 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA quantum chemical study on the mechanism and energetics of the direct esterification, thioesterification and amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide
Zoltán Mucsi,
Nóra Zsuzsa Kiss and
György Keglevich*
Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: gkeglevich@mail.bme.hu; Fax: +36 1 463 3648; Tel: +36 1 463 1111/5883
First published on 25th February 2014
Abstract
The direct esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (1) with 1-butanol is an interesting model, as it fails to occur upon conventional heating, but does, however, take place upon microwave (MW) irradiation. To be able to explain this phenomenon, high level quantum chemical calculations were carried out, considering all relevant intermolecular interactions and applying the explicit and implicit solvent model. The precise energetics (thermodynamic and kinetic data) were in accordance with our experimental observations, and on this basis, we could explain the beneficial effect of MW irradiation. The direct thioesterification and amidation of hydroxy-phospholene oxide 1, occurring only with incomplete conversions under MW irradiation, were also evaluated.
1. Introduction
It is well-known that phosphinic acids do not undergo direct esterification with alcohols upon conventional heating, and for this reason, phosphinates are usually prepared by the reaction of the corresponding phosphinic chloride with an alcohol.1,2 In this case, the hydrochloric acid has to be removed by a base. A further disadvantage is that the phosphinic chlorides cannot be regarded as inexpensive reagents and their reactions are not atom economical. We observed that the direct esterification of phosphinic acids does take place under microwave (MW) conditions.3–6 Moreover, partial thioesterification and amidation of a few phosphinic acids could also be accomplished on reaction with thioalcohols and primary amines, respectively, on MW irradiation.7,8 Of course, we wished to give an explanation of how it is possible that the direct esterification, impossible on conventional heating, takes place on MW irradiation. For this reason, gas phase B3LYP/6-31++G(d,p) calculations were carried out to evaluate the energetic background of the esterifications. The calculations were performed in the gas phase, assuming the interaction of an autoprotonated phosphinic acid molecule with an alcohol unit. The effect of the excess of alcohol applied as a solvent was ignored.5 Despite the above simplifications, the results of the calculations were useful in the interpretation of the experimental experiences. It is worth noting that a 4-membered transition state (TS, A) coming from the attack of the alcohol on the phosphorus atom of the protonated P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function of the phosphinic acid was indicated. In the TS (A), the hydrogen atom of the alcohol is half-way between the oxygen atom of the alkoxy group and that of the hydroxy group.5
O function of the phosphinic acid was indicated. In the TS (A), the hydrogen atom of the alcohol is half-way between the oxygen atom of the alkoxy group and that of the hydroxy group.5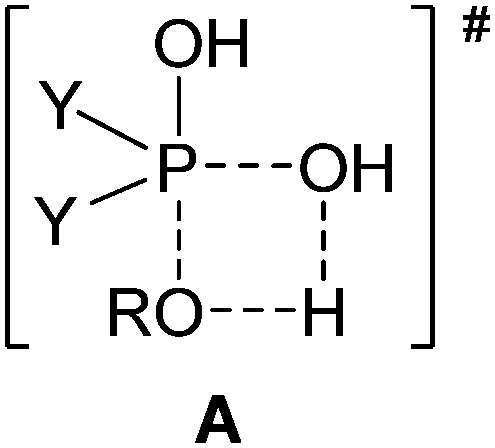
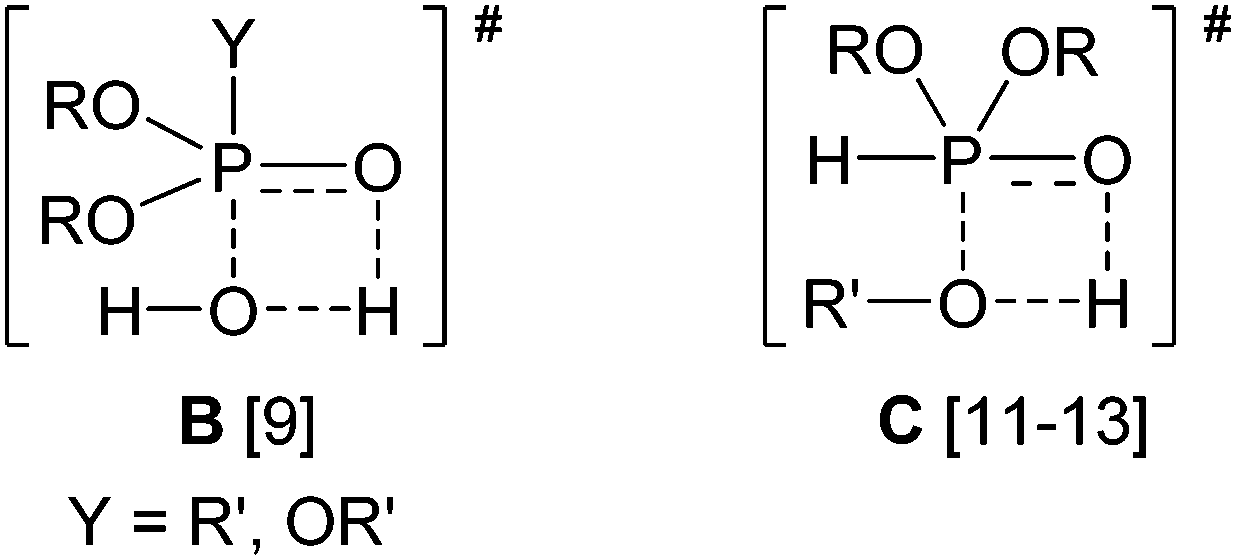
Interestingly, similar TSs were suggested in the hydrolysis of phosphinate and phosphate esters by thermodynamic calculations9 and by a comparative ab initio study,10 as well as in the transesterification of dialkyl phosphites, by other considerations,11,12 and by AM1 semiempirical calculations.13 The TSs of the two non-catalyzed reactions are shown as B and C, respectively.
