Reducing polyaromatic hydrocarbons: the capability and capacity of lithium†
Swati Panigrahi and
G. Narahari Sastry*
Centre for Molecular Modelling, CSIR-Indian Institute of Chemical Technology, Tarnaka, Hyderabad-500607, India. E-mail: gnsastry@gmail.com
First published on 4th February 2014
Abstract
Materials which are extensively and effectively reduced by lithium atoms are of outstanding importance in the lithium battery industry and carbonaceous materials show great promise in this area. Benzene (B), naphthalene (N), anthracene (A) and tetracene (T) are considered as prototypical carbonaceous materials, which mimic sp2 allotropical forms of carbon such as graphene, graphite, carbon nanotubes (CNT) and fullerenes. We have studied lithium adsorption in these carbonaceous materials and analyzed the reduction capability of lithium by different approaches. We observed that the binding energy of these polycyclic aromatic hydrocarbon (PAH)–lithium complexes highly depends on the positioning of the lithium atoms and the size of the PAH. The sequential binding energy data show that the adsorption of the second Li atom is more facile in nature as indicated by its higher binding strengths. The NBO charge analysis also reflects that neutral lithium acquires fractional positive charges which vary from 0.4 a.u. to 2.81 a.u upon interaction with the PAH, which is a clear indication of the ability of the lithium atoms to reduce the PAH molecules. A good correlation has been obtained between the extent of charge transfer, binding and deformation energies. The current study provides the first systematic and exhaustive analysis of the sequential addition of Li atoms to carbonaceous materials and computationally estimates the reduction abilities of this class of materials.
Introduction
Lithium batteries are arguably the most popular power supply gadgets in electronic devices due to their high efficiency, high energy density, high cell voltage and longer cycles.1,2 They have a range of applications starting from portable electronic devices such as watches and laptops to very powerful devices such as implantable electronic devices, air treatment etc. depending on their performance, weight, cost and efficiency.3 Traditionally the cathode of the lithium battery consists of transition metal oxides, such as LiNiO2, LiCoO2 and LiMn2O4, and the anode can be a lithium metal. However these kinds of arrangements have several drawbacks which lead to many safety issues, such as the formation of dendrites on the surface of the lithium electrode, which reduce the cyclic performance of the battery. Various types of carbonaceous materials have been proposed as the anode material for lithium batteries, starting from long range hydrocarbons, graphene nanosheets4 and graphite to disordered hydrocarbons,5 B- and BN-substituted hydrocarbons6–8 and carbon nanotubes (CNTs).9–11Several theoretical works have been carried out to study the non-covalent interaction of the metals and metal ions with ranges of polycyclic aromatic hydrocarbons to long range hydrocarbons such as graphene, CNTs etc.12–15 Lithium intercalation of a very high level has been achieved in polyaromatic hydrocarbons, graphene, graphite, CNTs etc.16–18 Friedlein et al.19 have studied Li intercalation in thick amorphous and polycrystalline forms of perylene and through photoelectron spectroscopy observed that the lithium perylene compound can be used as an energy storage device. The exact mechanism of the charge transfer and the interaction of the lithiated carbonaceous material is a topic of outstanding contemporary interest. Several theoretical and experimental works have been carried out to understand the mechanism.20,21 There are many reports on the interaction of Li and Li+ with polyaromatic hydrocarbons, graphene and carbon nanotubes.12,22–27 Pollak et al.28 through in situ spectroscopy have studied the interaction of Li+ with monolayer and multilayer graphene and observed that the amount of lithium adsorbed through single layer graphene is found to be greatly reduced.
The foregoing discussion illustrates the outstanding importance of these class of materials in gaining the attention of both experimentalists and theoreticians. Ishikawa et al.26 have studied the interaction of lithium dimers with pyrene, anthracene and phenanthrene. From the binding energy values, they observed that lithium dimers interact very strongly with these molecules. Lithium adsorption on graphene has been studied very extensively by Garay-Tapia et al.29 using density functional theory (DFT). They observed that with a higher level of Li concentration, lithium interacts covalently with the carbon substrate where as at a low concentration a strong ionic interaction is observed between the lithium and the graphene layer. The interactions of Li and Li+ with CNTs have been studied using the plane wave function,30 where it has been observed that Li/Li+ localized themselves at the side wall of the CNT, rather than at the centre. The earlier computational studies on lithium adsorption on carbonaceous materials are primarily centered on one or two lithium atoms adsorption.31–33 The mono lithium doped graphene and CNTs can increase the reactivity34 and also have several applications such as a use for the enhancement of hydrogen physisorption.35 A charge transfer between the alkali metals and polycyclic aromatic hydrocarbons is also reported.36
These studies lack information about the effect on the adsorption ability upon the sequential addition of Li, which is important to understand Li storage. The current study is a systematic attempt to understand the sequential lithium adsorption on carbonaceous materials which is expected to provide the utilities of these materials using DFT. We have considered polycyclic aromatic hydrocarbon (PAH) molecules such as benzene (B), naphthalene (N), anthracene (A) and tetracene (T) as the model pristine systems, which mimic the sp2 allotropical form of carbon such as graphite, graphene, CNTs etc. and carried out their interaction with Li atoms by varying the number of lithium atoms from 1 to 5 in a systematic manner. We have scanned all possible lithium adsorption sites to get the most favorable binding site for lithium adsorption. We have also shown how the adsorption of the second lithium atom is more feasible in all of the PAH molecules. Charge analysis also gave very important information about the role of the electrostatic interactions in stabilizing the PAH complexes. This article intends to explore the reduction ability of lithium in various carbonaceous materials.
