Preparation of nanostructured molybdenum carbides for CO hydrogenation
Changcheng Liuab,
Minggui Lina,
Kegong Fanga,
Yan Mengc and
Yuhan Sun*c
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, Shanxi, P.R. China
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P.R. China
cLow Carbon Conversion Center, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201203, P.R. China. E-mail: sunyh@sari.ac.cn
First published on 14th March 2014
Abstract
Novel Mo2C/C nano/microcomposites were prepared via a facile approach involving the hydrothermal carbonization of a solution of glucose as a carbon precursor in the presence of ammonium heptamolybdate tetrahydrate. The samples were subsequently characterized by X-ray diffraction, X-ray photoelectron spectroscopy, thermal gravimetric analysis, N2-physisorption, scanning electron microscopy and high-resolution transmission electron microscopy. The effect of the carbonization agents (Ar, CH4/H2 = 0.2, and H2) was investigated. In particular, the carbonization behaviours and the evolution of Mo species in the catalysts during the carbonization process and the effect on CO hydrogenation for higher alcohol synthesis were extensively studied. During the carbonization step, the catalyst structures experienced an extensive restructuring process, which in turn induced the different performances in the higher alcohol synthesis. Moreover, the nanostructured molybdenum carbides synthesized by this method exhibited great performances in CO hydrogenation for higher alcohol synthesis.
1. Introduction
Molybdenum carbide has been extensively studied due to its unique physical and chemical properties, including mechanical hardness, thermal stability, tolerance of carbon deposition and sulfur, and surface reactivity.1–3 It has also been identified as a promising catalyst that can substitute noble metals in several heterogeneous reactions,4–9 especially CO hydrogenation for obtaining higher alcohols.10,11So far, a variety of methods have been investigated for the synthesis of β-Mo2C nanoparticles, such as a sonochemical method,12 microwave-assisted method,13 organic–inorganic hybrid method,14 hydrothermal method,15 and a carbothermal hydrogenation reduction method.16 Recently, the one-pot saccharide hydrothermal-carburization process has been extensively studied for the preparation of metal–carbon materials .17–21 Due to the reducibility of saccharides, it is possible to achieve the preparation of homogeneously dispersed metal nanoparticles within certain porous solid matrixes or permeable shells, where active nanoparticles are largely isolated and their agglomeration under the reaction conditions can be prevented. The porous materials herein act as a physical barrier but still allow reactants and products to travel freely between the bulk reaction media and catalyst surface.19 Generally, carbon-dispersed nanoparticles are prepared through the direct carbonization of carbohydrates in the presence of a metal species using a hydrothermal process under mild conditions. Furthermore, this is an easy, economic and efficient strategy for the controllable preparation of nanostructured carbides, and can be used for large-scale manufacture and applications.
Herein, we have designed a one-pot hydrothermal cohydrolysis–carbonization process using glucose and ammonium molybdate as the starting materials and successfully fabricated carbonaceous spheres embedded with highly dispersed molybdenum carbide nanoparticles. The as-prepared materials were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), N2-physisorption, thermal gravimetric analysis (TGA), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Based on the data, the effect of the carburization agent upon the catalytic properties was evaluated by CO hydrogenation.
2. Experimental
2.1. Synthesis
Commercially available solvents and reagents were used as received. Highly dispersed molybdenum carbide nanoparticles embedded with carbon were prepared via a facile hydrothermal method followed by carbonization in a certain atmosphere. Typically, 2.88 g MoO3 [obtained from calcining (NH4)6Mo7O24·4H2O (AHM)] was dissolved in 22 mL 30 wt% aqueous H2O2 and 80 mL distilled water, followed by the addition of 19.82 g D-glucose (GL). Afterwards, the solution was transferred to a Teflon-lined stainless steel autoclave (150 mL in volume) and hydrothermally treated at 453 K for 6 h. After cooling, the dark product precipitate was filtrated, rinsed and dried. This as-synthesized sample was then loaded into a tube furnace and heated at 973 K for 4 h at a heating rate of 2 K min−1 in streams of Ar, 20 vol% CH4/H2, and H2 (GHSV = 6000 h−1), respectively. Finally, the samples were quenched to ambient temperature and passivated with 2.0 vol% O2/Ar (GHSV = 1000 h−1) before exposure to air. The carbonized samples are denoted as Mo–Ar, Mo–CH, and Mo–H, corresponding to treatment atmosphere of Ar, 20 vol% CH4/H2, and H2, respectively. The sample before carbonization is denoted Mo–Or.2.2. Characterization
The bulk structure of the samples was measured by XRD (Rigaku D/max-RA, Cu Kα radiation, λ = 1.542 Å). The dimensions and morphology of the samples were determined using SEM (Hitachi High-Technologies Co. Ltd. 2.0 kV) and TEM (JEM-2010 FEF, 200 kV). The specific surface area and pore volume of the samples were detected using N2 adsorption–desorption isotherms (Micromeritics ASAP-2000). Before analysis, the samples were degassed at 393 K for 12 h. The thermal behavior of the samples was monitored using TGA (Setaram, Setsys Evolution, 16/18). During the experiment, the temperature was raised from room temperature to 973 K at a heating rate of 10 K min−1 in Ar, H2 or 20 vol% CH4/H2. Ar was used as the carrier gas with a flow rate nonuple to that of H2 or 20 vol% CH4/H2. The content of carbon in the samples was measured with a Carlo Erba 1106 EL Microanalyzer. Surface compositions of the samples were investigated with XPS (PHI-5300 X, Perkin-Elmer Physical Electronics Co.) using an Al Kα primary radiation. All binding energies were referenced to C1s of 284.6 eV.2.3. CO hydrogenation for higher alcohols synthesis
1.0 mL sample (60–80 mesh) was equivalently diluted with quartz granules and evaluated in a pressurized fixed-bed reactor at 553 K, 7.0 MPa, 4000 h−1 with a H2/CO ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 without any pretreatment. Effluent gases were analyzed using a Carbosieve-packed column with TCD and a Al2O3 column with a flame ionization detector (FID). A Propake-Q column with TCD and FID was applied to the analysis of water, methanol and other alcohol products in the liquids.
:1 without any pretreatment. Effluent gases were analyzed using a Carbosieve-packed column with TCD and a Al2O3 column with a flame ionization detector (FID). A Propake-Q column with TCD and FID was applied to the analysis of water, methanol and other alcohol products in the liquids.
3. Results and discussion
3.1. Morphologies
In this method, highly dispersed molybdenum carbide nanoparticles embedded with carbon have been prepared in a large quantity with inexpensive ammonium molybdate and glucose as precursors. Fig. 1a gives the general morphology and structure of the sample prepared at 453 K for 6 h under hydrothermal conditions. As can be seen, the sample shows a porous structure comprised of small particles. After carbonization at 973 K for 4 h, there is almost no difference in the morphologies of these samples, while more tiny particles appear on the surface, they are probably due to the aggregation of the Mo species and gasification of carbon at high temperature. | ||
| Fig. 1 SEM images of (a) Mo–Or, (b) Mo–Ar, (c) Mo–CH, and (d) Mo–H. | ||
In order to investigate the microstructure features, the samples were further characterized by TEM. Shown in Fig. 2a and b, nanostructured molybdenum oxide embedded with carbon was readily formed after hydrothermal reaction but even prior to the carbonization treatment. The nanostructured molybdenum oxide particles were finely dispersed by the carbon matrix. Additional, it could be clearly seen from Fig. 2a and b that the size of the molybdenum oxide nanocrystals is of high degree of uniformity, of about 4 nm (Table 1). The lattice spacings of 0.34 nm and 0.24 nm could be assigned to the (−111) and (−211) planes of the MoO2 phase, which shall be further confirmed by XRD analysis. After carbonization, there is no significant change in the particle size of the encapsulated molybdenum species nanocrystals (Fig. 2c–h), elucidating the effectiveness of the carbon matrix in the Mo–Or to restrain the crystal growth during the carbonization treatment. The mean particle size of the Mo species synthesized under different carburization conditions are list in Table 1.
 | ||
| Fig. 2 TEM images of (a and b) Mo–Or, (c and d) Mo–Ar, (e and f) Mo–CH, and (g, h) Mo–H. | ||
| Sample | Carbon contenta (wt%) | Surface atom contentb (mol%) | ABET (m2 g−1) | TPV (cm3 g−1) | d (nm) | Dcc (nm) | Did | ||
|---|---|---|---|---|---|---|---|---|---|
| C | Mo | O | |||||||
| a Measured with Carlo Erba 1106 EL Microanalyzer.b Calculated from XPS data (Mo3d, C1s and O1s).c Crystalline size, obtained by Scherrer equation: Dc = 0.89λ/(βcosθ).d Mean particle size, observed from HRTEM.e ABET = BET surface area, TPV = total pore volume, d = average pore diameter. |
|||||||||
| Mo–Or | 42.73 | 68.84 | 2.39 | 28.77 | 38.8 | 0.27 | 25.9 | <4 | 4 |
| Mo–Ar | 39.80 | 80.75 | 4.59 | 14.66 | 305.6 | 0.48 | 6.28 | <4 | 5 |
| Mo–CH | 35.02 | 78.55 | 10.25 | 11.20 | 153.2 | 0.36 | 12.54 | <4 | 6 |
| Mo–H | 5.28 | 48.78 | 19.06 | 32.16 | 20.6 | 0.06 | 9.33 | 8 | 11 |
However, it could be seen that the mean particle size was still affected by the carbonization agents, which is 5 nm for Mo–Ar, 6 nm for Mo–CH, and 11 nm for Mo–H. Moreover, the crystal phase of the dispersed molybdenum oxide changed after carbonization as expected, the lattice spacings of 0.23 nm, 0.17 nm and 0.26 nm could be assigned to (101), (100), and (102) planes of Mo2C (Fig. 2d, f and h), as will be further discussed together with the XRD analysis below (Fig. 3).
 | ||
| Fig. 3 XRD patterns of (a) Mo–Or, (b) Mo–Ar, (c) Mo–CH, and (d) Mo–H. | ||
Besides, adventitious carbon is clearly observed in the figures and is verified by the element content analysis (Table 1), which is the product obtained from hydrothermal carbonization of D-glucose.22 Thus, it could be postulated that the unique combination of nano-sized Mo species particles and adventitious carbon might provide much more active sites than conventional molybdenum carbides. This approach is expected to offer an alternative opportunity for designing carbon supported nano-sized molybdenum carbide for CO hydrogenation due to some potential advantages of the carbon support.23
3.2. Phase and textural structure
The crystallographic structures of the samples was characterized by XRD. From the XRD pattern of Mo–Or (Fig. 3a), the poorly crystalline phase is assigned to MoO2 (2θ = 26.2°, 37.0°, 41.2°, 53.0°, 53.5°, 60.2° and 66.6°, JCPDS no. 32-0671), which agrees with the HRTEM in Fig. 2b. No additional peaks are found, confirming that the composite material corresponds to MoO2 embedded by carbon. In addition, the carbonaceous phase in these samples is amorphous, since no graphitic carbon peaks are observed. After carbonization in an Ar atmosphere, the major diffraction peaks of Mo–Ar are still assigned to MoO2. The peaks assigned to the peaks of the β-Mo2C (JCPDS no. 35-0787) crystalline phase are poor, indicating that not much MoO2 has been carbonized to β-Mo2C in Ar under the conditions of 973 K for 4 h. These results are confirmed by the HRTEM image in Fig. 2d, the largest lattice spacing is 0.34 nm and belongs to MoO2. As for the sample of MoC–H, the crystallinity of the β-Mo2C phase is significantly improved with major diffraction peaks such as 2θ = 34.4°, 38.0°, 39.4°, 52.1°, 61.5°, 69.6°, and 74.6° appearing and being enhanced. In addition, the peak assigned to MoO2 is too poor to be observed, which is verified by Fig. 3c. While for the sample of Mo–CH (Fig. 3d), the poorly crystalline peaks could not be distinguished, with no peaks even being assigned to MoO2, indicating that MoO2 in Mo–Or changed after carbonization in 20 vol% CH4/H2. Nevertheless, in Fig. 2f the lattice spacings of 0.24 nm and 0.23 nm could be assigned to (−211) plane of MoO2 and (101) plane of Mo2C. Therefore, these peaks could be assigned to β-Mo2C, though not all MoO2 was transformed to Mo2C under these conditions. Above all, it could be concluded that different atmospheres led to different carbonizations and crystallinity degrees of the molybdenum carbide. With reference to the work of the Wang4 and Wang24 groups, these results could be explained by the fact that the higher reactivity of H2 benefits the reduction of MoO2 and the formation of β-Mo2C at lower temperatures.The N2 adsorption–desorption isotherms for the samples are displayed in Fig. 4. The isotherms of all samples are of type IV with the hysteresis loops at relative pressure in the range of 0.4–1.0, confirming the mesoporous network. The corresponding pore size distribution calculated using the BJH method25 are shown in Fig. 5. These curves displayed a different pore distribution center for each sample. For Mo–Or, two peaks centered at 15.1 nm and 32.2 nm appear. After carbonization, the overall pore distribution center trends smaller. For Mo–CH, the pore distribution is centered at 11.3 nm, while for Mo–Ar, two peaks centered at 3.1 nm and 15.4 nm appear, and for the sample of Mo–H, the peak centers at about 6.9 nm. This probably results in the different amounts of carbon left in the samples which provides the porosity.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms (at 77 K) of (a) Mo–Or, (b) Mo–Ar, (c) Mo–CH, and (d) Mo–H. | ||
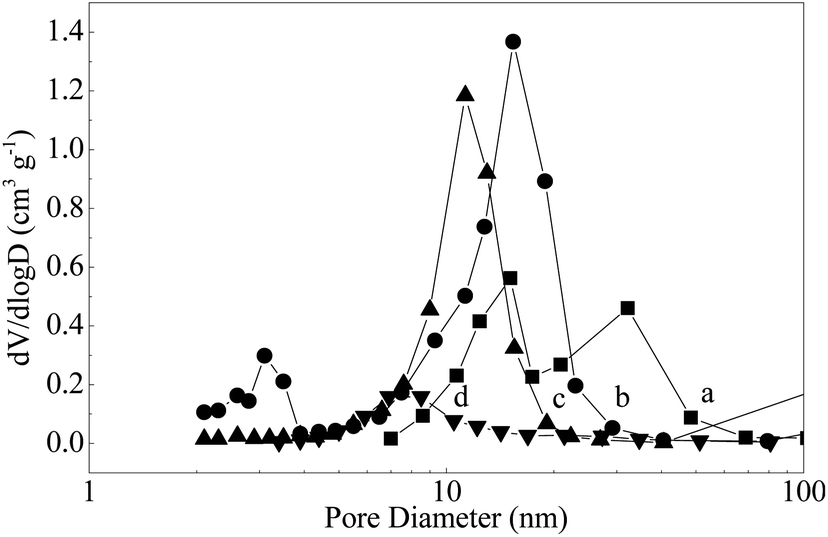 | ||
| Fig. 5 The pore size distribution patterns of (a) Mo–Or, (b) Mo–Ar, (c) Mo–CH, (d) Mo–H. | ||
The BET surface area (ABET), total pore volume (TPV) and average pore diameter (d) of the samples are presented in Table 1. The sample of Mo–Or obtained from hydrothermal treatment is porous, and the BET surface area is 38.8 m2 g−1. After carbonization, the BET surface area obviously increased, except for Mo–H. As tabulated in Table 1, Mo–H showed the lowest surface area (ABET = 20.6 m2 g−1) and the smallest total pore volume (TPV = 0.06 cm3 g−1) compared to the others. Mo–Ar showed the highest surface area (ABET = 305.6 m2 g−1) and largest total pore volume (TPV = 0.48 cm3 g−1), while for Mo–CH, the surface area is 153.2 m2 g−1, and total pore volume is 0.36 cm3 g−1. Combined with elemental analysis and TEM images, the differences of the texture can be derived from the carbon materials in the network of samples.
To gain a better understanding of the carbonization process, thermogravimetric analysis was further employed in this study. As shown in Fig. 6, the weight loss below 450 K is generally attributed to the evaporation of adsorbed water in all the cases.19 When the sample of Mo–Or carbonized in H2 or CH4/H2, the TG curve exhibited a main weight loss between 450 and 1000 K, which could be attributed to the decomposition of the carbonaceous material and the reduction–carbonization of molybdenum oxide. The decomposition of the carbonaceous material would result in the generation of the porosity, ensuring better accessibility of the encapsulated molybdenum carbide to the reaction.19 H2 would prefer to lead the carbon-source of precursors to flee in the carbonization process, evidenced by the fact that a brown tarry substance is present at the end of quartz tube after the carbonization process. In addition, CH4 could act as a carbon-donor at higher temperatures, for the sample carbonized in CH4/H2,26 as was verified by the carbon content in the samples. On the other hand, the weight loss in the range of 600–700 K could be ascribed to the reduction of MoO3 to MoO2, the presence of MoO3 could be from the oxidation of MoO2 and Mo2C, though no peaks were assigned to MoO3 for the samples of Mo–H and Mo–CH in the XRD spectra(Fig. 3). Moreover, the peaks at about 1000 K could be considered as the result of the carbonization of MoO2 to Mo2C. As for the TGA of pure Ar, the curve exhibited a gradual and unpronounced weight loss in the range of 450 and 1000 K, which could be attributed to the partial elimination of organic volatiles from the carbonaceous matrix and the transformation of MoO3 to MoO2. For Ar, the obvious weight-loss appeared at about 1050 K, which is much higher than the others. The different temperature of the samples at which the carbonization occurred implies that the presence of H2 apparently decreases the reduction temperature, which is consistent with the XRD results and other data.
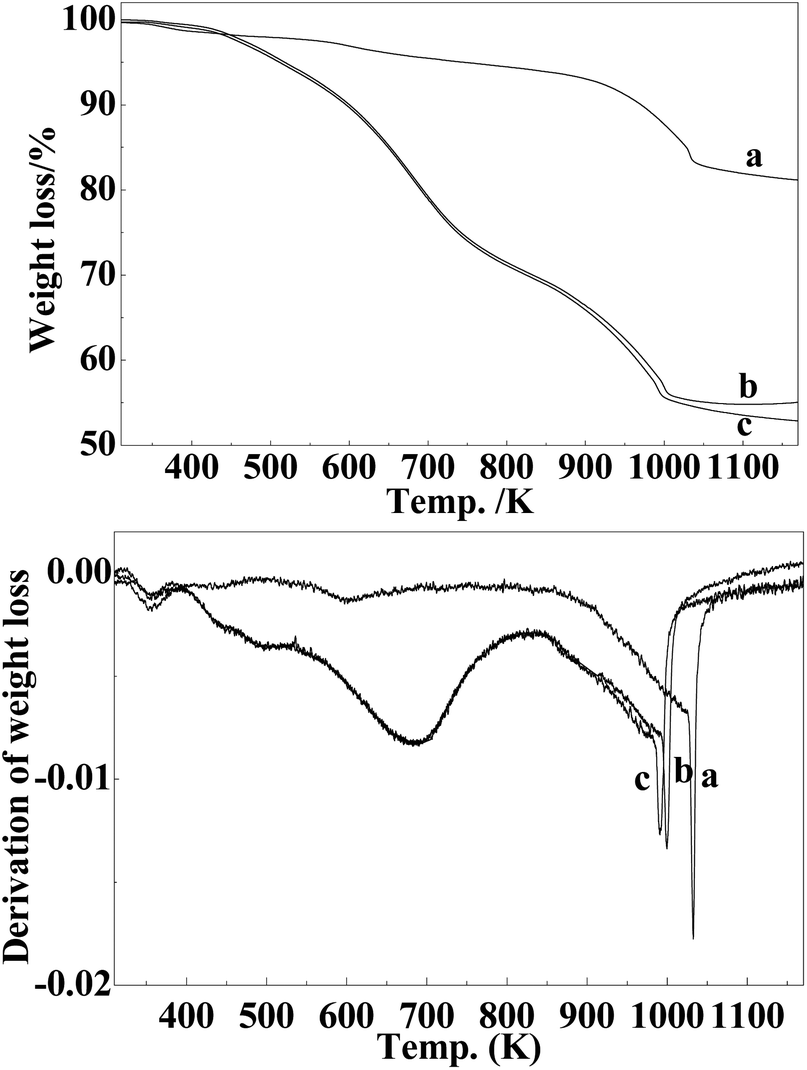 | ||
| Fig. 6 The TGA and DTG curves of (a) MoC–Ar, (b) MoC–CH4, and (c) MoC–H. | ||
3.3. Surface chemistry
The XPS measurement was conducted to investigate the surface chemistry of the carbonized samples. The Mo3d and C1s spectra with fitting curves are illustrated in Fig. 7. The surface compositions calculated from the XPS spectra for all samples are shown in Table 1 and the quantitative analysis of Mo3d signals for the synthesized samples are shown in Table 2. In Fig. 7, three peaks located at 226.0, 228.9, and 232.5 eV in the Mo3d spectra are observed for the samples, except Mo–Or. For the Mo–Or sample, the major molybdenum species on its surface are Mo6+ (3d5/2 = 234.6 eV) and Mo6+–Mo5+ (3d5/2 = 232.4 eV), yet the content of Mo5+–Mo4+ (3d5/2 = 230.8 eV) is low. However, our XRD results reveal only MoO2 phase in the sample (Fig. 3a). This conflict could be explained by the surface of the hydrothermally synthesized MoO2 nanocrystals with low crystallinity being easily oxidized to higher states during the XPS sample preparation and handling.19,27 After carbonization, the overall peaks of the Mo3d spectra move to lower states and the molybdenum species with low oxidation states become more observable, which coincides with the XRD analysis. For the Mo–Ar sample, the major molybdenum species, Mo6+ (3d5/2 = 233.1 eV), Mo5+ (3d5/2 = 231.8 eV) and Mo4+ (3d5/2 = 229.9 eV), are found on its surface. However, no peak belonging to carbidic molybdenum is observed, although our XRD and HRTEM results reveal a poorly crystallized β-Mo2C phase in this sample (Fig. 3a and 2d). It could be attributed to the ambient oxidation which turns the surface β-Mo2C nanocrystals to molybdenum oxides.19,27 For the Mo–CH sample, the molybdenum species with lower oxidation states, Mo2C (3d5/2 = 228.6 eV) and MoO2 (3d5/2 = 229.5 eV), become more observable (Table 2), which could be attributed to the carbonization in CH4/H2 improving the crystallinity. Nonetheless, the XRD analysis reveals that only poorly crystallized β-Mo2C is detected in the sample. Again, the Mo6+ (3d5/2 = 233.0 eV) and Mo5+–Mo4+components (3d5/2 = 230.6 eV) observed herein could be attributed similarly to the ambient oxidation which turns the surface MoO2 and β-Mo2C nanocrystals to α-MoO3 and HxMoO3 surface species, respectively.19,27 While, for the sample of Mo–H, the oxidation states of the molybdenum species are further lower than the others, even Mo0 (3d5/2 = 227.9 eV) appeared in the sample. Clearly, a higher reduction degree of the Mo species in the Mo–H sample is achieved.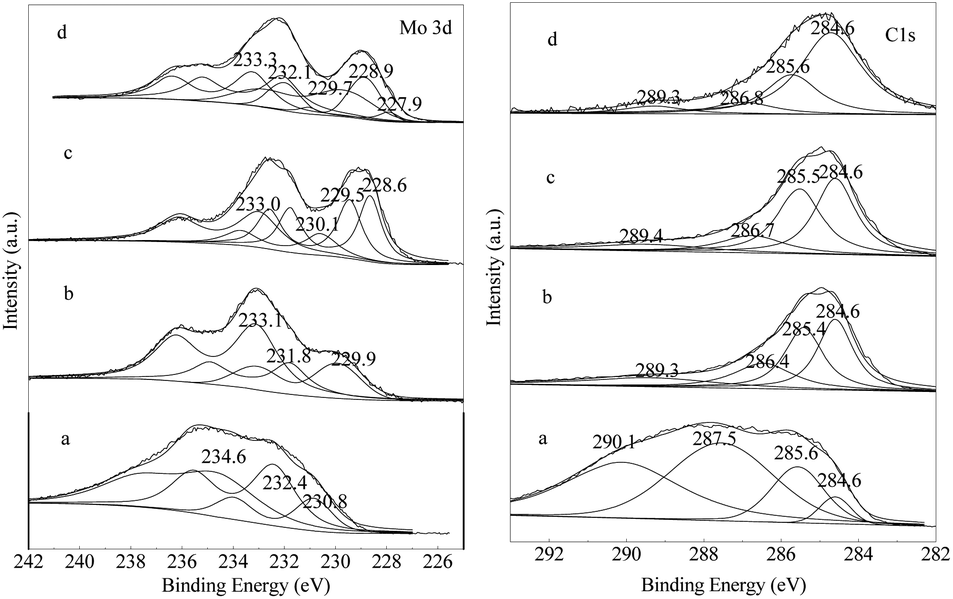 | ||
| Fig. 7 Mo3d and C1s XPS spectra of (a) Mo–Ar, (b) Mo–CH, (c) Mo–H. | ||
| Sample | Assignment | B.E./eV | Area/% |
|---|---|---|---|
| Mo–Or | Mo6+ | 234.6 | 43.7 |
| Mo6+–Mo5+ | 232.4 | 40.3 | |
| Mo5+–Mo4+ | 230.8 | 16.0 | |
| Mo–Ar | Mo6+ | 233.1 | 53.7 |
| Mo5+ | 231.8 | 23.5 | |
| Mo4+ | 229.9 | 22.8 | |
| Mo–CH | Mo6+ | 233.0 | 28.2 |
| Mo5+–Mo4+ | 230.6 | 13.6 | |
| Mo4+ | 229.5 | 28.4 | |
| Mo2+ | 228.6 | 29.8 | |
| Mo–H | Mo6+ | 233.3 | 24.6 |
| Mo5+ | 232.1 | 27.2 | |
| Mo4+ | 229.7 | 23.5 | |
| Mo2+ | 228.9 | 20.6 | |
| Mo0 | 227.9 | 4.1 |
The Cls spectrum shows the presence of four overlapping XPS peaks. For Mo–Or, the major peaks at 287.5 and 290.1 eV are attributed to surface carbonyl or quinone groups (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carboxylic groups, esters, or lactones (–COO–), probably due to the carbon materials produced from glucose.28–30 After carbonization, these peaks with higher BEs significantly decreased, and the major peak at 284.6 eV is assigned to the surface C–C bonds belonging to amorphous carbon, as reported by several groups.28–30 The other peaks located at around 285.5 and 286.7 eV are attributed to the hydroxyl groups or ethers (C–OH) and C–O–C, respectively. The signal for the carbon atoms in the carbide form is very weak and almost cannot be distinguished clearly, even though in Mo3d spectrum the peak belonging to carbidic molybdenum is observed. The surface compositions calculated from the XPS spectra are shown in Table 1, it could be expected that the various amounts of carbon on the surface will result in different catalytic performances in the CO hydrogenation reaction.
O) and carboxylic groups, esters, or lactones (–COO–), probably due to the carbon materials produced from glucose.28–30 After carbonization, these peaks with higher BEs significantly decreased, and the major peak at 284.6 eV is assigned to the surface C–C bonds belonging to amorphous carbon, as reported by several groups.28–30 The other peaks located at around 285.5 and 286.7 eV are attributed to the hydroxyl groups or ethers (C–OH) and C–O–C, respectively. The signal for the carbon atoms in the carbide form is very weak and almost cannot be distinguished clearly, even though in Mo3d spectrum the peak belonging to carbidic molybdenum is observed. The surface compositions calculated from the XPS spectra are shown in Table 1, it could be expected that the various amounts of carbon on the surface will result in different catalytic performances in the CO hydrogenation reaction.
4. Catalytic performance
Finally, to test the catalytic properties of the nanostructured β-Mo2C, a preliminary study on the CO hydrogenation reaction for obtaining higher alcohols using the as-prepared materials was carried out. For comparison, the β-Mo2C of the bulky counterpart obtained by the TPRe method (marked as MoC-TPRe) was also applied to the reaction.31,32 The detailed reaction results are summarized in Table 3 and 4. Generally, the performance of nanostructured molybdenum carbides synthesized by this hydrothermal carbonization method is better than that of the Mo2C obtained by TPRe method in the CO hydrogenation reaction. Moreover, the carbonization gases led to catalysts with different structural and surface properties, with the differences specifically manifesting in the CO hydrogenation performance of the catalysts. As shown in Table 3, the CO conversion of the catalysts varied across a wide range, for MoC–Ar it is only 12.19 C%, while it is 92.06 C% for MoC–H, 66.55 C% for MoC–CH4, and 55.79 C% for MoC-TPRe. As can be appreciated, transition metal carbides have been shown to possess better CO hydrogenation activity than the corresponding oxides, and the higher level of carbonization, the higher the activity of catalyst.1 Furthermore, the activity is closely related to the Mo species with a lower valence being on the surface of the catalysts.1,33,34 Moreover, the XPS analysis results (Fig. 7 and Table 2) show that the amount of carbon follows the sequence: MoC–H > MoC–CH4 > MoC–Ar. Combined with the XRD patterns (Fig. 3), it could be concluded that the CO conversion activity is in good accordance with the degree of carbonization. In addition, the textural properties of these materials could also play an important role, and the highly porosity of carbon materials would allow the reactants to reach the nanocrystals of the active molybdenum species. As a result, the higher CO conversion of MoC–H and MoC–CH4 compared to MoC-TPRe could be ascribed to the mesoporous structure and high surface area of the synthesized nanostructured catalysts, as the MoC-TPRe synthesized here is nonporous and the BET surface area is only 5 m2 g−1. Compared with Mo–CH, the lower amount of carbon in the Mo–H sample would favor more molybdenum species to react in the reaction and show higher activity. On the other hand, the selectivities towards the products exhibit different trends. It is known that the formation of alcohols is related to the surface stoichiometry and to the extent of the carbonization; whenever the level of carbonization is too high only hydrocarbons are formed.1 Combined with the XRD results, it is clearly seen that the trend of selectivity towards alcohols reverses with the degree of carburization of the samples. As seen in Table 3, the nanostructured catalysts give better selectivity towards alcohols, indicating the advantage of this method. As can be appreciated, Mo–Ar gives the highest selectivity towards alcohols (27.1%) though it performs with the lowest activity. Moreover, in the absence of MoC-TPRe, Mo–H shows the lowest selectivity towards alcohols (only 3.7%) even though it has the highest activity. As shown in Table 3 and 4, it can be seen that the major products over these catalysts are C1–C4 hydrocarbons, the yield of alcohols in the products is much lower than the amount of hydrocarbons for all the catalysts, and methanol is the main alcohol product, which is particularly characteristic of the action of β-Mo2C in CO hydrogenation.11 Combined with the XRD results, it could also be seen that the trend of producing methanol parallels with the carbonization degree of the catalysts, but the trend of methane is reversed. As for Mo2C, it is proposed to be a dual-site catalyst involving two types of molybdenum species,34 namely a low valence molybdenum (MoI, I = 0–2) and a high valence molybdenum (MoII, II = 4–5) species. The MoI species is the site for CO dissociation, hydrogenation and chain propagation to form surface alkyl species, while the MoII species is the site for molecularly adsorbed CO insertion and alcohol formation.32 Together with XPS and XRD, it could be noticed that the higher amount of MoI species and lower amount of MoII in the Mo–H sample, with the higher carbonization degree, favors the chain propagation of hydrocarbons and suppresses the chain propagation of alcohols, leading to more methanol and less methane in the catalyst. Therefore, to realize a desired Mo2C catalyst for higher alcohol synthesis it is necessary to precisely control the carbonization progress.| Cat. | CO conversion (C%) | Yield (g mL−1 h−1) | Selectivityb (wt%) | ||
|---|---|---|---|---|---|
| Alcohols | Hydrocarbon | ROH | CHn | ||
| a Reaction conditions: H2/CO = 2.0, T = 553 K, P = 7.0 MPa, GHSV = 4000 h−1.b Calculated on a CO2 free basis. | |||||
| Mo–Ar | 12.19 | 0.026 | 0.069 | 27.06 | 72.94 |
| Mo–CH | 66.55 | 0.038 | 0.405 | 8.67 | 91.33 |
| Mo–H | 92.06 | 0.026 | 0.676 | 3.71 | 96.29 |
| MoC-TPRe | 55.79 | 0.004 | 0.331 | 1.12 | 98.88 |
| Cat. | Alcohol distribution (wt%) | Hydrocarbon distribution (wt%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MeOH | EtOH | PrOH | BuOH | C5+OH | CH4 | C2H6 | C3H8 | C4H10 | C5+H | |
| a Reaction conditions: H2/CO = 2.0, T = 553 K, P = 7.0 MPa, GHSV = 4000 h−1. | ||||||||||
| Mo–Ar | 72.24 | 20.34 | 6.10 | 1.32 | 0.00 | 47.17 | 25.84 | 16.28 | 6.08 | 4.63 |
| Mo–CH | 72.58 | 17.43 | 7.89 | 1.99 | 0.12 | 50.36 | 25.73 | 15.20 | 5.48 | 3.23 |
| Mo–H | 87.04 | 7.65 | 3.28 | 1.14 | 0.88 | 41.52 | 27.75 | 17.72 | 6.38 | 6.63 |
| MoC-TPRe | 83.89 | 10.51 | 4.06 | 1.54 | 0.00 | 49.74 | 23.19 | 15.87 | 6.04 | 5.16 |
5. Conclusions
Highly dispersed β-Mo2C nanoparticles have been successfully synthesized by a facile hydrothermal method followed by a temperature-programmed carbonization process under different atmospheres. The subnanometer contact between the molybdenum oxides and D-glucose molecules in the hydrothermal treatment remarkably facilitates the achievement of Mo2C nanoparticles with large surfaces and enriched meso-porosity, which benefits the good performance in the production of alcohols through CO hydrogenation. In addition, various atmospheres led to different degrees of carbonization and matrix morphologies under the same conditions. The sample carbonized in Ar appeared to be only carbonized on the surface, but the surface-bulk of the sample carbonized in H2 is homogenous. The differences in structure and surface composition were revealed by the performance of the catalysts in CO hydrogenation.Acknowledgements
The authors acknowledge the financial support from the State Key Foundation Program for Development and Research of China (Contract no. 2005CB221402), the National High Technology Research and Development Program of China (nos 2012AA051002), the Strategic Priority Research Program of the Chinese Academy of Sciences (nos XDA01020304), and the projects of the National Natural Science Foundation of China (nos 21103217).Notes and references
- L. Leclercq, A. Almazouari, M. Dufour and G. Leclercq, in The Chemistry of Transition Metal Carbides and Nitrides, ed. S. T. Oyama, Blackie, Glasgow, 1996, pp. 345–361 Search PubMed.
- S. A. Jansen and R. Hoffmann, Surf. Sci., 1988, 197, 474 CrossRef CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185 CrossRef CAS PubMed.
- Z.-Q. Wang, Z.-B. Zhang and M.-H. Zhang, Dalton Trans., 2011, 40, 1098 RSC.
- A. Szymanska-Kolasa, M. Lewandowski, C. Sayag, D. Brodzki and G. Djéga-Mariadassoub, Catal. Today, 2007, 119, 35 CrossRef CAS PubMed.
- J. Han, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, Green Chem., 2011, 13, 2561 RSC.
- M. Zheng, Y. Shu, J. Sun and T. Zhang, Catal. Lett., 2007, 121, 90 CrossRef PubMed.
- A. R. S. Darujati and W. J. Thomson, Appl. Catal., A, 2005, 296, 139 CrossRef CAS PubMed.
- S. Liu, L. Wang, R. Ohnishi and M. Lchikawa, Kinet. Catal., 2000, 41, 132 CrossRef CAS.
- H. C. Woo, K. Y. Park, Y. G. Kim, I.-S. Nam, J. S. Chung and J. S. Lee, Appl. Catal., 1991, 75, 267 CrossRef CAS.
- S. Zaman and K. J. Smith, Catal. Rev., 2012, 54, 41 CAS.
- N. A. Dhas and A. Gedanken, Chem. Mater., 1997, 9, 3144 CrossRef CAS.
- M. Pang, C. Li, L. Ding, J. Zhang, D. Su, W. Li and C. Liang, Ind. Eng. Chem. Res., 2010, 49, 4169 CrossRef CAS.
- Q. Gao, C. Zhang, S. Xie, W. Hua, Y. Zhang, N. Ren, H. Xu and Y. Tang, Chem. Mater., 2009, 21, 5560 CrossRef CAS.
- B. Sun, L. Xu, K. Tang, L. Wang, Z. Ju and Y. Qian, Cryst. Res. Technol., 2012, 47, 467 CrossRef CAS.
- X. Li, D. Ma, L. Chen and X. Bao, Catal. Lett., 2007, 116, 63 CrossRef CAS PubMed.
- G. Yu, B. Sun, Y. Pei, S. Xie, S. Yan, M. Qiao and K. Fan, J. Am. Chem. Soc., 2010, 132, 935 CrossRef CAS PubMed.
- L. She, J. Li, Y. Wan, X. Yao, B. Tu and D. Zhao, J. Mater. Chem., 2011, 21, 795 RSC.
- J. Dou and H. C. Zeng, J. Phys. Chem. C, 2012, 116, 7767 CAS.
- C. Avendano, A. Briceno, F. J. Mendez, J. L. Brito, G. Gonzalez, E. Canizales, R. Atencio and P. Dieudonne, Dalton Trans., 2013, 42, 2822 RSC.
- W. Wang, G. Ding, T. Jiang, P. Zhang, T. Wu and B. Han, Green Chem., 2013, 15, 1150 RSC.
- M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195 CrossRef CAS PubMed.
- X. Li, L. Feng, Z. Liu, B. Zhong, D. B. Dadyburjor and E. L. Kugler, Ind. Eng. Chem. Res., 1998, 37, 3853 CrossRef CAS.
- H.-M. Wang, X.-H. Wang, M.-H. Zhang, X.-Y. Du, W. Li and K.-Y. Tao, Chem. Mater., 2007, 19, 1801 CrossRef CAS.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, J. Catal., 2002, 206, 230 CrossRef CAS.
- A. Hanif, T. Xiao, A. P. E. York, J. Sloan and M. L. H. Green, Chem. Mater., 2002, 14, 1009 CrossRef CAS.
- A. Cimino and B. A. De Angelis, J. Catal., 1975, 36, 11 CrossRef CAS.
- P. Delporte, F. D. R. Meunier, C. Pham-Huu, P. Vennegues, M. J. Ledoux and J. Guille, Catal. Today, 1995, 23, 251 CrossRef CAS.
- P. Delporte, C. Pham-Huu and M. J. Ledoux, Appl. Catal., A, 1997, 149, 151 CrossRef CAS.
- T. Miyao, I. Shishikura, T. M. Matsuoka and M. Nagai, Chem. Lett., 1996, 121, 561 CrossRef.
- T. Xiao, H. Wang, J. Da, K. S. Coleman and M. L. H. Green, J. Catal., 2002, 211, 183 CAS.
- N. Wang, K. Fang, D. Jiang, D. Li and Y. Sun, Catal. Today, 2010, 158, 241 CrossRef CAS PubMed.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133 CrossRef CAS PubMed.
- A. Muramatsu, T. Tatsumi and H. Tominaga, J. Phys. Chem., 1992, 96, 1334 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |