MgO-embedded fibre-based substrate as an effective sorbent for toxic organophosphates
Dong Jin Woo†
and
S. Kay Obendorf*
Fiber Science, Cornell University, Ithaca, NY 14853, USA. E-mail: sko3@cornell.edu
First published on 21st March 2014
Abstract
Magnesium oxide nanoparticles (MgO) were embedded in a cellulose acetate fibrous framework to provide self-decontaminating properties against toxic organophosphates. The concept of a co-continuous polymer blend structure coupled with selective polymer dissolution was used to develop electrospun fibres with novel morphology for application in chemical protective materials. An electrospinning solution built from 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 acetonitrile–acetone and 15 wt% of 60:40 cellulose acetate–PEO distinct fractions produced fibres that had a degree of continuity of about 0.77 relative to the PEO phase in the cellulose acetate matrix that led to intra-fibre pores with an average diameter of 89 nm, and a surface area of 21.8 m2 g−1. MgO was incorporated into the spinning solution for development of a fibre framework with self-decontamination properties for toxic organophosphates. In 100 min, the MgO-embedded fibres removed 33% of methyl parathion from a hexane solution while fibres with similar morphology and no MgO removed 14%.
:40 acetonitrile–acetone and 15 wt% of 60:40 cellulose acetate–PEO distinct fractions produced fibres that had a degree of continuity of about 0.77 relative to the PEO phase in the cellulose acetate matrix that led to intra-fibre pores with an average diameter of 89 nm, and a surface area of 21.8 m2 g−1. MgO was incorporated into the spinning solution for development of a fibre framework with self-decontamination properties for toxic organophosphates. In 100 min, the MgO-embedded fibres removed 33% of methyl parathion from a hexane solution while fibres with similar morphology and no MgO removed 14%.
Introduction
Self-decontaminating materials designed to deactivate pathogenic microorganisms or degrade chemical warfare agents are being developed. Protective clothing and membranes using catalytic or enzymatic degradation have been studied for use in water and air purification, chemical and biological defense, and personal protection.1,2 In addition to the use of nanofibrous frameworks with varying morphologies, metal oxide nanoparticles have been added to fibres to provide self-decontaminating properties.3,4 Nanocrystalline MgO, CaO, TiO2, and Al2O3 have high adsorption capacities for polar organics such as aldehydes and ketones with the potential to outperform activated carbon.5 TiO2 functions as a photocatalyst to oxidize organic materials particularly with exposure to UV radiation.3,6 Klabunde et al.7,8 reported that MgO functions by destructive adsorption, a mechanism that does not depend upon UV exposure. “Destructive adsorption” is initiated by surface adsorption of the toxic chemical on the MgO crystallites followed by chemical decomposition. Magnesium oxide is believed to carry out oxidative degradation by cleavage of the P–S bond or P–O bond of organophosphates.4,9In engineering self-contaminating protective materials, inclusion of metal oxides in fibres immobilizes the nanoparticles providing some potential advantage over powders or conglomerated materials. As the reaction between a toxic compound and metal oxide particles occurs on the surface of the crystallites, we previously observed a significant mass transfer limitation when nanoparticles were dispersed throughout the fibre structure.3 This means that modification of the morphology of fibre containing metal oxide particles could enhance reactivity.
Fibres containing continuous pores and channels have been used as sorbents, since they have large surface area.10,11 The interconnected microporous structures in fibres have been achieved based upon co-continuous structure of polymer blends, allowing control of pore size, pore size distribution and morphological change in external surface of fibre.12–16 While several existing techniques such as solvent casting, melt blending, emulsion phase separation, and nonwoven fiber bonding have been used to design a co-continuous blend, in this study, fibres with nanoscale morphologies of interconnected grooves and pores were produced from a co-continuous polymer blend formed in a binary solvent during electrospinning. This fibrous substrate was characterized as a sorbent, adding MgO nanoparticles to the fibre spinning solution to provide enhanced adsorption and degradation of organophosphates such as methyl parathion.
Experimental
Materials
Poly(ethylene oxide) (PEO) (Mv: 200000, Mn: 87000) and cellulose acetate (Mn: 30000, acetyl content = 39.8 wt%) were purchased from Sigma-Aldrich (St. Louis, MO). HPLC-grade acetonitrile (99.9%) was purchased from Fisher Chemical (Pittsburgh, PA), acetone (99.5%) from Spectrum Chemicals (New Brunswick, NJ), and HPLC grade water and sodium hydroxide from Mallinckrodt Laboratory Chemicals (Phillipsburg, NJ). Methyl parathion, O,O-dimethyl-O-4 nitrophenyl phosphorothioate, was purchased from Chem Service Incorporated (West Chester, PA). NanoActive Magnesium Oxide Plus® (600 m2 g−1, crystallite size: 4 nm, mean aggregate size: 12 μm) was purchased from NanoScale Corporation (Manhattan, KS).
Solution preparation
Electrospinning solutions were prepared using cellulose acetate and PEO polymers with acetonitrile and acetone as solvents. The desired weight percentage of cellulose acetate solution, (e.g. 15 wt%) was prepared in the desired ratio of acetonitrile and acetone as the solvent (e.g. 8/2 w/w) and sonicated for 1 h with stirring. Then, PEO with the desired weight ratio to cellulose acetate was added under constant stirring and ultrasonicated at 45 °C for 2 h and continuous stirring at 40 °C for 18 h. To study the self-decontaminating properties of metal oxide loaded fibre, a 10 wt% of MgO nanoparticles that had been vacuumed dried at ambient temperature for 24 h was prepared with solvents and sonicated for 20 min prior to addition of polymers. Cellulose acetate and PEO were added following the same procedure used for the solution containing no MgO.Viscosities of spinning solutions were measured using a TA Instruments AR2000 Advanced Rheometer (New Castle, DE) with cone geometry of 20 mm with a 4° angle and Rheology Advantage Instrument Control AR Version V5.7.2 software. The protocol used an initial temperature of 38 °C and equilibration time of 1 min. Temperature was decreased to 22 °C at the rate of 1 °C min−1 using a shear rate of 10.00 L s−1. Visual turbidity was recorded and used to develop a phase diagram in order to identify the transition region from a single phase to two phases (coexistence).
Electrospinning
Using a 5 mL syringe and syringe pump (PHD Ultra Pump; Harvard Apparatus, Holliston, MA), the polymer solution were infused at a speed of 0.04–0.08 mL min−1 through a stainless steel needle (Hamilton, Reno, NV) with an inner diameter of 0.016 inch (0.41 mm) (Hamilton N722). A 16–18 kV voltage was applied between the needle and a circular 20 cm diameter metal plate wrapped with aluminum foil located 15 cm from the needle tip. At room temperature and a 1 h collection time, a nanofibrous framework was deposited on the plate. The fibres were detached from the collection plate and vacuum dried at ambient temperature overnight. Published electrospinning research has reported solvent evaporation within a millisecond.17,18 For comparison to rapid solvent evaporation in electrospinning, a film was cast using the same composition as the spinning solution and dried slowly at ambient condition; this film had a thickness of about 300 μm.Selective dissolution
Co-continuous structures were prepared with the weight ratio of 60:40 cellulose acetate–PEO (80:20 mole) in binary solvents with solvent ratios of 20:80 to 80:20 acetonitrile–acetone. After drying, the fibrous frameworks were immersed in HPLC grade water using a Soxhlet extractor at a temperature above 80 °C for 10 h to selectively remove PEO and then dried for 6 h in a vacuum oven at ambient temperature. Masses before and after the solvent extraction were determined for calculation of the degree of continuity of the PEO phase.
The degree of continuity is the fraction of a phase that is continuous in the morphological structure and is calculated using the following equation:
| φi = (mio − mif)/mio | (1) |
Fibre characterization
The morphology of fibres with 50:50 weight ratio of cellulose acetate–PEO ratio (75:25 mole) was observed with a scanning electron microscope (LEICA 440 SEM, Wetzlar, Germany). Fibres were mounted on aluminum microscopy stubs using carbon tape and coated with gold–palladium (Au–Pd) for 30 s using an Edwards Auto 306 High Vacuum Evaporator (Edwards High Vacuum International, Wilmington, MA). All the electron microscopy images were obtained with an accelerating voltage of 10 keV. SEM images were analyzed to determine fibre diameters using ImageJ (National Institute of Health, USA).
Thermal properties of the electrospun fibres were evaluated using differential scanning calorimeter (DSC 2920; TA Instruments, New Castle, DE). Samples of 4–10 mg were crimped in an aluminum sample pan and scanned from 25 to 300 °C at a rate of 10 °C min−1 under a nitrogen purge (99.99% pure). Thermogravimetric analysis was performed at a heating rate of 10 °C min−1 between 25 and 700 °C in ambient air environment using a TGA 2950; TA Instruments (New Castle, DE).
Brunauer–Emmett–Teller (BET) surface area21 and intra-pore size were determined from nitrogen adsorption isotherm data at 77 K (liquid nitrogen temperature) using Micrometrics analyzer (ASAP 2020, Norcross, GA). Prior to measurement, fibre specimens (50–100 mg) were degassed for at least 12 h under vacuum at ambient temperature. BET surface areas were determined from 9-point adsorption isotherms that were completed using a 0.06–0.2 relative pressure range (p/p0). Pore-size and the distributions were calculated from Barrett–Joyner–Halenda (BJH)22 desorption data in 0.02–0.99 relative pressure range (p/p0).
Methyl parathion removal
Removal of methyl parathion with MgO-embedded fibre followed previously published procedures.4 Concentrations of methyl parathion were measured with a reverse-phase HPLC combined with a diode array UV-vis detector from the Agilent HP series 1200 (Santa Clara, CA). The injection volume was 20 μL. An Agilent XDB-C18 reversed phase column with 5 μm particle size and 4.6 × 150 mm dimension was used at 25 °C. The mobile phase consisted of 1:1 volume ratio of acetonitrile and water with 0.5 vol% formic acid, and the run time was 15 min. The UV-vis detector was set to scan from 280 to 320 nm. Standards of methyl parathion dissolved in hexane were run on the HPLC for use in determining the amount of methyl parathion in test solutions and to normalize for any drift of the HPLC instrument.
Degradation of methyl parathion were determined by exposing a 250 mg fibre specimen to 20 mL of a 62.5 mg L−1 methyl parathion–hexane solution with shaking for one of three reaction times (1, 10 and 100 min) using three replications. After reaction, 1.5 mL of the hexane solution was taken with a syringe and filtered by syringe filter with 25 mm diameter consisting of 0.2 μm pore size (Alltech Assoc. Inc., Deerfield, IL) and analyzed by HPLC. The amount of methyl parathion was calculated from the concentration determined by HPLC and original volume; no correction was made for any methyl parathion retained by the syringe filter.
Results and discussion
Development of co-continuous cellulose acetate–PEO in electrospun fibre
Poly(ethylene oxide) (PEO) and cellulose acetate were selected for development of co-continuous polymer blend due to their low miscibility. With this co-continuous blend, PEO can be selectively dissolved with hot water. Using this polymer blend in electrospinning can result in interesting fibre morphologies.17,23 For electrospinning, a binary solvent system consisting of acetonitrile and acetone was selected based upon a study of three-dimensional Hansen solubility parameters (δD, δP, δH) for these polymers.24 Rapid evaporation of the solvent as the jet surface in electrospinning results in phase separation and preservation of fine phase morphologies.25–27 Based upon these principles, we expect co-continuous structures rather than matrix-domain phase morphologies using the selected system of polymers and solvents. Viscosity of the spinning solution is another critical factor that influences the fibre morphology.28–31Varying the ratio of the two solvents or polymer in the spinning solution resulted in different fibre diameters (Fig. 1). Fibre diameter increased with increasing polymer concentration and increasing acetone content in the spinning solutions. Increase of fibre diameter with increasing polymer concentration is due to consequentially higher viscosity.29–33 The concentration of a polymer solution determines three important factors for electrospinning: viscosity, surface tension, and electric conductivity. Solution surface tension and viscosity play important roles in determining the range of concentrations from which continuous fibres can be obtained. At low concentrations, beads are formed instead of fibres, and at high concentrations, the formation of continuous fibres is prohibited because of the inability to maintain the flow of the solution at the tip of the needle. The polymer concentration with its corresponding viscosity is known to be one of the most effective variables to control fibre morphology. Increasing the relative amount of acetone to acetonitrile from 40 to 80% resulted in an increase in viscosity (Table 1) and fibre diameter (Fig. 1).
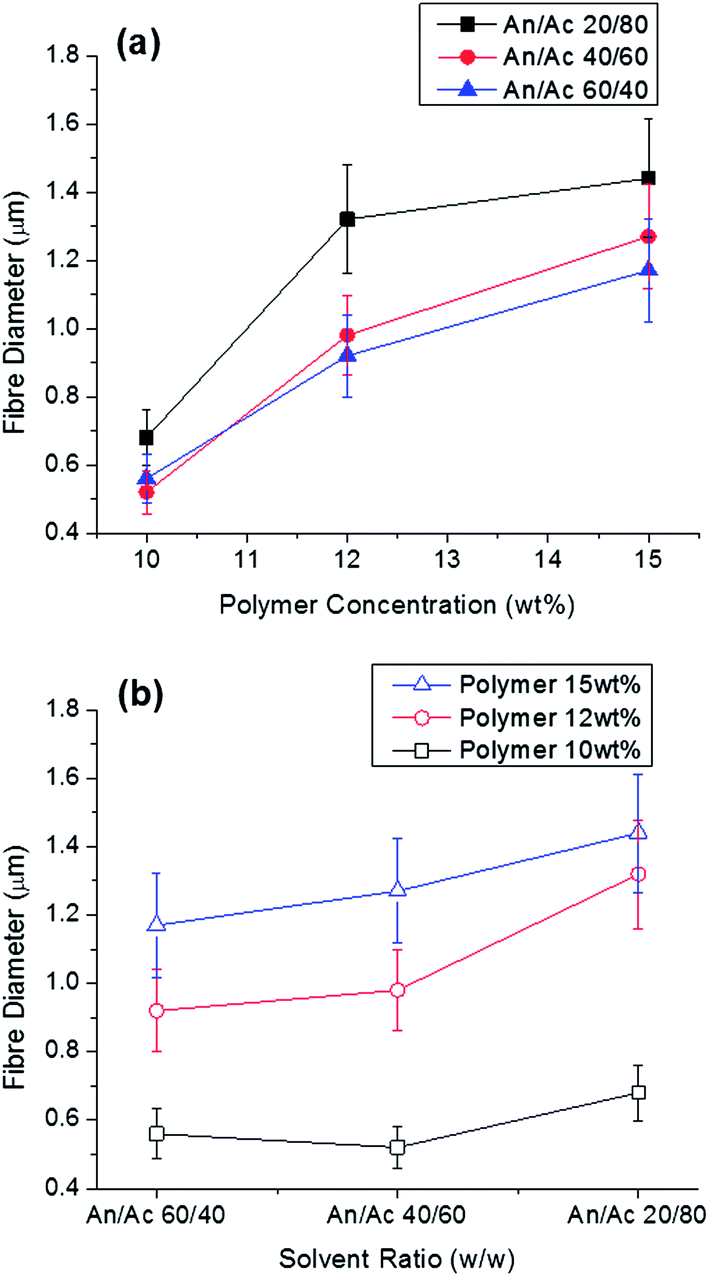 | ||
| Fig. 1 (a) Electrospun cellulose acetate–PEO fibre diameter with polymer content and solvent ratio and (b) electrospun cellulose acetate–PEO fibre diameter with solvent ratio and polymer content (50:50 cellulose acetate–PEO). Acetonitrile is labeled An, while acetone is Ac. | ||
:40 cellulose acetate–PEO (w/w)
| Polymer content (wt%) | Solvent ratio acetonitrile–acetone (w/w) | Viscosity at 22 °C (Pa s) |
|---|---|---|
| 10 | 60:40 |
0.07 |
| 10 | 20:80 |
0.11 |
| 15 | 60:40 |
0.24 |
| 15 | 20:80 |
1.36 |
A phase diagram was constructed with the turbidity data for 60:40 cellulose acetate–PEO using varying ratios of acetonitrile and acetone (Fig. 2). Solutions with high concentrations of acetonitrile resulted in homogeneous single phase solutions while high concentrations of polymer were not soluble. For some solutions near the phase boundary, it was ambiguous whether the specimen was one-phase or two-phase; these were denoted as coexisting. A narrow region with polymer content less than 18 wt% was observed to have a single phase. Solutions close to the phase boundary region which is expected to favor formation of co-continuous structures were selected for electrospinning. Fibres were obtained for 60:40 cellulose acetate–PEO with polymer concentration from 12–18 wt% with weight ratios of acetonitrile–acetone of 30:70, 40:60, 60:40, and 70:30.
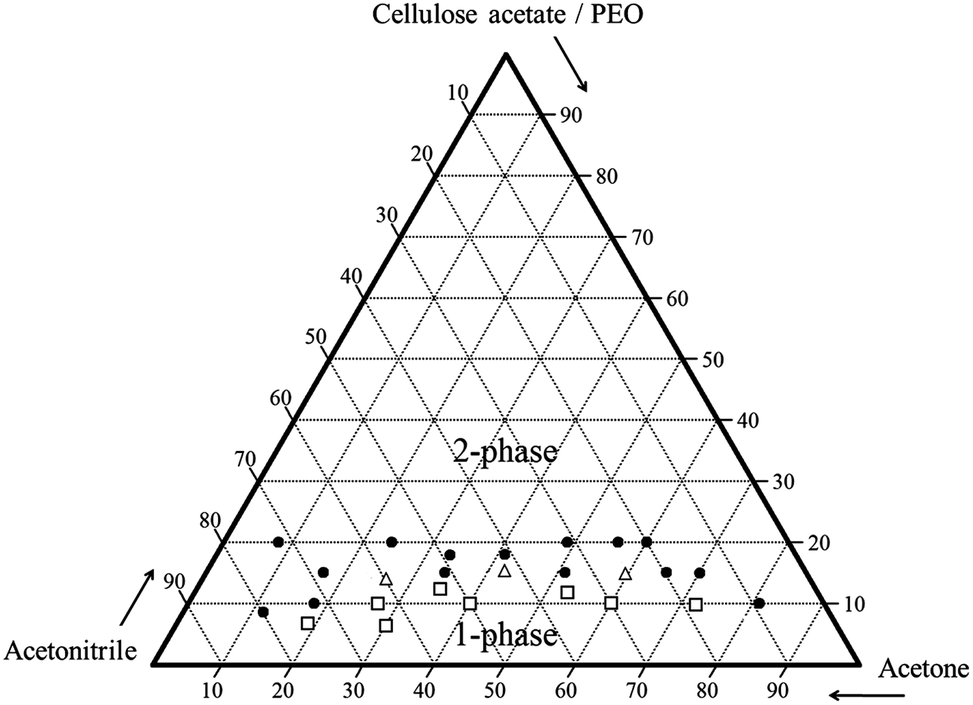 | ||
| Fig. 2 Ternary phase diagram with compositions (wt%) of solutions (solid circle: two-phase solution, hollow square: single-phase solution, hollow triangle: coexistence phase solution). | ||
Selective dissolution of PEO
The PEO component of fibres was selectively dissolved, and the degree of continuity of PEO was calculated (Table 2). Fibres spun from a solution with 60:40 acetonitrile–acetone had the highest degree of continuity of PEO (0.77). Lack of complete dissolution and extraction of PEO may be due a shielding effect by microencapsulation of PEO by cellulose acetate similar to that reported by other researchers.34–36
| Weight ratio of solvents (acetonitrile–acetone) | Fibre mass after water extraction (%) | Degree of continuity of PEO phase |
|---|---|---|
| a Electrospinning solutions of 60:40 cellulose acetate–PEO at 15 wt% and conditions of 0.08 mL min−1, 15–18 kV, 15 cm. |
||
| 30:70 |
95.8 | 0.11 |
| 40:60 |
89.0 | 0.28 |
| 60:40 |
69.1 | 0.77 |
| 70:30 |
75.7 | 0.61 |
| 0:100 |
98.9 | n/a |
Morphology of cellulose acetate fibre after PEO dissolution
Morphologies of fibres after water dissolution of PEO are shown in Fig. 3. Fibres spun from a 60:40 acetonitrile–acetone solvent had a unique morphology of surface grooves and elongated pores. These nano-structures had an average width of 130 nm (min: 90, max: 200, standard deviation: 32) and average depth of 1.3 μm.
 | ||
| Fig. 3 Morphology of 60:40 cellulose acetate–PEO electrospun fibres after water extraction electrospinning solvent ratio: acetonitrile–acetone (a) 30:70; (b) 40:60; (c) 60:40; (d) 70:30 in weight; polymer content: (a–c) 15 wt%, (d) 14 wt%. Scale bar is 1 μm. | ||
Fibres electrospun with 30:70 acetonitrile–acetone exhibited no significant porous structure or surface grooves and had a degree of continuity of only 0.11 (Table 2). This suggests that in the acetone-rich electrospinning solution the 60:40 cellulose–PEO blend was highly miscible resulting in little phase separation of PEO. In addition, the evaporation rate of acetone during electrospinning was rapid allowing little or no phase separation. With 70:30 acetonitrile–acetone, fibres with surface roughness and pores and a degree of continuity of 0.61 were obtained (Fig. 3 and Table 2). These observations agree with those of Megelski et al.11 who reported that electrospinning solutions with relatively low vapor pressure resulted in fibre morphologies with increased surface roughness and pores. Decreased viscosity promotes the development of a finely dispersed structure38 while differences in solubility of the polymer components in the common solvent leads to the formation of interconnected, co-continuous structure.39
Since strongly immiscible behavior in thermal analysis has been shown to relate to matrix-domain dispersed morphology and phase separation of co-continuous blends,26,40 DSC thermal analysis was conducted (Fig. 4). The glass transition temperature (Tg) of cellulose acetate is 198–205 °C, and melting temperature (Tm) is 224–230 °C.17 In the electrospun cellulose acetate fibre, the Tg transition is not obvious while a broad low endothermic peak centered at 224 °C corresponds to Tm. Electrospun PEO fibre showed a strong melting endotherm at 64 °C, and all PEO-containing fibres or film showed this typical melting transition. Different thermal behavior was observed for 60:40 cellulose acetate–PEO fibres electrospun with solvent systems with different ratios of acetonitrile and acetone. Fibres electrospun using 30:70 acetonitrile–acetone exhibited a strong peak at 172 °C between that melting temperatures of cellulose acetate and PEO. In contrast, use of 60:40 acetonitrile–acetone resulted in fibres that exhibited individual peaks at 50 and 220 °C, corresponding to PEO and cellulose acetate, respectively; these conditions produced fibres with the highest degree of continuity of the PEO phase (0.77).
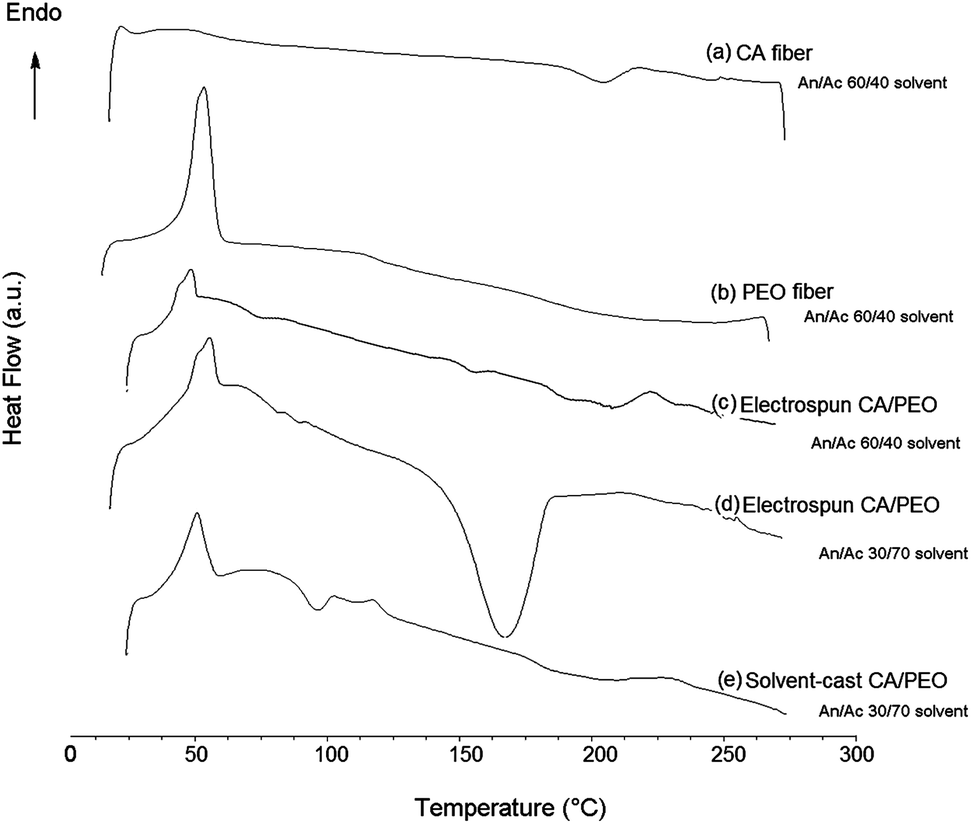 | ||
| Fig. 4 DSC of fibres electrospun from (a) cellulose acetate, (b) PEO, (c) 60:40 cellulose acetate–PEO with 60:40 acetonitrile–acetone, (d) 60:40 cellulose acetate–PEO from 30:70 acetonitrile–acetone, and (e) solvent-cast film from 60:40 cellulose acetate–PEO with 30:70 acetonitrile–acetone. Acetonitrile is labeled An, while acetone is Ac. | ||
Using 60:40 acetonitrile–acetone, the effect of polymer concentration on fibre morphology was investigated (Fig. 5). Fibres spun from 12 wt% of 60:40 cellulose acetate–PEO had tiny knots and blossom-like structures on their surfaces. Using a polymer concentration of 15 or 18 wt%, fibres with larger diameters and morphologies with grooves and pores were obtained.
 | ||
| Fig. 5 Morphology of electrospun 60:40 cellulose acetate–PEO fibres with electrospinning polymer concentrations: (a) 12 wt% and (b) 18 wt% with 60:40 acetonitrile–acetone; before PEO extraction (upper) and after PEO extraction (lower). Scale bar is 1 μm. | ||
After selective dissolution of PEO, the surface area of fibres spun using 12 wt% 60:40 cellulose acetate–PEO was 19.7 m2 g−1 compared to 6.5 m2 g−1 for the 100% cellulose acetate control fibres (Table 3). Furthermore, the fibre spun using 15 wt% polymer had a surface area 21.8 m2 g−1 and average fibre diameter of 1.3 μm. The surface area of this fibre with unique morphology was about three times larger than the control fibre, which if adjusted for fibre diameter would be six to seven times that of the control 100% cellulose acetate fibre with an average fibre diameter of 0.57 μm. The surface area obtained for these fibres with the unique morphology is higher than those reported by other researchers.41,42
:40 acetonitrile–acetone
| Polymer ratio (w/w) | Polymer wt% | Water extracted | BET surface area (m2 g−1) | BJH pore size (nm) | Fibre diameter (μm) |
|---|---|---|---|---|---|
| 100 cellulose acetate | 15 | No | 6.5 | 4.1 | 0.57 ± 0.17 |
| 60:40 cellulose acetate–PEO |
12 | Yes | 19.7 | 23.3 | 0.55 ± 0.20 |
| 60:40 cellulose acetate–PEO |
15 | No | 21.8 | 89.0 | 1.33 ± 0.28 |
Based upon an analysis of nitrogen adsorption–desorption isotherms (BET method) shown in Fig. 6, the control cellulose acetate fibres exhibited Type II behavior according to IUPAC classification.21 This physisorption behavior is consistent with that of nonporous materials adsorbents with very small average pore size and distribution and suggests a homogeneous state for the spinning solution. However, the isotherms for fibres electrospun from cellulose acetate–PEO showed Type IV adsorption with hysteresis loops of a Type H2, tending to saturate at high pressures, which means the fibres are associated with capillary condensation taking place in mesopore (2–50 nm). The desorption hysteresis is narrower for the fibre spun from 12 wt% polymer versus the 15 wt% polymer solution (Fig. 6c compared with 6e). This isotherm behavior indicates that the fluffy morphology fibre made using 12 wt% cellulose acetate–PEO has a relatively simple pore structure and a more uniform size distribution (Fig. 6d). In contrast, longer desorption tail in hysteresis of the isotherm (Fig. 6e) of the fibre with unique morphology made using 15 wt% cellulose acetate–PEO suggests a more complex porous structure. BET/BJH measurements show the smallest intra-fibre pore size of 4.1 nm for the control cellulose acetate fibre. The cellulose acetate–PEO fibre from a spin solution with the same polymer concentration had a bimodal pore size distribution as well as the largest and more complex porous structure with the most frequent pore diameter being 89.0 nm. These observation are consistent with porous structures reported by Han et al.27
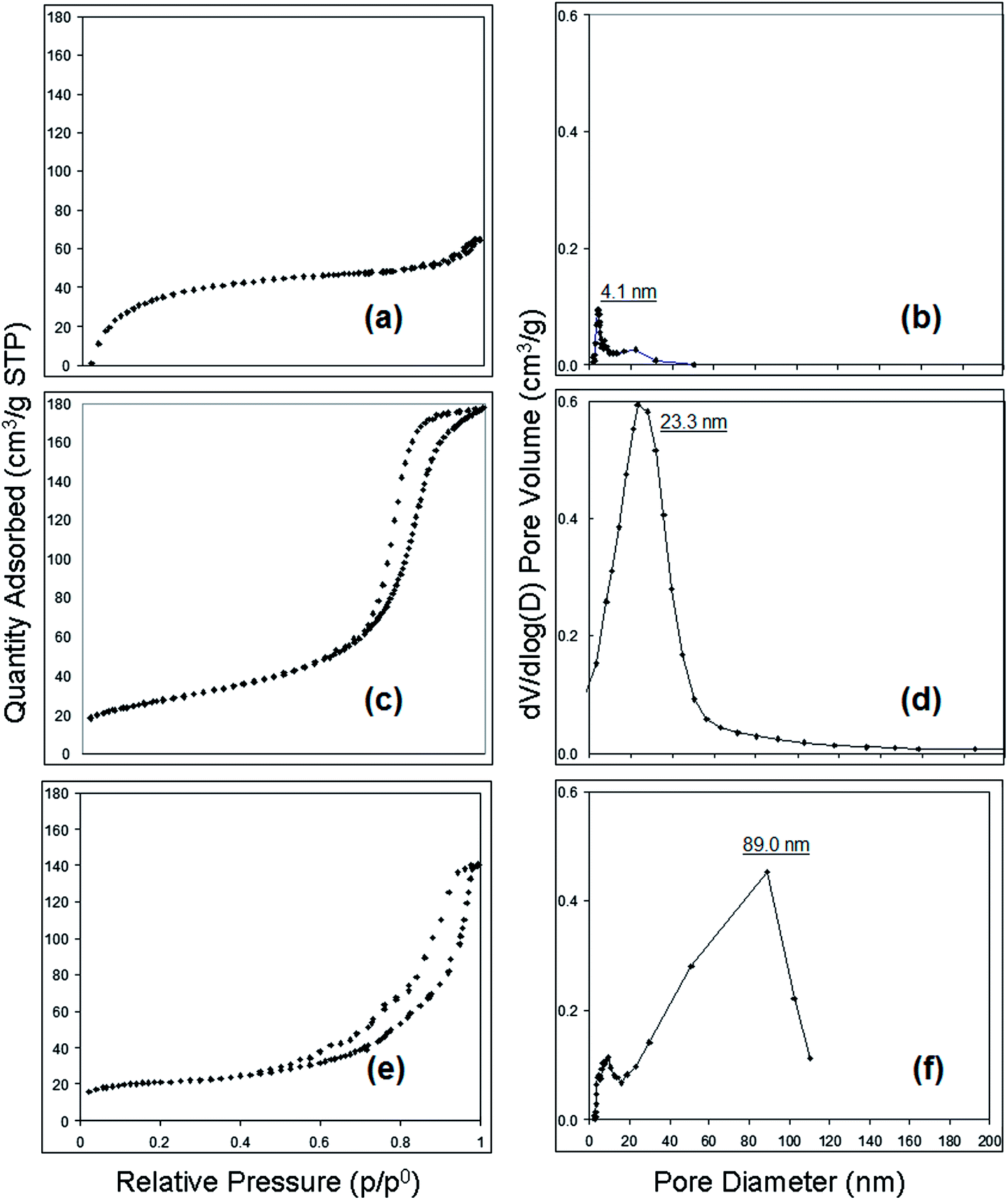 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherm (a, c, and e) and BJH pore size distribution (b, d, and f); (a) and (b) cellulose acetate fibre from 15 wt% polymer; (c) and (d) cellulose acetate–PEO fibre from 12 wt% polymer; (e) and (f) cellulose acetate–PEO fibre from 15 wt% polymer (solvent: 60:40 acetonitrile–acetone). | ||
Methyl parathion removal by MgO-embedded fibre
Fibres were loaded with MgO nanoparticles to provide self-decontaminating properties (Fig. 7). Observation of nanoparticles embedded in the fibres is consistent with published literature.3,43–45 There was approximately 8 wt% MgO in the fibres, and the thermal decomposition temperature at 5% weight loss was higher for fibres containing MgO (Table 4). MgO nanoparticle embedded in fibres are known to degrade organophosphate by a stoichiometric reaction that has been termed “destructive adsorption”, and the degradation is dependent upon the accessibility of the MgO microcrystalline surfaces4,16 (Fig. 8). Therefore, fibre morphology that had a large surface area had increased degradation of methyl parathion.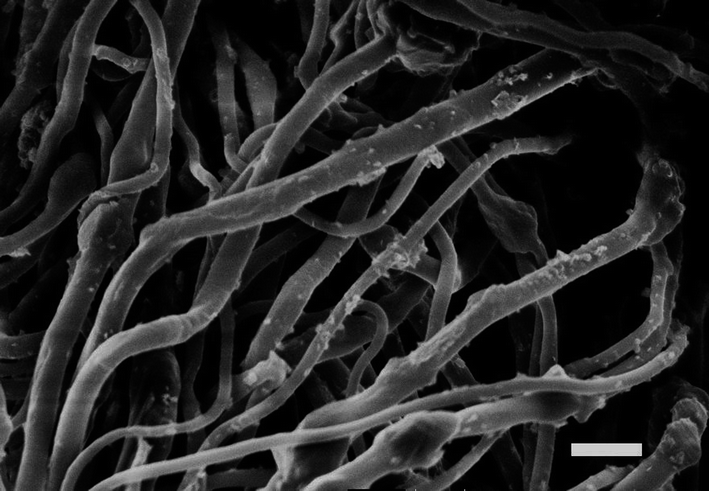 | ||
| Fig. 7 MgO-embedded fibre electrospun from 60:40:06 cellulose acetate–PEO–MgO before water extraction. Scale bar is 3 μm. | ||
:40 acetonitrile–acetone
| Polymer ratio (w/w) | MgO loading | PEO water extracted | Td (°C) at 5% wt loss | Weight residue at 700 °C, (wt%) |
|---|---|---|---|---|
| 100 cellulose acetate | No | No | 289 | 0.9 |
| 100 cellulose acetate | Yes | No | 297 | 11.2 |
| 60:40 cellulose acetate–PEO |
No | Yes | 290 | 0.7 |
| 60:40 cellulose acetate–PEO |
Yes | Yes | 295 | 9.1 |
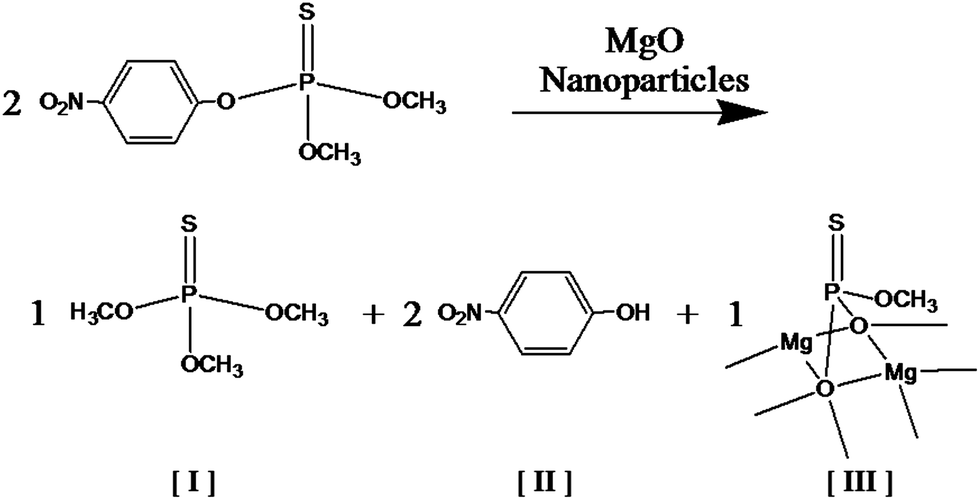 | ||
Fig. 8 Scheme for methyl parathion degradation in the presence of MgO nanoparticles where [I] is O,O,O-trimethyl phosphoric thiourate, [II] is 4-nitrophenol, and [III] is a degraded and adsorbed compound with P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond on MgO microcrystalline surface.4 S bond on MgO microcrystalline surface.4 | ||
Degradation products of O,O,O-trimethyl phosphoric thiourate and 4-nitrophenol were observed in addition to residual methyl parathion after exposure to the MgO-embedded fibrous membranes for 10 and 100 min in a hexane solution. The main degradation product, 4-nitrophenol, has been shown to be transformed into hydroquinone, which is later oxidized to 1,2,4-benzenetriol and then to 5-hydroxymethyl-5H-furfuran-2-one.37 The literature also reports that the organic intermediate species degrade at a slower rate than methyl parathion with mineralization of methyl parathion occurring in 6–8 h. Since our experiment was much shorter (less than 2 h), complete mineralization was not expected.
Methyl parathion is toxic to several target organs, predominately the central nervous system, especially when metabolized to methyl paraoxon which is more toxic than methyl parathion. Neurotoxicity is primarily caused by the inhibition of acetylcholinesterase which results in the accumulation of acetylcholine.45 We did not observe methyl paraoxon when methyl parathion was degraded by MgO-embedded fibres in agreement with our previous research with MgO-embedded fibres.4 The main degradation product of 4-nitrophenol is rated moderately hazardous by WHO while methyl parathion is rated as extremely hazardous. All degradation products that we observed in samples treated for up to 100 min have lower toxicity than methyl parathion. Cleavage of methyl parathion via dearylation to 4-nitrophenol and dimethyl thiophosphoric acid promotes detoxification (Fig. 8).
Amounts of residual methyl parathion for control fibres (S1) and fibres with the unique morphology created by co-continuous structure and selective dissolution (S2) are presented in Fig. 9 and (Table 5). The results confirmed that both MgO-embedded fibres enhanced methyl parathion removal. Overall, MgO-embedded fibre with the unique grooved morphology showed larger decrease of methyl parathion (33.6% removal in 100 min compared to 13.6%) and higher reaction rate than the control MgO-embedded fibres. This difference in performance is believed to be due to the large surface area of the grooved-fibre morphology (21.8 m2 g−1). Furthermore, the pores in the fibre (average intra-fibre pore width: 89 nm) are thought to contribute to the sorption of methyl parathion. Fibres with grooved morphology and no MgO reduced the methyl parathion in the solution after 100 min (7.2%) while no removal was observed for the unloaded control fibre. Results suggest that more than 100 min and/or more than 8 wt% MgO would be required to remove all of the methyl parathion from the solution by destructive adsorption and/or physical adsorption into the fibre pores. Since destructive adsorption by MgO crystallites is stoichiometric, removal of methyl parathion by MgO-embedded fibres will be related to the initiate load of methyl parathion relative to the amount of MgO.
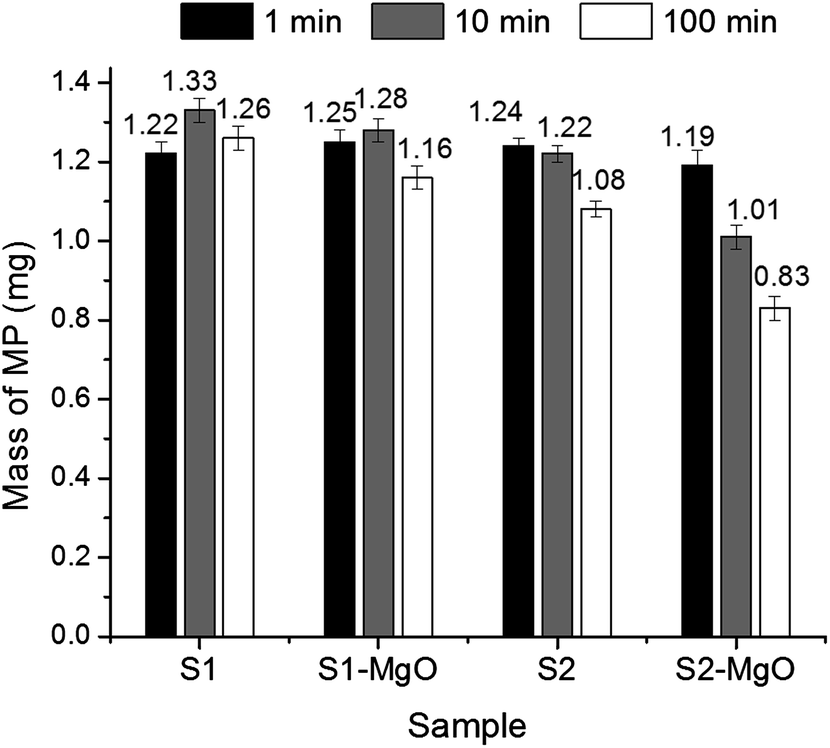 | ||
| Fig. 9 Amount of methyl parathion after reaction for 1, 10, and 100 min with electrospun fibrous membrane with conventional morphology (S1) and grooved morphology (S2). The starting mass of methyl parathion was 1.25 mg. | ||
:40 acetonitrile–acetone with 15 wt% polymer with and without loading with MgO nanoparticles used in analysis of methyl parathion removal
| Fibre morphology | Polymer ratio | MgO loaded | Water extracted | Fibre diameter (μm) |
|---|---|---|---|---|
| Control (S1) | 100 cellulose acetate | No | No | 0.57 ± 0.17 |
| Control (S1) | 100 cellulose acetate | Yes | No | 0.86 ± 0.21 |
| Grooved (S2) | 60:40 cellulose acetate–PEO |
No | Yes | 1.33 ± 0.28 |
| Grooved (S2) | 60:40 cellulose acetate–PEO |
Yes | Yes | 1.35 ± 0.39 |
Conclusions
Pores and grooved morphology were formed in fibre electrospun from cellulose acetate and polyethylene oxide using a binary solvent of acetonitrile and acetone. Polymer–polymer, polymer–solvent, and solvent–solvent interactions resulting from varying the ratios led to different morphologies in electrospun fibres. A 15 wt% polymer solution consisting of 60:40 cellulose acetate–PEO with 60:40 acetonitrile–acetone produced fibres with the highest degree of continuity of the PEO phase (0.77). Interconnected structures, intra-fibre pores (89 nm wide), and large surface area (21.8 m2 g−1) were observed for fibres fabricated based on the concepts of co-continuous structure formation followed by selective dissolution. MgO-embedded in a fibrous framework with unique morphology removed 33% of methyl parathion from a solution by physical adsorption and destructive adsorption while the fibre with similar morphology and no MgO removed 13.6%. The electrospun fibrous framework will have limited mechanical properties due to small, submicron fibre diameters; thus, they are used in a layered system with other conventional textiles providing mechanical strength while the electrospun fibrous framework provides functionality such as self-decontamination, filtration efficiency, and surface sorbency.46
Acknowledgements
This research was funded by grants from the National Textile Center (C05-CR01); the Cornell Agricultural Experiment Station, North Central Regional Research Project NC 170 federal formula funds, Project NYC329407 received from Cooperative State Research, Education, and Extension Service, U.S. Department of Agriculture; College of Human Ecology; and the American Association of Textile Chemists and Colorists. This work was performed in part at the Cornell Center for Materials Research Shared Experimental Facilities, supported through the NSF MRSEC program (DMR-0079992). The authors appreciate helpful discussion with Professor Y. L. Joo in the Department of Chemical Engineering.References
- H. L. Schreuder-Gibson, Q. Truong, J. E. Walker, J. R. Owens, J. D. Wander and W. E. Jones, MRS Bull., 2003, 28, 574–578 CrossRef CAS.
- A. Singh, W. J. Dressick and Y. Lee, Adv. Mater., 2004, 16, 2112–2115 CrossRef CAS.
- D. J. Woo, N. Hansen, Y. L. Joo and S. K. Obendorf, Text. Res. J., 2012, 82, 1920–1927 CrossRef.
- L. E. Lange and S. K. Obendorf, Arch. Environ. Contam. Toxicol., 2012, 62, 185–194 CrossRef CAS PubMed.
- A. Khaleel, P. N. Kapoor and K. J. Klabunde, Nanostruct. Mater., 1999, 11, 459–468 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- O. Koper, Y. Li and K. J. Klabunde, Chem. Mater., 1993, 5, 500–505 CrossRef CAS.
- G. W. Wagner, L. R. Procell, T. J. O'Connor, S. Munavalli, C. L. Carnes, P. N. Kapoor and K. F. Klabende, J. Am. Chem. Soc., 2001, 123, 1636–1644 CrossRef CAS PubMed.
- S. Rajagopalan, O. Koper, S. Decker and K. J. Klabunde, Chem.–Eur. J., 2002, 8, 2602–2607 CrossRef CAS.
- P. Gibson, H. Schreuder-Gibson and D. Rivin, Colloids Surf., A, 2001, 187–188, 469–481 CrossRef CAS.
- S. Megelski, J. S. Stephens, D. B. Chase and J. F. Rabolt, Macromolecules, 2002, 35, 8456–8466 CrossRef CAS.
- H. Pernot, M. Baumert, F. Court and L. Leibler, Nat. Mater., 2002, 1, 54–58 CrossRef CAS PubMed.
- C. Tang, P. Chen and H. Liu, Polym. Eng. Sci., 2008, 48, 1296–1303 CAS.
- V. Kalra, J. H. Lee, J. H. Park, M. Marquez and Y. L. Joo, Small, 2009, 5, 2323–2332 CrossRef CAS PubMed.
- D. Yao, W. Zhang and J. G. Zhou, Biomacromolecules, 2009, 10, 1282–1286 CrossRef CAS PubMed.
- W. Zhang, D. Yao, Q. Zhang, J. G. Zhou and P. I. Lelkes, Biofabrication, 2010, 2, 1–10 CrossRef PubMed.
- H. Q. Liu and Y. L. Hsieh, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2119–2129 CrossRef CAS.
- C. Chen, L. Wang and Y. Huang, AIChE J., 2009, 55, 820–827 CrossRef CAS.
- J. Li, P. L. Ma and B. D. Favis, Macromolecules, 2002, 35, 2005–2016 CrossRef CAS.
- J. Lyngaae-Jørgensen and L. A. Utracki, Polymer, 2003, 44, 1661–1669 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- L. Wannatong, A. Sirivat and P. Supaphol, Polym. Int., 2004, 53, 1851–1859 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, New York, 2007, ISBN: 978-1-4200-4931-2 Search PubMed.
- J. A. Galloway, K. J. Koester, B. J. Paasch and C. W. Macosko, Polymer, 2004, 45, 423–428 CrossRef CAS.
- M. Bognitzki, T. Frese, M. Steinhar, A. Greiner and J. H. Wendorff, Polym. Eng. Sci., 2001, 41, 982–989 CAS.
- S. O. Han, W. K. Son, J. H. Youk, T. S. Lee and W. H. Park, Mater. Lett., 2005, 59, 2998–3001 CrossRef CAS.
- D. H. Reneker and A. L. Yarin, Polymer, 2008, 49, 2387–2425 CrossRef CAS.
- M. Chowdhury and G. K. Stylios, J. Text. Inst., 2012, 103, 124–138 CrossRef CAS.
- C. J. Thompson, G. G. Chase, A. L. Yarin and D. H. Reneker, Polymer, 2007, 48, 6913–6922 CrossRef CAS.
- H. Fong, I. Chun and D. H. Reneker, Polymer, 1999, 40, 4585–4592 CrossRef CAS.
- J. M. Deitzel, J. Kleinmeyer, D. Harris and B. Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- M. M. Demir, I. Yilgor, E. Yilgor and B. Erman, Polymer, 2002, 43, 3303–3309 CrossRef CAS.
- H. T. Oyama, D. J. Hemker and C. J. Frank, Macromolecules, 1989, 22, 1255–1260 CrossRef CAS.
- C. Alkan, A. Sari and O. Uzun, AIChE J., 2006, 52, 3310–3314 CrossRef CAS.
- C. Chen, L. Wang and Y. Huang, Polymer, 2007, 48, 5202–5207 CrossRef CAS.
- E. Moctezuma, E. Leyva, G. Palestino and H. de Lasa, J. Photochem. Photobiol., A, 2007, 186, 71–84 CrossRef CAS.
- J. W. S. Hearle, in Structure Formation in Polymeric Fibers, ed. D. R. Salem, Hanser Publishers, Munich, 2000, ch 14, pp. 521–552 Search PubMed.
- S. Walheim, M. Boltau, J. Mlynek, G. Krausch and U. Steiner, Macromolecules, 1997, 30, 4995–5003 CrossRef CAS.
- Y. You, J. H. Youk, S. W. Lee, B. M. Min, S. J. Lee and W. H. Park, Mater. Lett., 2006, 60, 757–760 CrossRef CAS.
- L. Zhang and Y. L. Hsieh, Nanotechnology, 2006, 17, 4416–4423 CrossRef CAS.
- L. Ji, C. Saquing, S. A. Khan and X. Zhang, Nanotechnology, 2008, 19, 085605 CrossRef PubMed.
- H. Kong and J. Jang, Chem. Commun., 2006, 3010–3012 RSC.
- B. Ding, J. Lin, X. Wang, J. Yu, J. Yang and Y. Cai, Soft Matter, 2011, 7, 8376–8383 RSC.
- S. J. Garcia, A. W. Abu-Qare, W. A. Meeker-O'Connell, A. J. Borton and M. B. Abou-Donia, J. Toxicol. Environ. Health, Part B, 2003, 6, 185–210 CAS.
- T. H. Grafe and K. M. Graham, Nonwovens in Filtration – Fifth International Nonwoven Technical Conference, Stuttgart, Germany, March 2003 Search PubMed.
Footnote |
| † Current address: Physics, Naval Postgraduate School, Monterey, CA 93943, USA. |
| This journal is © The Royal Society of Chemistry 2014 |