Computational details
To analyze the adsorption capacity of lithium on different polyaromatic hydrocarbons, a systematic computational study has been performed, by taking the model systems. We have considered tetracene as the standard model, and studied its interaction with Li to procure an understanding, at a reliable level, on the sequential Li adsorption capacity of carbonaceous materials. These results were also verified on smaller models such as the B, N and A lithium complexes to assess the generality of the trends.To study the reduction capability of the lithium atoms, we have sequentially increased the number of lithium atoms from 1 to 5. There are different possibilities for the adsorption of the lithium atoms on the PAH surface: (A) lithium atom/atoms can be adsorbed on the two consecutive rings of the same molecule, and on the same side of the plane of the PAH, (B) the adsorption of the lithium atoms can take place on two non-consecutive rings and on same side of the plane of PAH, (C) two lithium atoms can interact with the same ring of PAH simultaneously, (D) two lithium atoms interact on either side of the plane of the PAH through different hexagonal rings (Scheme 1).
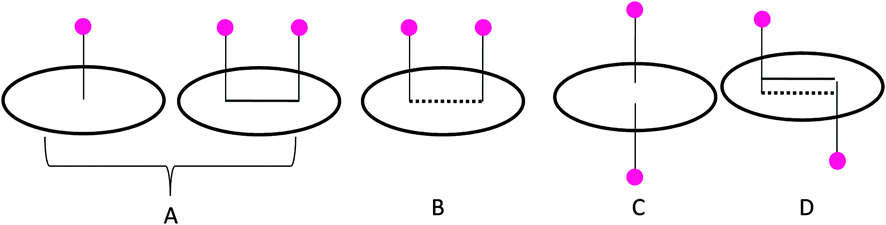 | ||
| Scheme 1 A schematic presentation of the adsorption of lithium atoms on the PAH represented by a circle, where the straight line indicates the adsorption on adjacent rings, and the dotted line indicates the adsorption of lithium in non-adjacent rings. | ||
Considering the size of the system, we undertook a systematic analysis on the performance of the various possible computational methods on the smaller models, B and N, where up to two Li atoms can be accommodated for a single ring. The energies were evaluated at many levels, viz., B3LYP/cc-pVTZ, M06-2X/cc-pVTZ, B3LYP/6-311++G(d), B3LYP/aug-cc-pVDZ, B3PW91/6-31G(d), MP2/cc-pVTZ, ωB97XD/6-31G(d), B2PLYP/6-311++G(d) and B3LYP/6-31G(d), by taking both the B3LYP/6-31G(d) (Table 1) and MP2/6-31G(d) (ESI Table I†) geometries. We have observed that the binding energies calculated by all the possible methods are in good agreement with the binding values obtained at the B3LYP/6-31G(d) level except in some cases, such as the M06-2X/cc-pVTZ method which overestimates the energy due to inclusion of long range dispersion interactions. We observed that the interaction energy calculated by the B3LYP/aug-cc-pVDZ, B3PW91/6-31G(d) and B3LYP/6-311++G(d) levels are in close proximity with that of the B3LYP/6-31G(d) level. Thus, B3LYP/6-31G(d) has been chosen as it is not only economic, but also reliable for calculations on the larger systems.