Returning to the esterification of phosphinic acids, we wished to evaluate the mechanism of the reaction including all elementary steps, taking into account the intermolecular interactions between the reactants and the solvent molecules, and the effect of the solvent. It is a typical situation that what at first sight appears to be a simple mechanism turns out to be quite complex. Each individual elementary step may involve reactants, solvent molecules and catalysts. All these components should be considered in order to investigate the real mechanism and to get a correct view of the reaction in a real medium. The determination of the minimal size of the appropriate chemical model (number of explicit solvent molecules and other species) is rather difficult, and it is time-consuming and requires a serious computational capacity. Reactions taking place in solution usually require the consideration of a catalyst together with quite a few solvent molecules in an appropriate 3D arrangement. The absence of a solvent model or an inappropriate approach may result in not only inaccurate energy values, but also completely wrong data. Taking into consideration all necessary factors, it seems nearly impossible to model even a simple acylation reaction. In the literature,14,15 one of the most frequently used methods to model the surrounding media properly is the implicit–explicit solvent model, where the most important association of species is considered only in the closest layer as an explicit model and the remaining part of the bulk is considered as an average by the relative permittivity (ε) as an implicit model. It is like a “nanodrop” surrounded by media.
In addition to the aims outlined above, we wished to obtain precise energetics for the course of the esterification, thioesterification and amidation of a selected phosphinic acid. For this, high level B3LYP/6-31G(d,p)16 calculations were planned with the application of the explicit–implicit solvent model,14,15 by Gaussian 09.17
2. Results and discussion
2.1 Esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide by 1-butanol
The model reaction selected as the subject for the quantum chemical calculations was the esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (1) with 1-butanol (used also as the solvent) to afford phosphinate (2) (Scheme 1). | ||
| Scheme 1 | ||
This reaction did not take place on conventional heating at 200 °C, but could be performed under microwave (MW) irradiation at the same nominal temperature in a closed vessel to give phosphinate 2 in a yield of 58% after a reaction time of 2 h.4 The use of alcohols with a longer carbon atom chain, which allowed somewhat higher reaction temperatures of 220–235 °C, resulted in yields of around 80–90%.5,6
Three thermodynamic parameters, the enthalpy (ΔH), Gibbs free energy (ΔG) and entropy (ΔS) values were computed at the B3LYP/6-31G(d,p) level of theory.16 According to the enthalpy (ΔH) values, the esterification reaction of cyclic phosphinic acid 1 with 1-butanol is slightly endothermic (Table 1).
| Thermodynamic data | Kinetic data | |||||
|---|---|---|---|---|---|---|
| 1 → 2 | 4 | 5 | 6 | 7 | 8 | |
| ΔH | 0.8 | 134.6 | 131.1 | 133.0 | 133.5 | 51.6 |
| ΔG | 6.14 | 165.20 | 159.01 | 154.99 | 170.35 | 41.67 |
| ΔS | −11.3 | −64.7 | −59.0 | −46.5 | −77.9 | 21.0 |
Several possible routes were considered as the mechanism for the esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (1) with 1-butanol, but for the sake of simplicity, only the most advantageous route is discussed. The simplest possibility for the ester formation is the direct reaction between a phosphinic acid and an alcohol molecule. However, this process cannot be evaluated without the consideration of the participation of solvent molecules and another molecule of the reagent (1), as the simple approach of an alcohol molecule toward the PO function of the hydroxy-phospholene oxide (1) cannot lead to any TS, or stable intermediate. Using numerous modelling efforts, the necessity of the hydrogen bond acceptor solvent network and the participation of another P-acid molecule (1) as the catalyst were proved by our high level calculations. It was found that the sophisticated explicit–implicit solvent model14,15 is suitable to compute the kinetic parameters accurately, where the relative permittivity of the implicit polarizable continuum model (PCM)18 was set to ε = 17.33 corresponding to 1-butanol. The proposed mechanism and the calculated enthalpies are shown in Scheme 2 and Table 1, respectively. In the starting molecular complex (3), a second molecule of phosphinic acid 1 establishes a H bond with the PO unit of the reacting hydroxy-phospholene oxide (1), hence the second P-acid plays an important role as the acid catalyst. A BuOH solvent molecule is also involved in the formation of the starting complex (3). The reacting nucleophilic species (BuOH) is accompanied by another BuOH molecule. One may see, that two BuOH solvent molecules form an important hydrogen bond network for the reaction. The esterification starts with the nucleophilic attack of the oxygen of BuOH on the PO moiety of the starting phospholene oxide (1), resulting in an unstable intermediate state (5) via high energy TS 4 (Table 1). Then, the 5 → 6 multistep transformation connected with proton transfers involving two alcohol molecules takes place via TSs with low energies, for which reason, the corresponding intermediate steps were omitted from our study. In the final step, the phosphinic acid catalyst protonates the leaving OH group, which leaves the P atom via TS 7, giving rise to associate 8, and finally product 2. The values for the enthalpies of the intermediates and TSs are quite comparable during the elemental steps of the mechanism. The overall process is entropically driven by the elimination of water, but the non-beneficial enthalpy is not compensated by the beneficial entropy change.
