-(2-Chloro-7-methylquinolin-3-yl)hepta-4,6-diene-2,3-diol (3d)
According to the procedure 3c, the sulphone 6 (0.10 g, 0.27 mmol) and requisite aldehyde 5c (0.07 g, 0.35 mmol) gave the compound 3d (0.06 g, 71%, for two steps) as white solid; mp 130–131 °C; [α]24D − 91.18 (c 0.024, CHCl3); IR (KBr): 3397, 2925, 1623, 1049, 756 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.19 (s, 1H), 7.73 (br s, 1H), 7.68 (d, 1H, J = 8.3, Hz), 7.37 (dd, 1H, J = 8.3, 1.13 Hz), 6.99 (d, 1H, J = 15.5 Hz), 6.83 (dd, 1H, J = 15.5, 10.3 Hz), 6.60 (dd, 1H, J = 15.3, 10.3 Hz), 5.91 (dd, 1H, J = 15.3, 6.8 Hz), 4.01 (t, 1H, J = 6.8 Hz), 3.73 (quin, 1H, J = 6.4 Hz), 2.54 (s, 3H), 1.24 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3, 100 MHz): δ 149.8, 146.9, 141.0, 134.5, 133.3, 132.4, 131.7, 129.5, 128.8, 127.8, 127.2, 127.1, 125.4, 77.1, 70.8, 21.9, 19.0; MS (ESI): m/z 304 (M + H)+; HRMS (ESI): m/z 304.1096 [M + H]+ (calcd for C17H19O2NCl 304.1098).(4R,5R,E)-Ethyl 5-((tert-butyldimethylsilyl)oxy)-4-hydroxyhex-2-enoate (12)
To a solution of the diol 9 (3.0 g, 17.5 mmol) in CH2Cl2 (30 mL) at 0 °C, Et3N (4.1 mL, 29.8 mmol), DMAP (1.06 g, 0.88 mmol) and TBSCl (3.96 g, 26.3 mmol) were added simultaneously, and the mixture was stirred for 24 h at rt. The reaction mixture was diluted with CH2Cl2, successively washed with 10% aq. HCl and sat. NaHCO3, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting oil was purified by silicagel column chromatography (EtOAc–hexane, 1:4) to afford the TBS ether (12) (3.29 g, 65%) as a colorless oil; [α]25D − 0.71 (c 0.051, EtOH); IR (Neat): 3475, 2932, 2858, 1721, 1257, 838 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.88 (dd, 1H, J = 15.8, 4.5 Hz), 6.10 (dd, 1H, J = 15.8, 1.8 Hz), 4.19 (q, 2H, J = 7.1 Hz), 4.02–3.98 (m, 1H), 3.79–3.71 (m, 1H), 2.57 (brs, 1H), 1.29 (t, 3H, J = 7.1 Hz), 1.20 (d, 3H, J = 6.0 Hz), 0.88 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 166.2, 147.2, 121.9, 75.2, 71.0, 60.3, 25.7, 20.1, 17.9, 14.2, −4.3, −4.9; MS (ESI): m/z 289 (M + H)+.
(4S,5R,E)-Ethyl-5-((tert-butyldimethylsilyl)oxy)-4-hydroxyhex-2-enoate (13)
Under inert atmosphere, to a solution of PPh3 (0.62 g, 2.38 mmol) in 5 mL benzene at 0 °C, DEAD (0.16 mL, 2.38 mmol), TBS ether (12) (0.274 g, 0.95 mmol) and formic acid (0.11 mL, 2.85 mmol) were added simultaneously and the mixture was stirred for 2 h at rt. The solvent was evaporated in vacuo, and the crude residue was dissolved in NH4OH–MeOH (1:1, 3.0 mL) at 0 °C and stirred for 3 h at rt. This reaction mixture was diluted with Et2O, successively washed with 10% HCl solution and sat. NaHCO3, dried over Na2SO4, and concentrated in vacuo. The resulting oil was purified by silica gel column chromatography (EtOAc–hexane, 1:4) to afford alcohol 13 (0.15 g, 55%) as a colorless oil; [α]25D − 27.1 (c 0.051, EtOH); IR (Neat): 3466, 2930, 2857, 1720, 1259, 836 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.87 (dd, 1H, J = 15.6, 4.6 Hz), 6.08 (dd, 1H, J = 15.6, 2.3 Hz), 4.24–4.21 (m, 1H), 4.19 (q, 2H, J = 7.0 Hz), 3.93–3.88 (m, 1H), 2.43 (brs, 1H), 1.28 (t, 3H, J = 7.0 Hz), 1.08 (d, 3H, J = 6.2 Hz), 0.89 (s, 9H), 0.07 (s, 6H); 13C NMR (CDCl3, 70 MHz): δ 166.3, 145.5, 121.7, 74.8, 70.7, 60.3, 25.7, 17.9, 17.7, 14.2, −4.4, −4.9; MS (ESI): m/z 289 (M + H)+.
(4S,5R,E)-Ethyl-4,5-dihydroxyhex-2-enoate (14)
To a solution of 13 (0.5 g, 1.73 mmol) in 10 mL THF at 0 °C, was added TBAF (1.0 M, 5.2 mL, 5.19 mmol) and the resulting mixture was stirred for 1 h. The reaction was quenched with water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic extracts were dried over Na2SO4, concentrated in vacuo. The crude reside was purified by silica gel column chromatography to yield diol 14 (0.25 g, 85%); [α]24D − 9.15 (c 0.047, CHCl3); IR (Neat): 3429, 2981, 2931, 1714, 1659, 1278, 984 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.94 (dd, 1H, J = 15.1, 4.5 Hz), 6.11 (d, 1H, J = 15.1 Hz), 4.30 (brs, 1H), 4.20 (q, 2H, J = 6.8 Hz), 3.99–3.91 (m, 1H), 1.28 (t, 3H, J = 6.8 Hz), 1.16 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 75 MHz): δ 166.6, 145.8, 122.37, 74.5, 69.9, 60.6, 17.4, 14.1; anal. calcd for C8H14O4 (174.19): C, 55.16; H, 8.10. Found: C, 55.19; H, 8.14%.(E)-3-((4S,5R)-2,2,5-Trimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (15)
According to the procedure 10, compound 14 (2.4 g, 13.79 mmol) gave alcohol 15 (1.90 g, 80%); [α]24D − 15.1 (c 0.1, CHCl3); IR (Neat): 3431, 2984, 1380, 1219, 772 cm−1; 1H NMR (CDCl3, 500 MHz): δ 5.90 (td, 1H, J = 15.4, 5.1 Hz), 5.68 (tdd, 1H, J = 15.4, 7.7, 1.5 Hz), 4.52 (brt, 1H, J = 7.7 Hz), 4.32 (quin, 1H, J = 6.4 Hz), 4.17 (dd, 2H, J = 5.1, 1.5 Hz), 1.47 (s, 3H), 1.35 (s, 3H), 1.13 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3, 100 MHz): δ 133.5, 127.1, 108.0, 79.0, 74.0, 62.8, 28.2, 25.5, 16.1; anal. calcd for C9H16O3 (172.10): C, 62.77; H, 9.36. Found: C, 62.61; H, 9.59%.1-Phenyl-5-(((E)-3-((4S,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl)allyl)sulfonyl)-1H-tetrazole (7)
According to the procedure 6, the alcohol 15 (0.5 g, 2.9 mmol) gave sulphone 7 (0.86 g, 82%); [α]24D − 3.14 (c 0.05, CHCl3); IR (Neat): 2922, 1679, 1618, 1524, 1347, 741 cm−1; 1H NMR (CDCl3, 300 MHz): δ 7.68–7.56 (m, 5H), 5.96 (dd, 1H, J = 15.8, 6.8 Hz), 5.86–5.76 (m, 1H), 4.54–4.38 (m, 3H), 4.32 (dd, 1H, J = 12.8, 6.7 Hz), 1.45 (s, 3H), 1.34 (s, 3H), 1.03 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 75 MHz): δ 152.9, 140.3, 132.9, 131.4, 129.6, 125.1, 116.1, 108.5, 78.1, 73.8, 59.0, 28.0, 25.3, 15.8; MS (ESI): m/z 365 (M + H)+; HRMS (ESI): m/z [M + H]+ 387.1094 (M + Na)+ (calculated for C16H20N4O4NaS 387.1097).3-Methyl-2-((3E)-4-((4S,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl)buta-1,3-dien-1-yl)pyridine (16)
According to the procedure 11, the sulphone 6 (0.38 g, 1.04 mmol) and 5a (0.13 g, 1.14 mmol), gave 16 (0.22 g, 81%) (inseparable cis, trans mixture, 4:6) as colourless, viscous oil; IR (Neat): 2923, 2851, 1667, 1382, 1048, 771 cm−1; 1H NMR (CDCl3, 300 MHz): δ 8.45 (d, 0.4H, J = 4.5 Hz), 8.42 (d, 0.6H, J = 4.5 Hz), 7.48–7.36 (m, 2H), 7.08–7.02 (m, 1H), 6.81 (d, 0.6H, J = 15.1 Hz), 6.56–6.39 (m, 1.6H), 5.96–5.81 (m, 1H), 4.65–4.59 (m, 1H), 4.41–4.30 (m, 1H), 2.36 (s, 1.8H), 2.30 (s, 1.2H), 1.53 (s, 1.8H), 1.50 (s, 1.2H), 1.39 (s, 1.8H), 1.36 (s, 1.2H), 1.16 (t, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 75 MHz): δ 154.7, 153.2, 146.9, 146.4, 138.1, 137.6, 133.3, 132.8, 132.6, 132.3, 130.9, 128.3, 126.3, 122.0, 121.7, 108.0, 107.9, 79.4, 79.1, 74.37, 74.32, 28.2, 25.4, 19.0, 18.7, 16.17; MS (ESI): m/z 260 (M + H)+; HRMS (ESI): m/z 260.1640 (M + H)+ (calcd for C16H22O2N 260.1645).
(2R,3S,4E,6E)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (4a)
According to the procedure 3a, compound 16 (0.1 g, 0.38 mmol) gave fuzanin D (4a) (0.05 g) as viscous, colour less oil, and cis isomer 4b (0.02 g) as viscous, colour less oil, (total yield 82%); [α]24D + 29.47 (c 0.045, CHCl3); IR (Neat): 3424, 2924, 2854, 1621, 1458, 1078, 771 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.40 (d, 1H, J = 4.1 Hz), 7.45–7.36 (m, 2H), 7.04 (dd, 1H, J = 7.2, 4.1 Hz), 6.79 (d, 1H, J = 14.5 Hz), 6.54 (dd, 1H, J = 14.5, 10.4 Hz), 6.01 (dd, 1H, J = 14.5, 6.2 Hz), 4.20 (dd, 1H, J = 6.2, 4.1 Hz), 3.90 (dd, 1H, J = 6.2, 4.1 Hz), 2.34 (s, 3H), 1.16 (d, 1H, J = 6.2 Hz); 13C NMR (CDCl3, 100 MHz): δ 153.3, 146.9, 138.2, 134.6, 133.2, 132.3, 130.7, 128.1, 122.0, 75.9, 70.3, 18.7, 17.6; MS (ESI): m/z 242 (M + Na)+; HRMS (ESI): m/z 242.1148 (M + Na)+ (calcd for C13H17O2NNa 242.1151).(2R,3S,4E,6Z)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (4b)
Yield: (0.02 g, 90% overall yield); [α]24D + 59.15 (c 0.04, CHCl3); IR (Neat): 3405, 2924, 1449, 995 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.45 (d, 1H, J = 5.2 Hz), 7.46 (d, 1H, J = 7.2 Hz), 7.39 (dd, 1H, J = 15.6, 11.4 Hz), 7.07 (dd, 1H, J = 7.2, 5.2 Hz), 6.52 (d, 1H, J = 11.4 Hz), 6.44 (t, 1H, J = 11.4 Hz), 5.97 (dd, 1H, J = 15.6, 7.2 Hz), 4.18 (dd, 1H, J = 7.27, 4.1 Hz), 3.9 (dd, 1H, J = 6.2, 4.1 Hz), 2.31 (s, 3H), 1.16 (d, 3H, J = 6.2 Hz); 13C NMR (CDCl3, 100 MHz): δ 154.5, 146.2, 138.0, 135.7, 133.1, 132.0, 129.9, 125.9, 121.9, 75.9, 70.1, 19.0, 17.7; MS (ESI): m/z 220 (M + H)+; HRMS (ESI): m/z 220.1329 (M + H)+ (calcd for C13H18O2N 220.1332).(2R,3S,4E,6E)-7-(2-Chloro-6-isopropylquinolin-3-yl)hepta-4,6-diene-2,3-diol (4c)
According to the procedure 3c, the sulphone 7 (0.10 g, 0.27 mmol) and aldehyde 5b (0.07 g, 0.32 mmol), gave compound 4c (0.07 g, 70% overall yield); mp 121–122 °C; [α]24D − 24.0 (c 0.03, CHCl3); IR (KBr): 3423, 2962, 2924, 1585, 1046, 757 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.22 (S, 1H), 7.90 (d, 1H, J = 8.9 Hz), 7.60–7.58 (m, 2H), 7.01 (d, 1H, J = 15.9 Hz), 6.87 (dd, 1H, J = 15.9, 6.9 Hz), 6.59 (dd, 1H, J = 15.9, 9.8 Hz), 5.99 (dd, 1H, J = 15.9, 6.7 Hz), 4.24 (dd, 1H, J = 6.7, 4.5 Hz), 3.98–3.94 (m, 1H), 3.08 (sep, 1H, J = 6.9 Hz), 1.34 (d, 6H, J = 6.9 Hz), 1.20 (d, 3H, J = 6.9 Hz); 13C NMR (CDCl3, 100 MHz): δ 149.1, 148.0, 145.6, 133.4, 133.3, 132.6, 132.0, 130.3, 129.6, 128.0, 127.8, 127.4, 123.5, 75.9, 70.3, 34.0, 23.7, 17.7; MS (ESI): m/z 332 (M + H)+; HRMS (ESI): m/z 332.1409 (M + H)+ (calcd for C19H23O2NCl 332.1411).(2R,3S,4E,6E)-7-(2-Chloro-7-methylquinolin-3-yl)hepta-4,6-diene-2,3-diol (4d)
According to the procedure 3c, the sulphone 7 (0.10 g, 0.27 mmol) and aldehyde 5c (0.06 g, 0.32 mmol) gave compound 4d (0.07 g, 70% overall yield); mp 134–136 °C; [α]24D − 75.14 (c 0.03, CHCl3); IR (Neat): 3418, 2922, 1622, 1337, 986 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.20 (s, 1H), 7.73 (brs, 1H), 7.68 (d, 1H, J = 8.3 Hz), 7.36 (d, 1H, J = 8.3 Hz), 6.99 (d, 1H, J = 15.1 Hz), 6.84 (dd, 1H, J = 15.1, 10.5 Hz), 6.57 (dd, 1H, J = 15.1, 10.5 Hz), 5.98 (dd, 1H, J = 15.1, 6.7 Hz), 4.24 (dd, 1H, J = 6.7, 3.0 Hz), 4.01–3.92 (m, 1H), 2.54 (s, 3H), 1.20 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 100 MHz): δ 149.9, 147.0, 141.0, 133.3, 132.6, 131.8, 129.5, 128.8, 127.8, 127.7, 127.2, 127.1, 126.0, 75.9, 70.3, 21.9, 17.7; MS (ESI): m/z 304 (M + H)+; HRMS (ESI): m/z 304.1097 (M + H)+ (calculated for C17H18N3O6 304.1098).(E)-Ethyl-3-((2S,3S)-3-methyloxiran-2-yl)acrylate (19)
To a solution of oxalyl chloride (0.87 mL, 9.96 mmol) in 30 mL of CH2Cl2 at −78 °C, DMSO (1.41 mL, 19.92 mmol) was added dropwise. The solution was stirred for 10 min and a solution of epoxy alcohol 18 (0.73 mg, 8.3 mmol) in 20 mL of CH2Cl2 was added. After 20 min at same temperature, triethylamine (3.47 mL, 24.7 mmol) was added. After 5 min, the reaction mixture of was allowed to warm to rt during 30 min. A solution of (carbethoxymethylene)triphenylphosphorane (7.63 g, 20.8 mmol) in 10 mL of CH2Cl2 was added and the reaction mixture was stirred for 24 h. Reaction mixture was quenched with aq. NH4Cl, and extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was dried over Na2SO4, crude residue was purified by silica gel column chromatography (EtOAc–hexane, 1:4) to afford epoxide 19 (0.55 g, 56%); [α]24D − 15.54 (c 0.04, CHCl3); IR (Neat): 2927, 1721, 1455, 1272, 1178, 772 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.67 (dd, 1H, J = 15.6, 7.0 Hz), 6.12 (d, 1H, J = 15.6 Hz), 4.20 (q, 2H, J = 7.17 Hz), 3.18 (dd, 1H, J = 8.7, 7.0 Hz), 2.97 (qd, 1H, J = 5.1, 2.07 Hz), 1.39 (d, 3H, J = 5.1 Hz), 1.28 (t, 3H, J = 7.17 Hz); 13C NMR (CDCl3, 75 MHz): δ 165.5, 144.5, 123.5, 60.4, 57.2, 57.1, 17.3, 14.0; anal. calcd for C8H12O3 (156.07): C, 61.52; H, 7.74. Found: C, 61.62; H, 7.82%.
(4R,5S,E)-Ethyl-4-(benzyloxy)-5-hydroxyhex-2-enoate (20)
To a solution of epoxide ester 19 (3.14 g, 20.15 mmol), benzyl alcohol (4.19 mL, 40.3 mmol) in CH2Cl2 (40 mL) at −20 °C, BF3·OEt2 (2.5 mL, 40.3 mmol) was added and whole mixture was stirred at rt for 1 h, then diluted with H2O and extracted with ether. The ether layer was washed with saturated brine and dried over Na2SO4. The organic layer was concentrated under vacuo. Crude residue was purified by silica gel column chromatography (EtOAc–hexane, 3:2) to afford 20 (3.03 g, 60%) as colorless oil; [α]25D − 51.51 (c 1.0, CHCl3); IR (Neat): 3508, 2983, 1721, 1264, 836 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.40–7.30 (m, 5H), 6.91 (dd, 1H, J = 15.8, 6.8 Hz), 6.08 (d, 1H, J = 15.8 Hz), 4.66 (d, 1H, J = 11.3 Hz), 4.42 (d, 1H, J = 11.3), 4.23 (q, 2H, J = 7.5 Hz), 4.01–3.90 (m, 2H), 1.31 (t, 3H, J = 7.5 Hz), 1.16 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 75 MHz): δ 165.7, 143.8, 137.6, 128.4, 127.8, 127.7, 124.7, 81.9, 71.3, 69.2, 60.6, 17.9, 14.2; anal. calcd for C15H20O4 (264.13): C, 68.16; H, 7.63. Found: C, 68.22; H, 7.69%.
(4R,5S,E)-Ethyl 4,5-dihydroxyhex-2-enoate (21)
To a well-stirred solution of AlCl3 (0.99 g, 7.5 mmol) in 10 mL CH2Cl2 at 0 °C was added a solution of 20 (0.40 g, 1.5 mmol) in 5 mL m-xylene and the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was poured in ice cold water and extracted with ether. The ether layer was washed with saturated NaCl solution and dried over Na2SO4. The ether layer was concentrated under vacuo. Crude residue was purified by silica gel column chromatography (EtOAc–hexane, 2:1) to give 21 (0.29 g, 76%) as a colorless viscous oil; [α]25D + 9.01 (c 0.09, CHCl3); IR (Neat): 3417, 2981, 1706, 1276 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.94 (dd, 1H, J = 15.6, 5.0 Hz), 6.11 (dd, 1H, J = 15.6, 1.5 Hz), 4.31–4.27 (m, 1H), 4.20 (q, 2H, J = 7.1 Hz), 3.98–3.90 (m, 1H), 1.29 (t, 3H, J = 7.1 Hz), 1.14 (d, 3H, J = 6.6 Hz); 13C NMR (CDCl3, 50 MHz): δ 166.5, 145.8, 122.2, 74.5, 69.9, 60.5, 17.2, 14.0; anal. calcd for C8H14O4 (174.19): C, 55.16; H, 8.10. Found: C, 55.12; H, 8.19%.
(E)-3-(4R,5R)-2,2,5-(E)-3-((4R,5S)-2,2,5-Trimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (22)
According to the procedure 10, diol 21 (0.26 g, 1.49 mmol) gave alcohol 22 (0.21 g, 86%) as colourless, viscous liquid; [α]24D + 21.20 (c 0.1, CHCl3); IR (Neat): 3422, 2985, 1376, 1217, 772 cm−1; 1H NMR (CDCl3, 300 MHz): δ 5.92 (td, 1H, J = 15.4, 5.1 Hz), 5.70 (tdd, 1H, J = 15.4, 7,7, 1.3 Hz), 4.53 (t, 1H, J = 7.7 Hz), 4.34 (quin, 1H, J = 6.4 Hz), 4.19 (m, 2H), 1.49 (s, 3H), 1.36 (s, 3H), 1.15 (d, 3H, J = 6.4 Hz); 13C NMR (CDCl3, 100 MHz): δ 133.5, 127.1, 108.0, 79.0, 74.0, 62.8, 28.2, 25.5, 16.1.1-Phenyl-5-(((E)-3-((4R,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)allyl)sulfonyl)-1H-tetrazole (17)
According to the procedure 6, compound 22 (0.1 g, 0.58 mmol) gave sulphone 17 (0.17 g, 81%); [α]24D + 1.12 (c 0.1, CHCl3); IR (Neat): 2984, 1497, 1347, 1153, 763 cm−1; 1H NMR (CDCl3, 300 MHz): δ 7.69–7.56 (m, 5H), 5.95 (dd, 1H, J = 15.1, 6.0 Hz), 5.85–5.75 (m, 1H), 4.52–4.42 (m, 3H), 4.32 (t, 1H, J = 6.7 Hz), 1.44 (s, 3H), 1.33 (s, 3H), 1.02 (d, 3H, 6.7 Hz); 13C NMR (CDCl3, 100 MHz): δ 152.9, 140.3, 131.4, 129.6, 125.1, 116.1, 108.5, 78.0, 73.8, 59.0, 28.0, 25.3, 15.8; MS (ESI): m/z 364 (M + H)+; HRMS (ESI): m/z 387.1093 [M + Na]+ (calculated for C16H20N4O4NaS 387.1097).(2S,3R,4E,6E)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (11R,12S)-4a
According to the procedure 3a, compound 23 (0.05 g, 0.19 mmol) gave (11R,12S)-4a (0.02 g), and cis isomer (11R,12S)-4b (0.01 g) as viscous, color less oils, (total yield 82%); [α]24D − 26.9 (c 0.04, CHCl3); IR (Neat): 3414, 2924, 2853, 1731, 1617, 995 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.42 (d, 1H, J = 4.3 Hz), 7.49–7.40 (m, 2H), 7.06 (dd, 1H, J = 7.5, 4.3 Hz), 6.80 (d, 1H, J = 15.1 Hz), 6.56 (dd, 1H, J = 15.1, 11.1 Hz), 6.02 (dd, 1H, J = 15.4, 6.2 Hz), 4.23 (dd, 1H, J = 6.2, 3.3 Hz), 3.93 (dd, 1H, J = 6.2, 3.3 Hz), 2.36 (s, 3H), 1.16 (d, 3H, J = 6.2 Hz); 13C NMR (CDCl3, 100 MHz): δ 153.2, 146.9, 138.2, 134.3, 133.0, 132.4, 130.8, 128.2, 122.1, 75.9, 70.2, 18.7, 17.6; MS (ESI): m/z 220 (M + H)+; HRMS (ESI): m/z 220.1327 [M + H]+ (calculated for C13H18O2N 220.1332).(2S,3R,4E,6Z)-7-(3-Methylpyridin-2-yl)hepta-4,6-diene-2,3-diol (11R,12S)-4b
Yield: 0.01 g, as viscous, colour less oil; [α]24D − 59.6 (c 0.03, CHCl3); IR (Neat): 3419, 2924, 1628, 1457, 762 cm−1; 1H NMR (CDCl3, 500 MHz): δ 8.43 (d, 1H, J = 4.0 Hz), 7.46 (d, 1H, J = 7.1 Hz), 7.30 (dd, 1H, J = 15.3, 11.2 Hz), 7.07 (dd, 1H, J = 7.1, 4.0 Hz), 6.50 (d, 1H, J = 11.2 Hz), 6.43 (t, 1H, J = 11.2 Hz), 5.97 (dd, 1H, J = 7.1, 15.3 Hz), 4.16 (dd, 1H, J = 6.1, 4.0 Hz), 3.89 (dd, 1H, J = 4.0, 6.1 Hz), 2.30 (s, 3H), 1.15 (d, 3H, J = 6.1 Hz); 13C NMR (CDCl3, 100 MHz): δ 154.6, 146.4, 137.8, 135.3, 132.8, 130.2, 128.5, 126.3, 121.9, 76.0, 70.1, 19.0, 17.8; Mass (ESI-MS): m/z 220 (M + H)+; HRMS (ESI): m/z 220.1327 (M + H)+ (calculated for C13H18O2N 220.1332).Molecular modeling
The calculations of this investigation have been carried out by following the procedure given below. Fuzanin C (3a), fuzanin D (4a) and (11R,12S)-4a were considered for computational prediction of optical rotations. Initially, geometry optimization was performed at B3LYP/6-31G (d) basis set and frequency calculations were done for lowest energy conformer in order to ascertain the minima. Optical rotation was calculated at 589.3 nm using B3LYP/Aug-CC-pVDZ method in gas phase for lowest energy conformer of respective molecules.24 B3LYP/Aug-CC-pVDZ method was opted based on the literature.22 The solvent effect has been omitted for geometry optimizations and optical rotation calculations, since a methodology does not yet exist which predicts the solvent effects with uniform reliability.25 All these calculations were performed using Gaussian 09 software.26Biological activity
Preparation of samples for cytotoxicity
20 mM stock solution for compounds was prepared in DMSO, from the above stock various dilutions were made with sterile PBS to get required concentration.Cytotoxicity screening using MTT assay
MTT assay was performed according to the method of Naidu et al.27 MTT assay is a standard colorimetric assay for measuring cellular proliferation. MTT is a tetrazolium salt, which is yellow in color and is photosensitive. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] is taken by the living cells and reduced by a mitochondrial dehydrogenase enzyme to a purple formazan product that is impermeable to the cell membrane. Solubilisation with solvents like DMSO leads to liberation of product and amount of purple formazan product is directly related to the cell viability. 1 × 104 cells (counted by trypan blue exclusion dye method) in 96-well plates were incubated with series of concentrations of compounds for 48 h at 37 °C in DMEM with 10% FBS medium. Then the above media was replaced with 90 μL of fresh serum free media and 10 μL of MTT reagent (5 mg mL−1) and plates were incubated at 37 °C for 4 h, there after the above media was replaced with 200 μL of DMSO and incubated at 37 °C for 10 min. The absorbance at 570 nm was measured on a spectrophotometer (spectra max, Molecular devices). IC50 values were determined from plot: % cell viability (from control) versus concentration.Acknowledgements
The authors thank the Director CSIR-IICT and Project Director NIPER-Hyderabad for support. VJR thank ORIGIN-0108 for funding. SNK, CH.N.S.S.P.K, E.S thank CSIR-New Delhi, for the fellowships. This is main lab project (MLP) of IICT.References
- (a) L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058 RSC; (b) M. C. Bagley, C. Glover and E. A. Merritt, Synlett, 2007, 2459 CrossRef CAS; (c) T. Laird, Org. Process Res. Dev., 2006, 10, 851 CrossRef CAS; (d) G. D. Henry, Tetrahedron, 2004, 60, 6043 CrossRef CAS; (e) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC.
- (a) Pharmaceutical Chemistry. Drug Synthesis, ed. H. J. Roth and A. Kleemann, Prentice Hall Europe, London, 1988, vol. 1, p. 407 Search PubMed; (b) MDDR: MDL Drug Data Registry, by MDL Informations Systems, Inc; San Leandro, California, USA.
- G. Matolcsy, Pestic. Chem., 1988, 427 Search PubMed.
- (a) C. C. Musonda, V. Yardley, R. C. Carvalho de Souza, K. Ncokazi, T. J. Egan and K. Chibale, Org. Biomol. Chem., 2008, 6, 4446 RSC; (b) E. Srihari, G. S. Kumar, C. H. N. S. S. Pavan kumar, R. K. Seth, S. Biswas, B. Sridhar and V. Jayathirtha Rao, Heterocycl. Commun., 2011, 17, 111 CrossRef CAS.
- (a) N. C. Desai, V. V. Joshi and K. M. Rajpara, Med. Chem. Res., 2013, 22, 3663 CrossRef CAS; (b) P. Narendar, U. Srinivas, M. Ravinder, B. Anand Rao, Ch. Ramesh, K. Harakishore, B. Gangadasu, U. S. N. Murthy and V. Jayathirtha Rao, Bioorg. Med. Chem., 2006, 14, 4600 CrossRef PubMed.
- (a) V. R. Soloman and H. Lee, Curr. Med. Chem., 2011, 18, 1488 CrossRef; (b) C. H. Tseng, Y. L. Chen, K. Y. Chung, C. H. Wang, S. I. Peng, C. M. Cheng and C. C. Tzeng, Org. Biomol. Chem., 2011, 9, 3205 RSC.
- R. Musiol, M. Serda, S. Hensel-Bielowka and J. Polanski, Curr. Med. Chem., 2010, 17, 1960 CrossRef CAS PubMed.
- S. N. Suryawanshi, C. Naveen, P. Kumar, P. Jyoti and G. Suman, Eur. J. Med. Chem., 2008, 43, 2473 CrossRef CAS PubMed.
- S. Mukherjee and M. Pal, Drug Discovery Today, 2013, 18, 389 CrossRef CAS PubMed.
- R. E. Khidre, B. F. Abdel-Wahab and F. B. Abdel-Rehem, Lett. Drug Des. Discovery, 2011, 8, 640 CrossRef CAS.
- W. Aida, T. Ohtsuki, X. Li and M. Ishibashi, Tetrahedron, 2009, 65, 369 CrossRef CAS.
- (a) C. H. Srinivas, C. H. N. S. S. Pavan kumar, B. China Raju, V. Jayathirtha Rao, V. G. M. Naidu, S. Ramakrishna and P. V. Diwan, Bioorg. Med. Chem. Lett., 2009, 19, 5915 CrossRef CAS PubMed; (b) M. Ravinder, P. S. Sadhu, P. Narender, G. Y. S. K. Swamy, K. Ravi Kumar and V. Jayathirtha Rao, Synthesis, 2010, 573 CAS; (c) P. A. Kumar, D. Raman, U. S. N. Murthy and V. Jayathirtha Rao, Bioorg. Chem., 2009, 37, 46 CrossRef PubMed; (d) P. A. Kumar, D. Raman, U. S. N. Murthy and V. Jayathirtha Rao, Synth. Commun., 2010, 40, 686 CrossRef; (e) C. H. Srinivas, C. H. N. S. S. Pavan Kumar, B. China Raju and V. Jayathirtha Rao, Helv. Chim. Acta, 2011, 94, 669 CrossRef CAS.
- (a) D. Xu, G. A. Crispino and K. B. Sharpless, J. Am. Chem. Soc., 1992, 114, 7570 CAS; (b) S. D. Garaas, T. J. Hunter and G. A. O'Doherty, J. Org. Chem., 2002, 67, 2682 CrossRef CAS PubMed.
- A. D. Dunn, Org. Prep. Proced. Int., 1999, 31, 120 CrossRef CAS.
- (a) J. J. Peter and R. C. Liam, J. Org. Chem., 2008, 73, 7616 CrossRef PubMed; (b) D. Takano, T. Nagamitsu, H. Ui, K. Shiomi, Y. Yamaguchi, R. Masuma, I. Kuwajima and S. Ōmura, Org. Lett., 2001, 3, 2289 CrossRef CAS PubMed.
- (a) K. Sugimoto, Y. Kobayashi, A. Hori, T. Kondo, N. Toyooka, H. Nemoto and Y. Matsuya, Tetrahedron, 2011, 67, 7681 CrossRef CAS; (b) K. B. Sharpless, W. Amberg, Y. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa and Z. M. Wang, J. Org. Chem., 1992, 57, 2768 CrossRef CAS.
- (a) O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS; (b) T. Sunazuka, T. Hirose, N. Chikaraishi, Y. Harigaya, M. Hayashi, K. Komiyama, P. A. Sprengeler, A. B. Smith and S. Ōmura, Tetrahedron, 2005, 61, 3789 CrossRef CAS; (c) S. Hanessian, S. P. Sahoo and M. Botta, Tetrahedron Lett., 1987, 28, 1143 CrossRef CAS.
- (a) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS; (b) H. Okamura, S. Kuroda, K. Tomita, S. Ikegami, Y. Sugimoto, S. Sakaguchi, T. Katsuki and M. Yamaguchi, Tetrahedron Lett., 1991, 32, 5137 CrossRef CAS.
- P. Wipf and P. C. Fritch, J. Org. Chem., 1994, 59, 4875 CrossRef CAS.
- M. Ono, C. Saotome and H. Akita, Tetrahedron: Asymmetry, 1996, 7, 2595 CrossRef CAS.
- C. Saotome, M. Ono and H. Akita, Tetrahedron: Asymmetry, 2000, 11, 4137 CrossRef CAS.
- (a) C. O. da Silva, B. Mennucci and T. Vreven, J. Org. Chem., 2004, 69, 8161 CrossRef CAS PubMed; (b) P. J. Stephens, D. M. Mccann, J. R. Cheeseman and M. J. Frisch, Chirality, 2005, 17, S52 CrossRef CAS PubMed; (c) J. R. Cheeseman, M. J. Frisch, F. J. Delvin and P. J. Stephens, J. Phys. Chem. A, 2000, 104, 1039 CrossRef CAS; (d) P. J. Stephens, F. J. Delvin, J. R. Cheeseman and M. J. Frisch, J. Phys. Chem. A, 2001, 105, 5356 CrossRef CAS.
- B. Gangadas, P. Narendar, S. Bharat Kumar, M. Ravindar, B. Anand Rao, Ch. Ramesh, B. China Raju and V. Jayathirtha Rao, Tetrahedron, 2006, 62, 8398 CrossRef.
- (a) P. L. Polavarapu, Mol. Phys., 1997, 91, 551 CrossRef CAS; (b) P. J. Stephens, F. J. Delvin, J. R. Cheeseman, M. J. Frisch, B. Mennucci and J. Tomasi, Tetrahedron: Asymmetry, 2000, 11, 2443 CrossRef CAS; (c) P. L. Polavarpu, Chirality, 2002, 14, 768 CrossRef PubMed; (d) P. L. Polavarapu, Chirality, 2012, 24, 909 CrossRef CAS PubMed.
- (a) B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102 CrossRef CAS; (b) A. Lattanzi, A. Russo, P. Rizzo, G. Monaco and R. Zanasi, Chirality, 2010, 22, E130 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- V. G. M. Naidu, U. M. Bandari, A. K. Giddam, K. R. D. Babu, J. Ding, K. S. Babu, B. Ramesh, R. R. Pragada and P. Gopalakrishnakone, Asian Pac. J. Trop. Med., 2013, 6, 337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra and 13C NMR spectra for all final compounds, molecular modeling studies, Cartesian coordinates of fuzanins C, D, (11R,12S)-fuzanin D. See DOI: 10.1039/c3ra47263a |
| This journal is © The Royal Society of Chemistry 2014 |