| B-1A | B-2C | N-1A | N-2A | N-2C | N-2D | N-3C | N-4C | |
|---|---|---|---|---|---|---|---|---|
| B3LYP/cc-pVTZ | −5.75 | −51.61 | −21.02 | −43.17 | −60.80 | −72.02 | −63.71 | −57.25 |
| M062X/cc-pVTZ | −9.78 | −62.64 | −28.81 | −55.95 | −74.62 | −87.54 | −81.16 | −77.33 |
| B3LYP/6-311++G(d) | −5.63 | −50.97 | −20.81 | −42.60 | −60.31 | −71.34 | −62.87 | −55.96 |
| B3LYP/aug-cc-pVDZ | −5.01 | −49.67 | −19.99 | −41.13 | −59.05 | −69.83 | −61.29 | −53.80 |
| B3PW91/6-31G(d) | −5.78 | −50.24 | −19.42 | −44.31 | −60.13 | −72.37 | −65.52 | −57.45 |
| MP2/cc-pVDZ | −4.27 | −58.88 | −17.09 | −52.31 | −69.47 | −79.64 | −66.83 | −80.80 |
| ωB97XD/6-31G(d) | −4.55 | −52.75 | −20.80 | −45.82 | −64.27 | −78.32 | −71.58 | −57.96 |
| B2PLYP/6-311++G(d) | −4.80 | −52.12 | −19.66 | −44.47 | −61.57 | −72.26 | −66.60 | −62.36 |
| B3LYP/6-31G(d) | −5.78 | −49.19 | −18.47 | −41.44 | −57.40 | −69.14 | −62.32 | −54.81 |
Therefore, a full geometry optimization of all possible orientations of the PAH–Li complexes were performed with the B3LYP/6-31G(d) level using the Gaussian 09 suite of programs.37 The full binding energy (B.E.) was calculated using the following equation:
| B.E. = EXY − EX − EY | (1) |
The sequential binding energy was also calculated for the PAH–Li complexes, which gives information about the energy released/required on the sequential adsorption of the lithium atoms. We have considered the complexes with the highest binding energy as our standard model when more than one structures are possible for the similar kind of geometry for a particular PAH complex. ΔE2, ΔE3 and ΔE4 are the differences in the interaction energy of the model systems upon the sequential adsorption of the 2nd, 3rd, 4th and 5th lithium atoms to the PAH molecule. For example ΔE2 has been calculated using the following equation:
| ΔE2 = B.E.(PAH_2Li) − B.E.(PAH_1Li) | (2) |
The adsorption of the lithium atoms can undergo structural and energetic changes in the parent geometry, so the deformation energy (D.E.) has been calculated on the model systems using the following equation:
| D.E. = Eoptx − Eisox | (3) |
We have also calculated the total interaction energy of the lithium complexes (IE2) which is the sum of the deformation energy and the binding energy of the model systems using the following equation:
| IE2 = B.E. + D.E. | (4) |
To study the amount of charge transfer and orbital interaction, natural bond orbital (NBO) analysis has been performed.38–40 The NBO analysis includes all possible interactions between the donor Lewis-type NBOs and the acceptor non-Lewis NBOs. We have calculated the cumulative natural charges on the lithium atoms in the optimized complexes and analyzed them to study the amount of reduction.
Results and discussion
In this section we have started with a discussion on the model system tetracene, as it provides an ideal platform to systematically analyze the sequential Li adsorption capacity up to 5 atoms. After discussing the factors responsible for the variations, the results on the smaller model systems will be analyzed.Tetracene–Li complexes
The B3LYP/6-31G(d) optimized geometry of the tetracene–Li complexes are presented in Fig. 1, along with the Li to ring centroid distances from the nearest hexagonal ring of the tetracene molecule. The Li⋯ring centroid distances are found to be around 2 Å for all of the PAH complexes with an exception for 5C, where the Li⋯π bond length is found to be around 4 Å, which signifies a very weak interaction. In cases where the lithium atoms position themselves symmetrically on the PAH, such as in T-2B′, T-2C′, T-2D′′ and T-4C′′′, we observed that the Li to ring centroid distances are also similar.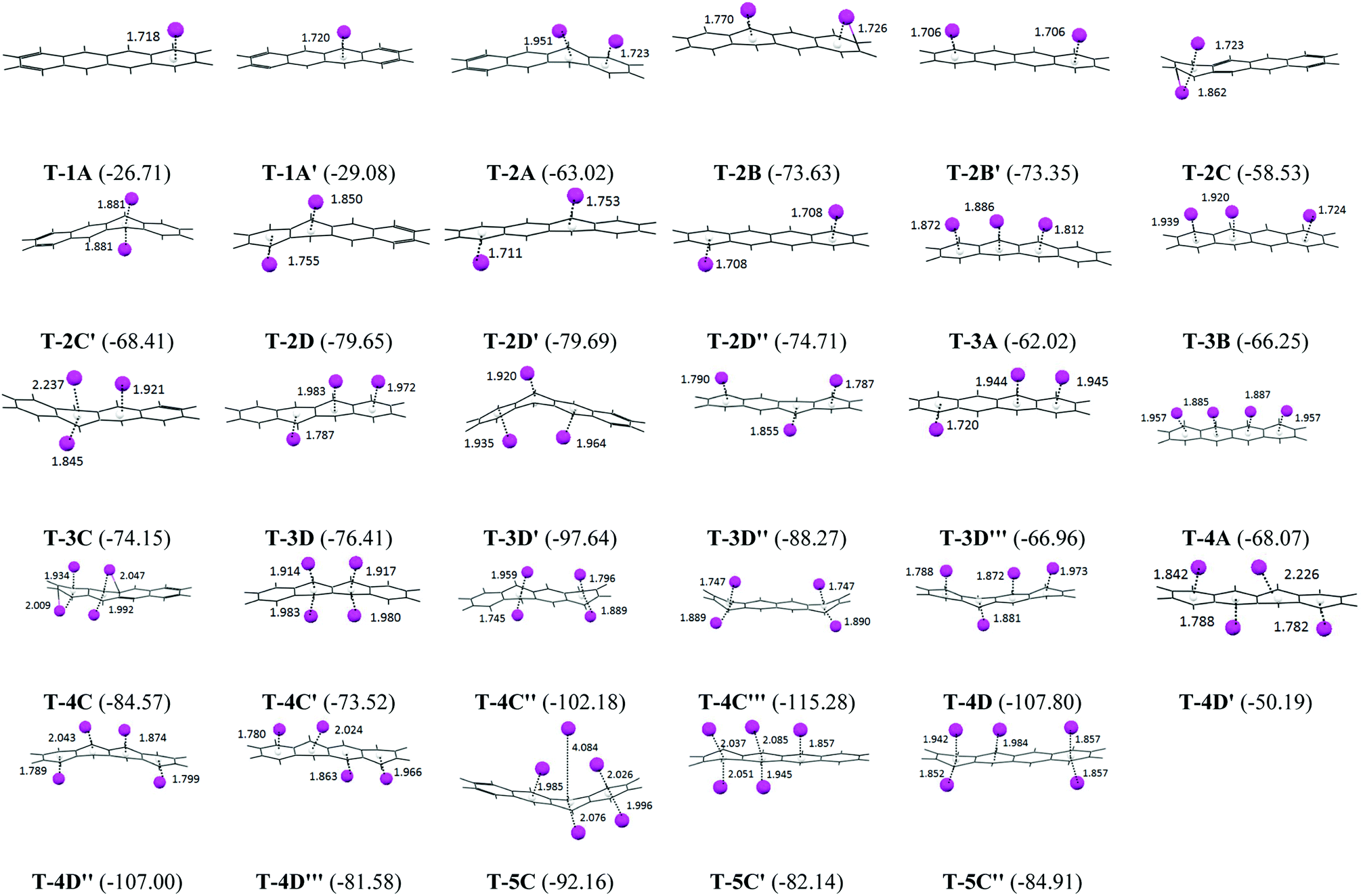 | ||
| Fig. 1 The B3LYP/6-31G(d) optimized geometries of the mono, di, tri and tetra lithium tetracene complexes. The binding energy values are in kcal mol−1 and the Li⋯ring centroid distances are in Å. | ||
There are two possibilities for the mono lithium atom to interact with the tetracene, it can interact through the terminal hexagonal ring (1A) or it can interact with the non-terminal hexagonal rings (1A′) as shown in Fig. 1. From Fig. 2, for the mono-lithium tetracene complex, the maximum binding energy is observed for the 1A′ orientation (B.E. = −29.08 kcal mol−1), when the lithium atom adsorbs at the non-terminal hexagonal ring of the tetracene, while the binding energy of the 1A orientation is calculated as −26.71 kcal mol−1. Therefore, we observed that mono-lithium adsorption at the non-terminal hexagonal ring of the tetracene molecule is more feasible in nature. Among the possible di-lithium complexes, the 2D and 2D′ orientations are found to be more feasible with B.E. values of −79.65 and −79.69 kcal mol−1, respectively. Among the tri-lithium tetracene complexes, the 3D′ orientation has a maximum B.E. (−97.64 kcal mol−1), while among the tetra-lithium complexes, a maximum B.E. is observed for the 4C′′′ orientation (−155.28 kcal mol−1). The B.E. of 4C′′ (−102.18 kcal mol−1), 4D (−107.80 kcal mol−1) and 4D′′ (−107.00 kcal mol−1) are also found to be comparable to that of 4C′′′. From Fig. 2, we can infer that the B.E. increases upon the sequential adsorption of the lithium atoms, attaining its peak for the 4C′′′ orientation. The average binding energies of the mono, di, tri and tetra lithium tetracene complexes are observed to be −27.90, −71.37, −75.96, −92.14 and −86.40 kcal mol−1, respectively. Among all of the tetracene–Li complexes, the maximum binding energy is observed for the 4C′′′ orientation, where the lithium atoms are separated from each other to their maximum extent to minimize the electrostatic repulsion. The Li⋯Li distances are found to be 6.62 Å and 9.42 Å, respectively. The sequential binding energy for the tetracene molecule as given in Table 2 shows that the adsorption of the second Li atom is more feasible in nature (ΔE2 = −50.61 kcal mol−1). The adsorption of the mono lithium atom induces charge redistribution in the PAH molecule, which may be traced to the higher binding energy for the subsequent addition or the addition of the next Li atom.
 | ||
| Fig. 2 The B3LYP/6-31G(d) binding energy (B.E.), deformation energy (D.E.) and cumulative natural charges of the Li atoms (qe) in the T–Li complexes. The B.E. and D.E. are in kcal mol−1, while the natural charges are in a.u. | ||
| System | ΔE2 | ΔE3 | ΔE4 | ΔE5 |
|---|---|---|---|---|
| Tetracene | −50.61 | −17.95 | −17.64 | 23.12 |
| Anthracene | −52.18 | −23.55 | 1.67 | 5.69 |
| Naphthalene | −50.67 | 6.82 | 7.51 | |
| Benzene | −43.41 |
The average of total interaction energy (IE2) of the mono, di tri, tetra and penta lithium complexes were calculated as −24.66, −55.15, −59.27, −60.54 and −63.12 kcal mol−1, respectively (see ESI Fig. I†). Unlike the binding energy strength, we observed that the total interaction energy (IE2) shows a slightly contrasting trend, which is obviously due to the variation of the extent of the deformation energy. The 4D′′ geometry shows the maximum binding energy (−75.19 kcal mol−1) among all of the tetracene–Li complexes. Among the di lithium complexes, the maximum total IE2 is observed for 2D′ (−69.45 kcal mol−1), among the tri lithium complexes, 3D′′ has the maximum IE2 (−67.06 kcal mol−1), among the tetra lithium complexes, 4D′′ (−75.19 kcal mol−1) and 4D (−73.10 kcal mol−1) have the maximum IE2 values. In all of these optimized geometries with maximum IE2 values, we observed that the lithium atoms occupy the positions on both sides of the plane of the PAH molecules, so the adsorption of lithium on both sides of the plane of the PAH is more favorable in nature.
The adsorption of the lithium atoms induces structural and energetic variations in the PAH molecules. We have calculated the deformation energy for the complex systems to understand the structural variations (Fig. 2). The binding energy and deformation energy graphs of the T–Li complexes almost follow a similar trend. Among the mono, di, tri, tetra and penta lithium complexes, maximum deformation energies are observed for 1A′ (3.68 kcal mol−1), 2C′ (27.85 kcal mol−1), 3D′ (35.46 kcal mol−1), 4C′′′ (57.65 kcal mol−1) and 5C′ (24.57 kcal mol−1) for the mono, di, tri, tetra and penta lithium complexes, respectively. The deformation energy is found to be at a maximum in case of the T-4C′′′ orientation (57.63 kcal mol−1), which is associated with the maximum binding energy also. So, the sequential adsorption of lithium atoms induces deformation in the planar PAH molecule and the amount of deformation is found to be highly dependent on the dimension of the PAH and adsorption sites.
The natural charges on the lithium atoms in the optimized geometry for the tetracene lithium complex are presented graphically in Fig. 2. We observed that the natural charges on the lithium atoms vary from 0.4 to 2.81 a.u. for the T–Li complexes, and this signifies the reduction ability of the Li, which results in the charge distribution in the conjugated aromatic PAH molecules. The NBO charges on Li atoms are found to be at a maximum in the 4C′′′ orientation (2.81 a.u.). The same geometry has been observed to have a maximum binding energy and maximum deformation energy. So we observed a very strong correlation between the binding energy, deformation energy and electrostatic distribution. The average and standard deviation of the NBO charges of the di, tri, tetra, and penta lithium complexes are calculated as 1.501 a.u. (0.317), 1.282 a.u. (0.024), 2.204 a.u. (0.334) and 2.079 a.u. (0.482), respectively, where the standard deviation values are given in the parenthesis. It has been observed that for the tetra lithium complexes, the charge distribution spans a wide range. The maximum charge transfer indicates that the two components interact strongly via electrostatic interaction, which indicates that the reduction ability of Li is found to be site specific.
Other PAH–Li complexes
The smaller models of the PAH molecules such as B, N and A were also subjected to a very systematic computational study. We have followed the same protocol which we have employed for the analysis of tetracene. The B3LYP/6-31G(d) optimized geometry of the anthracene, and naphthalene lithium complexes, the binding energy, and the Li to ring centroid distances for all of the orientations are given in the ESI Fig. II† and the most stable orientations (the complexes with the maximum binding energy) for the mono, di, tri, tetra, and penta lithium atoms are given in Fig. 3. It has been observed that the D-orientations are the most favorable orientations, indicating that the adsorption of the Li atoms on both sides of the plane of PAH is highly stabilizing. The graphical presentations of the binding energy, deformation energy and NBO charge analysis are given in Fig. 4. We observed that the binding energy increases upon the sequential adsorption of the lithium atoms. In the case of anthracene, the maximum binding energies are observed for the 3D′ orientation (−99.93 kcal mol−1) and the 4C′′ orientation (−98.26 kcal mol−1), and in the case of naphthalene the maximum binding energy is obtained for the 2D orientation (−69.14 kcal mol−1). So, the adsorption of the lithium atoms through both sides of the plane of the PAH molecule is found to be more favorable. The sequential binding energy value shows that the adsorption of the second lithium atom is found to be feasible in the case of benzene, naphthalene and anthracene (Table 2). The deformation energy follows an almost similar trend as that of the binding energy as shown in Fig. 4. In the case of anthracene, the deformation energy is found to be at a maximum for the the 4C′′ orientation (51.68 kcal mol−1), the charges on the lithium atom are observed to be at a maximum for this orientation (2.46 a.u.) and the same structure is also found to have a maximum binding energy value. However in the case of naphthalene, the maximum charge transfer is observed for the 4C orientation (1.967 a.u.). The distribution of the total interaction energy (IE2) of the anthracene and naphthalene lithium complexes is shown in the ESI Fig. I.† We observed that the 5C orientation shows a maximum IE2 value (−61.88 kcal mol−1) among the A–Li complexes and the IE2 is at a maximum for the 2D orientation (42.24 kcal mol−1) among the N–Li complexes, where the adsorption of lithium atoms takes place on both sides of the plane of the PAH molecule. So we have analyzed the importance of lithium in reducing the PAH molecule.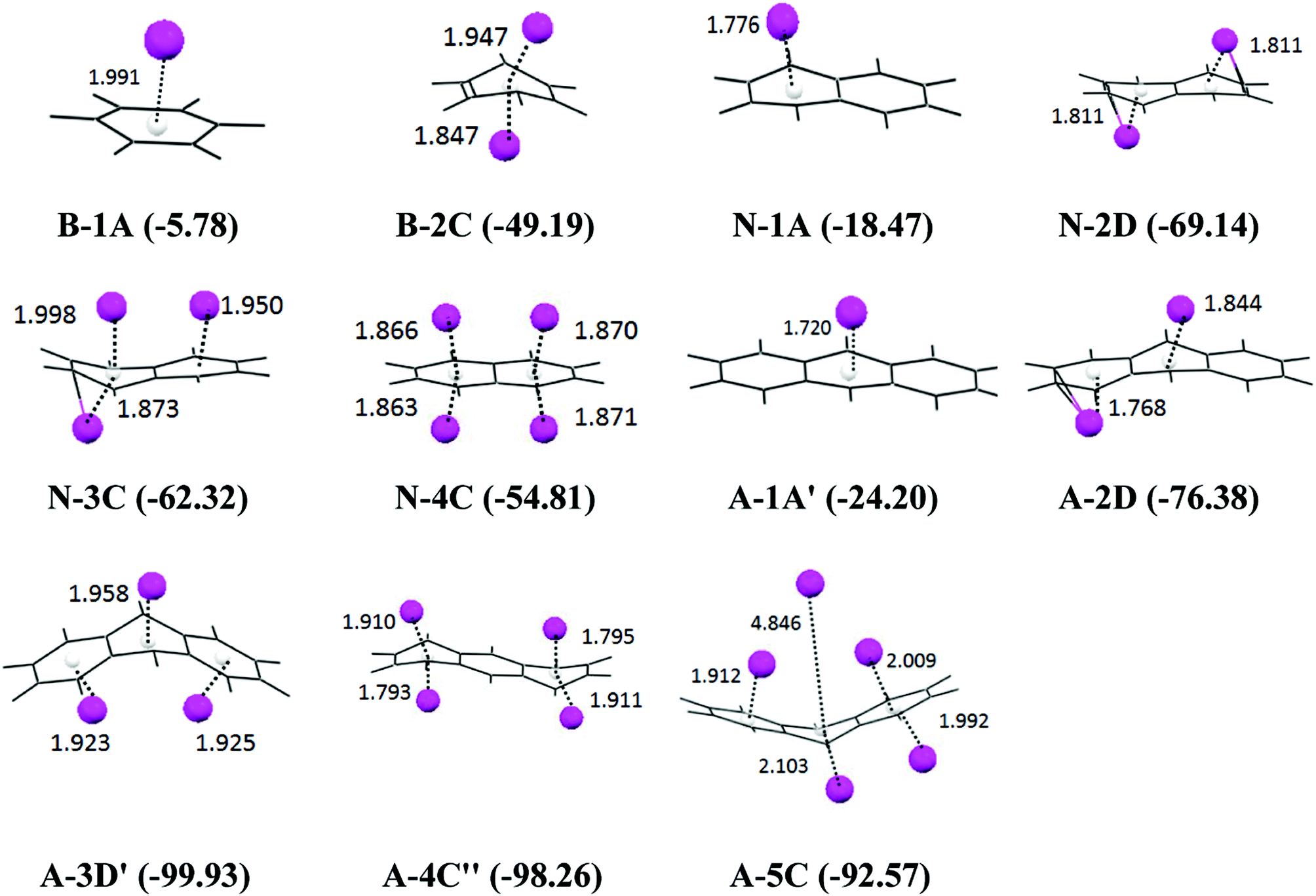 | ||
| Fig. 3 The B3LYP/6-31G(d) optimized geometries of the most favorable orientations of the B, N and A Li complexes upon the sequential adsorption of Li atoms. The B.E. values are in kcal mol−1 and the Li⋯ring centroid distances are in Å. The other possible orientations are provided in the ESI Fig. II.† | ||
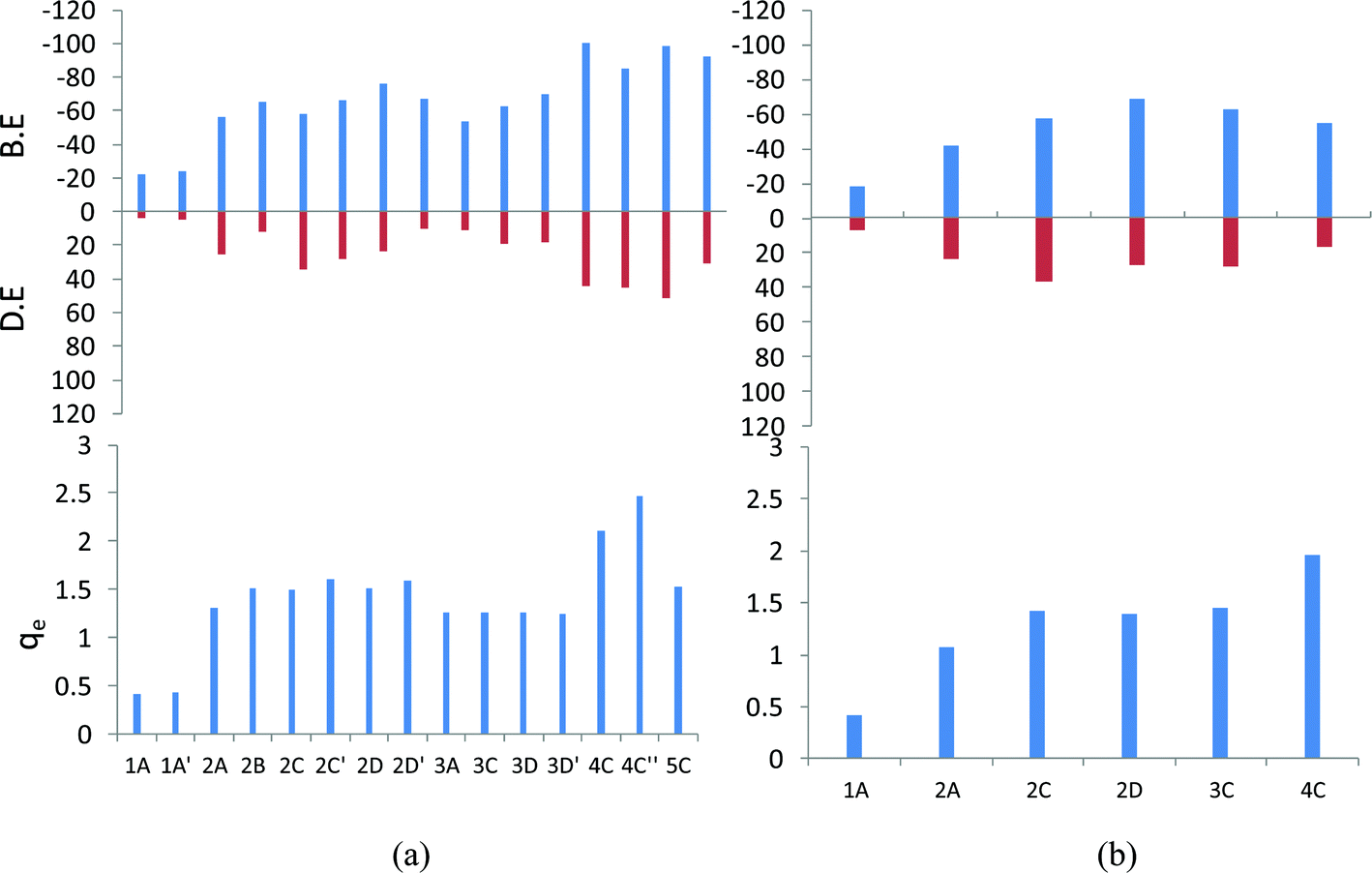 | ||
| Fig. 4 The B3LYP/6-31G(d) binding energy (B.E.), deformation energy (D.E.) and cumulative natural charges on the Li atoms (qe) of (a) the A–Li complexes and (b) the N–Li complexes. The B.E. and D.E. are in kcal mol−1, while the natural charges are in a.u. | ||
Conclusions
A systematic study has been carried out to understand the adsorption of lithium atoms on some of the prototypes of polyaromatic hydrocarbons by density functional theory to explore some of the fundamentals. Lithium adsorption has been carried out in all of these acenes/polyacenes complexes with all possible orientations. We can infer that lithium shows a strong propensity to become adsorbed on the carbonaceous materials, and the sequential adsorption of the second lithium atom is found to be very feasible. We observed that the stabilities of the PAH–Li complexes were largely affected by the positioning of the lithium atoms. The sequential binding energy data also reflect that the lithium atoms found their most stable position when they become adsorbed through either the side of the plane of the molecule. The reduction of carbonaceous materials is often accompanied by structural distortions, which are also site specific. When two lithium atoms bind to the same ring through opposite faces, one often finds higher binding and deformation energies. These observations provide quantitative and qualitative insights on the reduction feasibility of carbonaceous materials by lithium.Acknowledgements
SP thanks CSIR for funding the research in the form of the Nehru Science Post doctoral Fellowship. We thank CSIR-New Delhi for the support in the form of the 12th five year projects, INELCOT and MSM.References
- G. Pistoia, Lithium batteries: new materials, developments, and perspectives, Elsevier Science Ltd, 1994, vol. 5 Search PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- F. Bonino, S. Brutti, P. Reale, B. Scrosati, L. Gherghel, J. Wu and K. Müllen, Adv. Mater., 2005, 17, 743–746 CrossRef CAS.
- M. Endo, C. Kim, K. Nishimura, T. Fujino and K. Miyashita, Carbon, 2000, 38, 183–197 CrossRef CAS.
- M. Velinova, G. Madjarova, A. Ivanova and A. Tadjer, THEOCHEM, 2010, 955, 109–122 CrossRef CAS.
- X. Wang, Z. Zeng, H. Ahn and G. Wang, Appl. Phys. Lett., 2009, 95, 183103 CrossRef.
- M. Khantha, N. A. Cordero, J. Alonso, M. Cawkwell and L. A. Girifalco, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 115430 CrossRef.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 CAS.
- J. Zheng, S. Nai, M.-F. Ng, P. Wu, J. Wei and M. Gupta, J. Phys. Chem. C, 2009, 113, 14015–14019 CAS.
- D. Umadevi and G. N. Sastry, J. Phys. Chem. C, 2011, 115, 9656–9667 CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2012, 113, 2100–2138 CrossRef PubMed.
- D. Umadevi and G. N. Sastry, Chem. Phys. Lett., 2012, 549, 39–43 CrossRef CAS.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4186 CrossRef CAS PubMed.
- F. Valencia, A. H. Romero, F. Ancilotto and P. L. Silvestrelli, J. Phys. Chem. B, 2006, 110, 14832–14841 CrossRef CAS PubMed.
- R. Friedlein, X. Crispin and W. R. Salaneck, J. Power Sources, 2004, 129, 29–33 CrossRef CAS.
- M. Broussely, P. Biensan and B. Simon, Electrochim. Acta, 1999, 45, 3–22 CrossRef CAS.
- R. Friedlein, X. Crispin, M. Pickholz, M. Keil, S. Stafström and W. R. Salaneck, Chem. Phys. Lett., 2002, 354, 389–394 CrossRef CAS.
- G. E. Froudakis, Nano Lett., 2001, 1, 531–533 CrossRef CAS.
- G. E. Froudakis, Rev. Adv. Mater. Sci., 2003, 5, 259–264 CAS.
- G. Mpourmpakis, E. Tylianakis, D. Papanikolaou and G. E. Froudakis, J. Nanosci. Nanotechnol., 2006, 6, 3731–3735 CrossRef CAS PubMed.
- J.-F. Gal, P.-C. Maria, M. Decouzon, O. Mó, M. Yáñez and J. L. M. Abboud, J. Am. Chem. Soc., 2003, 125, 10394–10401 CrossRef CAS PubMed.
- J. S. Rao, H. Zipse and G. N. Sastry, J. Phys. Chem. B, 2009, 113, 7225–7236 CrossRef CAS PubMed.
- D. Vijay and G. N. Sastry, J. Phys. Chem. A, 2006, 110, 10148–10154 CrossRef CAS PubMed.
- S. Ishikawa, G. Madjarova and T. Yamabe, J. Phys. Chem. B, 2001, 105, 11986–11993 CrossRef CAS.
- D. Vijay, H. Sakurai, V. Subramanian and G. N. Sastry, Phys. Chem. Chem. Phys., 2012, 14, 3057–3065 RSC.
- E. Pollak, B. Geng, K.-J. Jeon, I. T. Lucas, T. J. Richardson, F. Wang and R. Kostecki, Nano Lett., 2010, 10, 3386–3388 CrossRef CAS PubMed.
- A. M. Garay-Tapia, A. H. Romero and V. J. Barone, J. Chem. Theory Comput., 2012, 8, 1064–1071 CrossRef CAS.
- A. Udomvech, T. Kerdcharoen and T. Osotchan, Chem. Phys. Lett., 2005, 406, 161–166 CrossRef CAS.
- K. Rana, G. Kucukayan-Dogu, H. S. Sen, C. Boothroyd, O. Gulseren and E. Bengu, J. Phys. Chem. C, 2012, 116, 11364–11369 CAS.
- J. I. Martinez, I. Cabria, M. Lopez and J. Alonso, J. Phys. Chem. C, 2008, 113, 939–941 Search PubMed.
- M. Zhao, Y. Xia and L. Mei, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 165413 CrossRef.
- P. A. Denis, J. Phys. Chem. C, 2011, 115, 13392–13398 CAS.
- I. Cabria, M. J. Lopez and J. A. Alonso, J. Chem. Phys., 2005, 123, 204721 CrossRef CAS PubMed.
- T. A. Baker and M. Head-Gordon, J. Phys. Chem. A, 2010, 114, 10326–10333 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. E. Carpenter and F. J. Weinhold, J. Mol. Struct. (Theochem), 1988, 169, 41–62 CrossRef.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed, L. A. Curtsiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47326k |
| This journal is © The Royal Society of Chemistry 2014 |