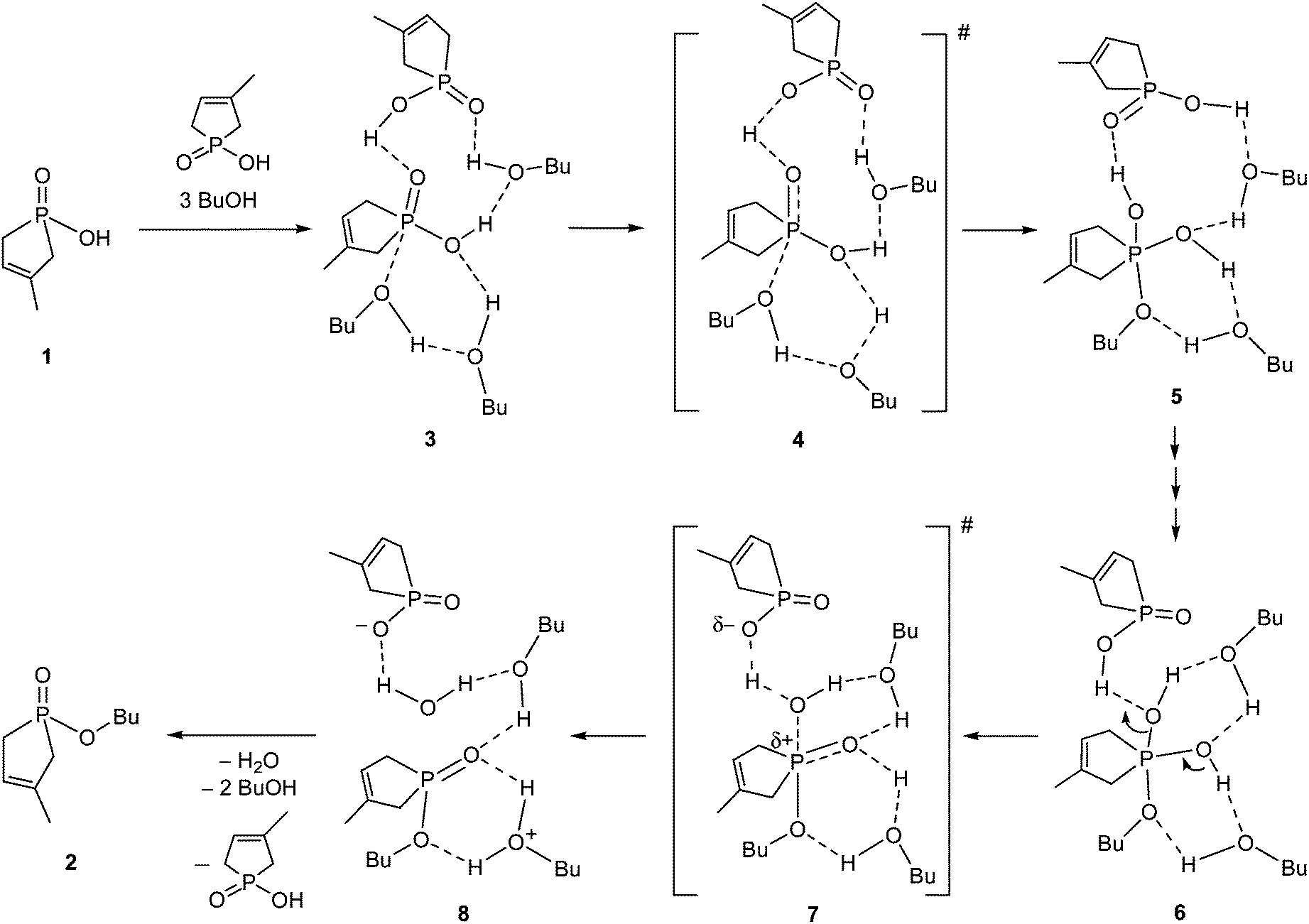 | ||
| Scheme 2 | ||
In the light of the activation enthalpies computed, an elevated temperature is necessary to overcome the high activation enthalpy of ca. 135 kJ mol−1. At a nominal temperature of 200 °C, the statistically occurring local overheating effect may ensure a significantly higher temperature.19 It is known that the extent of local overheating may be in the range of up to 60 °C, which is very efficient.
It is recalled that the earlier gas-phase B3LYP/6-31++G(d,p) calculations suggested a reaction enthalpy (ΔH0), of 3.3 kJ mol−1 for the 1 → 2 transformation, while the enthalpy of activation (ΔH#) for the TS of the rate determining step was 101.7 kJ mol−1.5 Now, the B3LYP/6-31G(d,p) calculations provided a ΔH0 of 0.80 kJ mol−1 and, applying the explicit and implicit solvent model, a ΔH# of 134.6 kJ mol−1. According to the new calculations, the ΔH0 value remained quite similar and predicts a slightly endothermic reaction. However, taking into account the solvent model and other interactions, the ΔH# value was somewhat higher, as compared to that obtained by the calculations ignoring intermolecular interactions. When intermolecular H-bond formations are considered, the alcohol is of decreased nucleophilicity, compared with the instance, where it is “nude”. Moreover, the earlier calculations ignored the enthalpy demand for the protonation of the phosphinic acid. This could explain the somewhat higher ΔH# obtained with the solvent model. Independently of the methods, both kinds of calculations predict an esterification with a relatively high barrier that prevents the esterification on conventional heating.
One can see that the high enthalpy of activation of ca. 135 kJ mol−1 calculated for the esterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide (1) with 1-butanol justifies the experience that this phosphinic acid (1), together with other analogues, cannot undergo reaction with alcohols on conventional heating. It was shown, however, that on MW irradiation the esterification becomes possible. Our explanation is that the statistically occurring local overheating effect19 is beneficial in overcoming the relatively high activation barrier.
2.2 Thioesterification of 1-hydroxy-3-methyl-3-phospholene 1-oxide by thiobutanol
The MW-assisted direct esterification of hydroxy-phospholene oxide 1 with thiobutanol used in a 15-fold quantity was reluctant, and after an irradiation at 210 °C for 6 h, thiophosphinate 9 was obtained in a conversion of only 50% and in a yield of 38%7 (Scheme 3).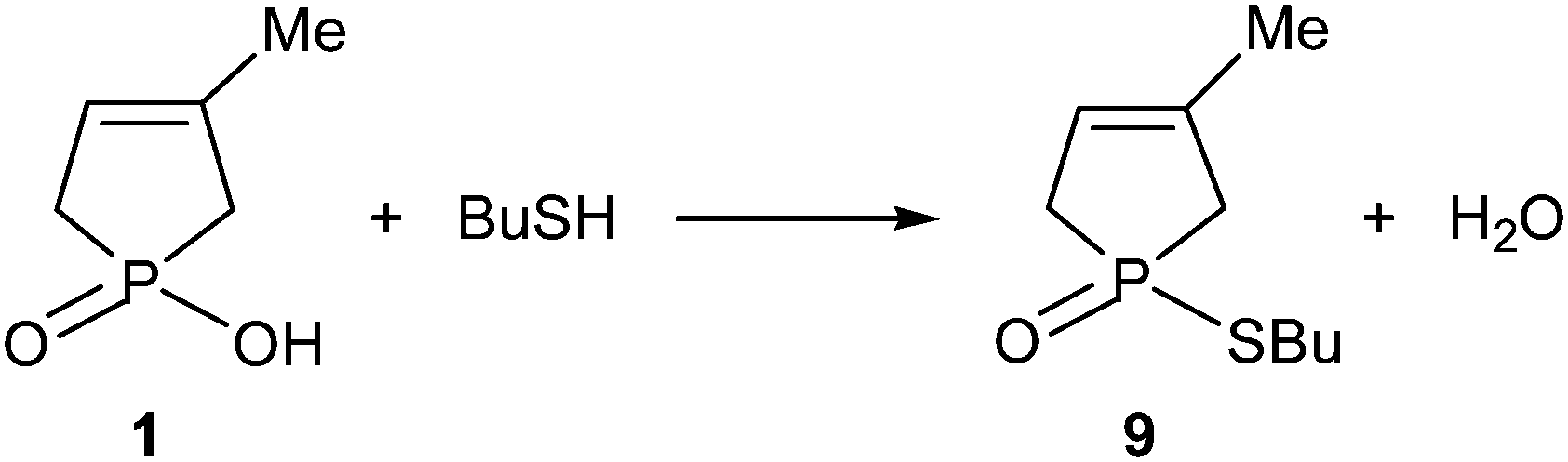 | ||
| Scheme 3 | ||
The energetics and mechanism of this reaction was also investigated using the B3LYP/6-31G(d,p) calculation, and where it was appropriate, the solvent model was also considered. The energetic data are listed in Table 2.
| Thermodynamic data | Kinetic data | |||
|---|---|---|---|---|
| 1 → 9 | 10 | 11 | 12 | |
| ΔH | 47.9 | 216.7 | 230.5 | 222.5 |
| ΔG | 50.64 | 219.16 | 226.48 | 233.85 |
| ΔS | −5.8 | −5.2 | 8.5 | −24.0 |
From thermodynamic point of view, the thioesterification differs significantly from the previous esterification, as the reaction with thiobutanol goes with an enthalpy change of 47.9 kJ mol−1, meaning a significant degree of endothermicity. The mechanism of the thioesterification, substantiated by the calculations, is also different from that of the previous example. Considering the implicit–explicit solvent model, the direct interaction between the sulfur atom of BuSH and the P atom of the PO function in hydroxy-phospholene oxide 1 does not result in any stable intermediate. The failure of this route is the consequence of the lesser extent of basicity of the sulfur atom, which is not able to take part in interactions that are possible with oxygen. In order to find a realistic reaction route, the hydroxy-phospholene oxide (1) protonated by another molecule of starting material (1) was chosen as the starting species (10), and only the implicit solvent model was used. It is noteworthy that although the formation of the species protonated on the PO group (10A) is ca. 104.4 kJ mol−1 more stable than that protonated on the OH unit (10B), it is still the latter form, that is present only in a small quantity, that may be attacked by a thiobutanol molecule to form protonated thioester 12 via a quite high activation barrier (230.5 kJ mol−1) belonging to TS 11. The deprotonation of intermediate 12 leads to thiophosphinate 9 (Scheme 4).
 | ||
| Scheme 4 | ||
The mechanism represented by Scheme 4 seems to be realistic for the thioesterification. The computed high activation enthalpy value and the endothermicity are in accord with the poor preparative experiences under MW irradiation. It is recalled that the earlier B3LYP/6-31++G(d,p) calculations suggested a reaction enthalpy (ΔH0), of 48.5 kJ mol−1 for the 1 → 9 transformation, while the enthalpy of activation (ΔH#) for the TS of the rate determining step was 145.4 kJ mol−1.7 Now, the B3LYP/6-31G(d,p) calculations led to a ΔH0 of 47.9 kJ mol−1 and, applying the implicit solvent model, to a ΔH# of 230.5 kJ mol−1. The thermodynamics is in agreement, however, the ΔH# was probably underestimated by the previous method.7 Beside the simplifications, another reason, i.e. the earlier calculations ignored the energy requirement for the protonation of the phosphinic acid, may cause the deviation.
The B3LYP/6-31G(d,p) calculations using the implicit solvent model may be expected to provide a more realistic ΔH# value, than the simplified calculations.
2.3 Amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide by n-hexylamine
The direct amidation of 1-hydroxy-3-phospholene oxide 1 with n-hexylamine (Scheme 5) was also incomplete under MW conditions used in a 15-fold quantity. Preparative experiments at 220 °C for 2 h provided phosphinic amide 13 in a yield of 26%.8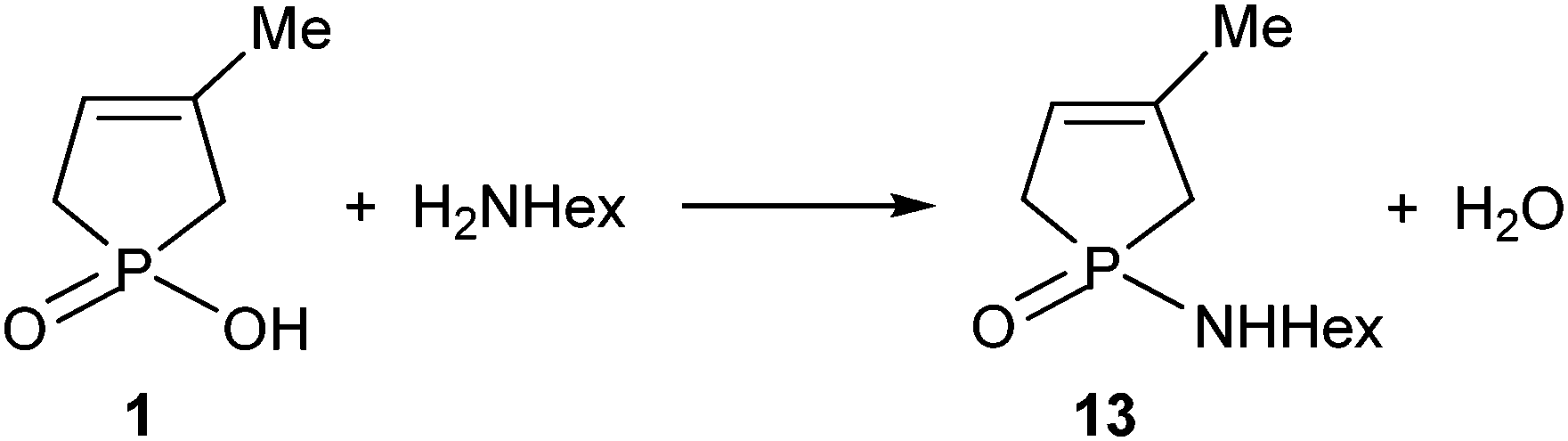 | ||
| Scheme 5 | ||
The energetics of the 1 → 13 conversion were evaluated as above. The thermodynamic and kinetic parameters are listed in Table 3.
| Thermodynamic data | Kinetic data | ||||
|---|---|---|---|---|---|
| 1 → 13 | 14 → 15 | 17 | 18 | 22 | |
| ΔH | 35.2 | −4.3 | 114.1 | 109.8 | 44.6 |
| ΔG | 41.77 | −2.74 | 140.68 | 132.65 | 40.72 |
| ΔS | −13.9 | −3.3 | −56.2 | −48.3 | 8.2 |
The amidation of phosphinic acid 1 also exhibits a rather endothermic enthalpy profile (35.2 kJ mol−1), which predicts an unfavourable course for the amidation. Interestingly, this reaction exhibits the most simple reaction mechanism, where the reaction sequence may start with a protonation equilibrium between phosphinic acid 1 and hexylamine. The equilibrium comprises a neutral (14) and an ionic (15) associate (Scheme 6). In 15, the amine is protonated by the acid. The equilibrium is slightly shifted towards the zwitterionic form (15), (as the enthalpy gain is −4.3 kJ mol−1), which is a crucial situation, as the protonated form of the amine is unable to make a nucleophilic attack. Consequently, in the case of stronger acids, the exclusive presence of the anionic form may prevent the amide formation. The situation is well-known for acids, like sulfuric or sulfonic acid, which cannot be converted to amide derivatives by reaction with amines.
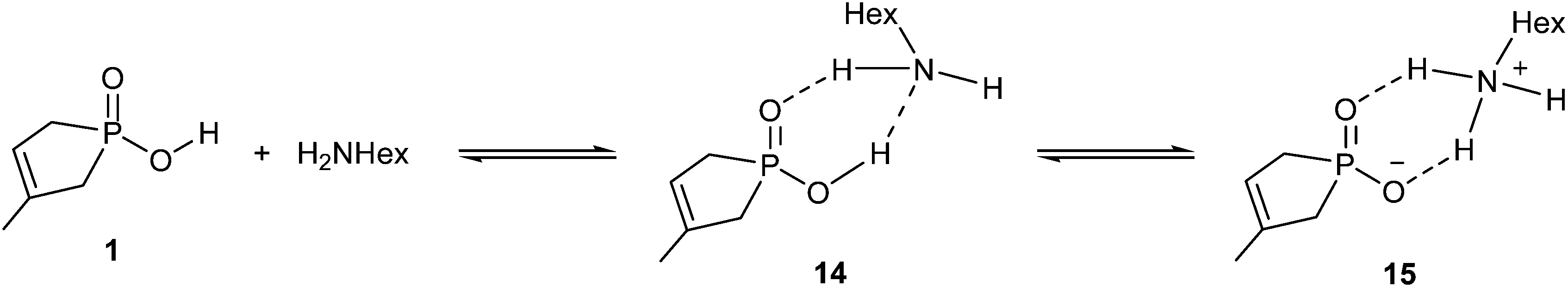 | ||
| Scheme 6 | ||
The rational mechanism for the amide formation is shown in Scheme 7, where hydroxy-phospholene 1-oxide 1 and the HexNH2 react with each other in their neutral forms. A protonated hexylamine molecule (HexNH3+) acts as an acid catalyst, while an additional HexNH2 molecule behaves as base catalyst in the course of the reaction. During the nucleophilic attack of the amine reagent, a protonated amine molecule transfers its proton to the PO group simultaneously, providing the first intermediate (18). Supposing a fast solvent rearrangement around the molecule, where the proton from the positively charged N atom migrates to the neighboring solvating amine, then to another (more distant) solvating amine moiety to furnish intermediates 19 and 20, respectively. The reaction sequence is completed by the elimination of a molecule of water to afford product complex 22 without any further barrier or intermediate, meaning that 19, 20 and 21 are not real stationary points. The first barrier height is corrected by the energy gain of the protolytic equilibrium. The energy content of TS 17 is rather moderate (114.1 kJ mol−1) as compared to that of the TSs belonging to the esterification and thioesterification discussed. The results show undoubtedly that the amidation under discussion is controlled by thermodynamic factors in accord with the rather high endothermicity.
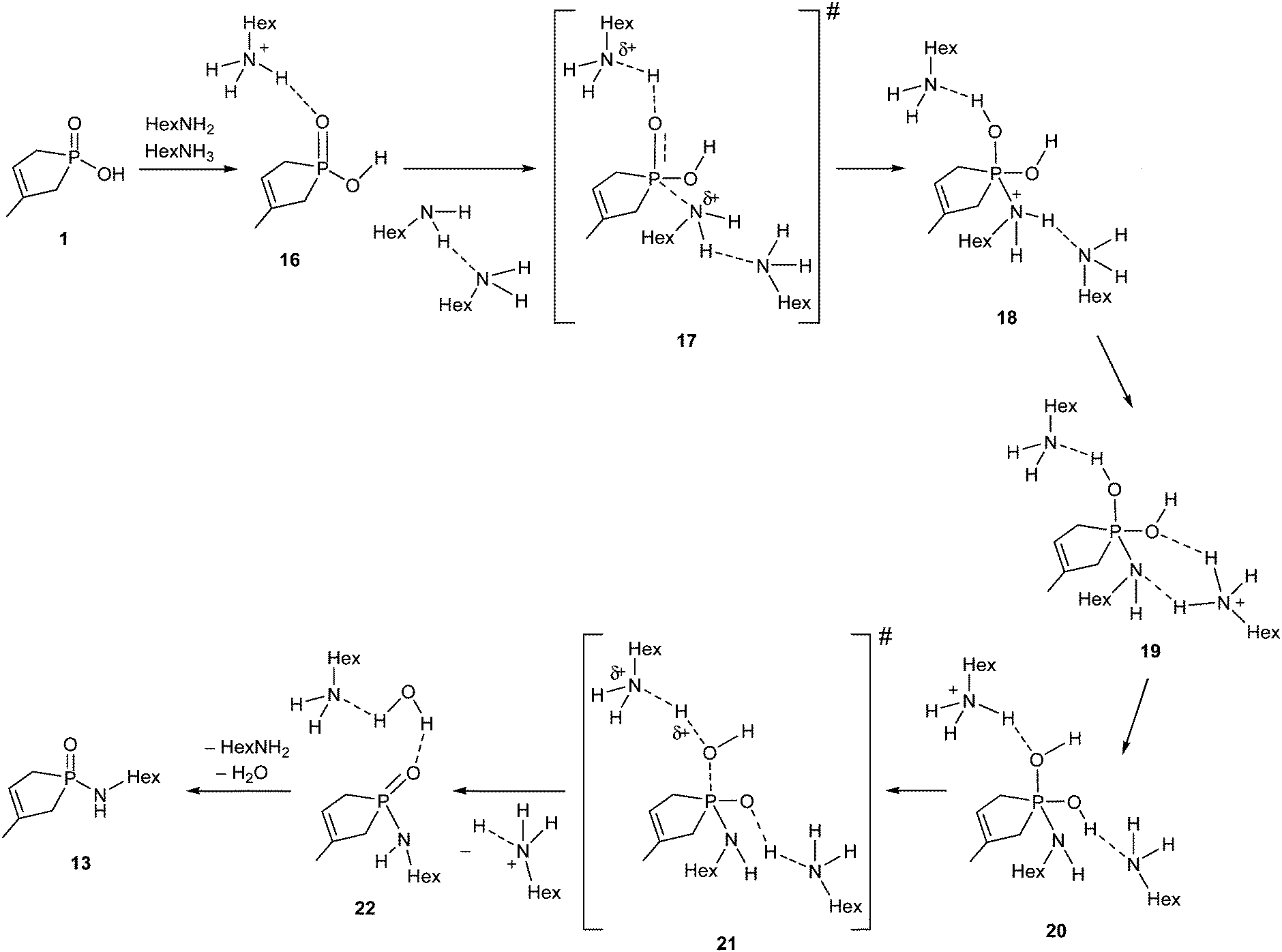 | ||
| Scheme 7 | ||
The earlier B3LYP/6-31++G(d,p) calculations suggested ΔH0 of 32.6 kJ mol−1 for the 1 → 13 transformation, while the ΔH# for the TS of the rate determining step was 79.4 kJ mol−1.7 Now, the B3LYP/6-31G(d,p) calculations led to a ΔH0 of 35.2 kJ mol−1 and, applying the implicit solvent model, to a ΔH# of 114.1 kJ mol−1. The thermodynamics is again in agreement, however, the ΔH# was probably underestimated by the previous method.8 It is needless to say that the results obtained by the application of the solvent model are more reliable, than those of the simplified calculations.
The three reactions studied by us can be explained well by the hard–soft-acid–base (HSAB or Pearson) theory.20 According to this concept, the phosphinic acid can be considered as a hard acid (due to the two hard oxygen atoms) that may be attacked either by hard bases, like alcohols and amines, or by soft bases like the thioalcohol. The computed and experimentally obtained results correlate well with the HSAB theory, namely the reaction of the phosphinic acid with the two hard bases is faster, than that with the soft base.
2.4 Conclusions
In summary, quantum chemical calculations were applied to assess the energetic profile and mechanism for the esterification, thioesterification and amidation reactions of 1-hydroxy-3-methyl-3-phospholene 1-oxide. All the relevant intermediates and TSs were identified and characterized in respect of structure and energy, making use of B3LYP/6-31G(d,p) calculations with the implicit solvent model and, where it was possible, taking into account also the intermolecular interactions and explicit solvent model. Each of the three reactions follows a different route. The computed energetics justified the observation that the reactions investigated do not take place on conventional heating. MW irradiation was, however, useful by enhancing the derivatizations in partial or complete conversions due to the beneficial effect of the statistically occurring local overheating. The results calculated were in agreement with the reactivity of 1-butanol, thiobutanol and hexylamine toward 1-hydroxy-3-methyl-3-phospholene 1-oxide. The summary of the energetics and experimental data are shown in Table 4.| Process | Thermodynamic characteristic | Kinetic characteristic | Experimental conditions/yield |
|---|---|---|---|
| a Using 1-octanol instead of 1-butanol, the yield was 95%. | |||
| Esterification (1 → 2) | Slightly endothermic | Medium TS | 200 °C/2 h/58%a |
| Thioesterification (1 → 9) | Strongly endothermic | High TS | 200 °C/2 h/18% |
| Amidation (1 → 13) | Strongly endothermic | Medium TS | 220 °C/2 h/26% |
3. Experimental
Quantum chemical investigations were carried out with electronic structure computations, using the B3LYP method, employing the 6-31G(d,p) basis16 set for all atoms and using the Gaussian09 (G09) program package.17 The implicit solvent environment was modelled by the standard default IEFPCM method of G09,14,18 using the relative permittivity of 1-butanol (ε = 17.33). All input structures were created using standard methodology. The TS structures are computed by the QST3 method followed by a frequency calculation at B3LYP/6-31G(d,p) level of theory from a preliminary scanning procedure of the heteroatom of the reagent and the P atom of the reactant from the optimized starting point at the HF/3-21G* level of theory. The maximum of the reaction coordinate was optimized at B3LYP/6-31G(d,p) level of theory with fixed O–P bond distance and followed by a frequency calculation at the same level of theory.Acknowledgements
The above project was supported by the Hungarian Scientific Research Fund (OTKA no. K83118) and by the New Széchenyi Development Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-0002). GK thanks Professor Harry R. Hudson (London Metropolitan University) for his advice.References
- L. D. Quin, A guide to organophosphorus chemistry, Wiley, New York, 2000, ch. 5B, p. 138 Search PubMed.
- R. S. Edmundson, in Comprehensive Organic Chemistry, ed. D. Barton, D. Ollis and I. O. Sutherland, Pergamon, Oxford, 1979, vol. 2, ch. 10.5.5, p. 1291 Search PubMed.
- N. Z. Kiss, K. Ludányi, L. Drahos and G. Keglevich, Synth. Commun., 2009, 39, 2392–2404 CrossRef CAS.
- G. Keglevich, E. Bálint, N. Z. Kiss, E. Jablonkai, L. Hegedűs, A. Grün and I. Greiner, Curr. Org. Chem., 2011, 15, 1802–1810 CrossRef CAS.
- G. Keglevich, N. Z. Kiss, Z. Mucsi and T. Körtvélyesi, Org. Biomol. Chem., 2012, 10, 2011–2018 CAS.
- N. Z. Kiss, É. V. Böttger, L. Drahos and G. Keglevich, Heteroat. Chem., 2013, 24, 283–288 CrossRef CAS.
- G. Keglevich, N. Z. Kiss, L. Drahos and T. Körtvélyesi, Tetrahedron Lett., 2013, 54, 466–469 CrossRef CAS PubMed.
- G. Keglevich, N. Z. Kiss and T. Körtvélyesi, Heteroat. Chem., 2013, 24, 91–99 CrossRef CAS.
- P. Guthrie, J. Am. Chem. Soc., 1977, 99, 3991–4001 CrossRef.
- K. D. Troev, Chemistry and Application of H-Phosphonates, Elsevier, Amsterdam, 2006, ch. 3.4, pp. 25–29 Search PubMed.
- W. Vogt and S. Balasubramanian, Macromol. Chem., 1973, 163, 111–134 CrossRef CAS.
- V. Vassileva and G. Lihina, Kinetic investigations of dimethylphosphite interchange reaction by alcohols of guaiacol and veratrine series, Izv. Tomsk. Politech. Inst., 1976, vol. 258, pp. 83–86 Search PubMed.
- K. D. Troev, Chemistry and Application of H-Phosphonates, Elsevier, Amsterdam, 2006, ch. 3.6, pp. 29–32 Search PubMed.
- Z. Mucsi, A. Szabó, I. Hermecz, Á. Kucsman and I. G. Csizmadia, J. Am. Chem. Soc., 2005, 127, 7615–7631 CrossRef CAS PubMed.
- Z. Mucsi, M. Porcs-Makkay, Gy. Simig, I. G. Csizmadia and B. Volk, J. Org. Chem., 2012, 77, 7282–7290 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- K. Kranjc and M. Kočevar, Curr. Org. Chem., 2010, 14, 1050–1074 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |