Azobenzene–siloxane hybrids with lamellar structures from bridge-type alkoxysilyl precursors†
Sufang Guoa,
Watcharop Chaikittisilpa,
Tatsuya Okuboa and
Atsushi Shimojima*b
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Ohkubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: shimojima@waseda.jp
First published on 29th May 2014
Abstract
Lamellar azobenzene–siloxane hybrids were prepared by controlled hydrolysis and polycondensation of three types of precursors, where azobenzene is sandwiched by mono-, di- and triethoxysilyl groups using propylene linkers. All precursors underwent reversible and fast trans–cis isomerization upon UV/Vis irradiation in dilute solution. Upon hydrolysis of the triethoxysilylated precursor in a homogeneous solution under acidic conditions, precipitation occurred by self-assembly of hydrolyzed monomers into a lamellar structure. Although di- and mono-ethoxysilylated precursors produced less ordered products under identical conditions, highly ordered lamellar films were obtainable either by evaporation induced self-assembly of the hydrolyzed monomers or by solid-state reactions of precursor films. The degree of trans–cis isomerization of azobenzene moieties in the hybrid films was enhanced by decreasing the cross-linking degree of siloxane networks using precursors with less condensable alkoxy groups.
1 Introduction
Constructing inorganic–organic hybrid nanoarchitectures requires the integration of inorganic and organic building blocks on molecular or nanometer-length scales.1,2 Organosiloxane is an excellent example of such hybrid materials, possessing the benefits of siloxane networks, such as good thermal/chemical stabilities and high transparency, along with diverse functionalities endowed by organic moieties. The preparation of organosiloxane-based materials with unique properties, structures and morphologies has been widely studied.2–4Sol–gel hydrolysis and polycondensation of organoalkoxysilanes provides a convenient route to obtaining organosiloxane hybrids; however, this process generally yields hybrids with amorphous structures. In recent years, careful precursor designing and optimization of the reaction conditions have produced organosiloxane hybrids with ordered structures, such as lamellar and two-dimensional hexagonal structures. Organoalkoxysilanes with pendant and bridging organic (R) groups (R–Si(OR′)3 and (R′O)3Si–R–Si(OR′)3, respectively) are commonly used. The components of organic moieties (R) are extremely important for self-assembly and therefore to direct the formation of ordered structures. Non-covalent interactions, such as hydrophobic interactions, strong H-bonds (e.g. urea and amide linkages) and π–π stacking interactions, often favour self-directed assembly.5–12 In general, bridge-type precursors have the ability to incorporate a wider variety of organic groups in the siloxane networks, providing greater opportunities to create novel functional hybrid materials.
Azobenzene is a typical photo-responsive molecule that undergoes photochemical conversion between trans and cis isomers, accompanied by large changes in molecular shape and size. The arrangement and mobility of azobenzene critically influence the photo-responsive properties of azobenzene-containing materials. Azobenzene–siloxane hybrids, produced by self-directed assembly of azobenzene-containing alkoxysily precursors, are a potential new class of photo-responsive materials; however, their synthesis and properties have not yet been fully investigated. Liu et al. reported the synthesis of a photoactive azobenzene-bridged alkoxysilane containing urea (–NH–CO–NH–) linkages.13 This precursor can self-assemble into lamellar structures through H-bonding interaction between urea groups and π–π interaction between azobenzene groups. However, trans–cis isomerization is almost inhibited in the solids possibly due to intense H-bonds. Azobenzene-bridged alkoxysilanes containing simple alkylene linkers are worth investigating as a means of improving their photo-responsive properties. Such precursors were used to produce photo-responsive mesoporous silica, where azobenzenes were randomly arranged in the pore walls, by surfactant-directed self-assembly with additional silica source.14,15 As yet, their self-directed assembly, as well as photo-responsive properties of the products, are unexplored.
In this study, ordered azobenzene–siloxane hybrid materials were prepared by self-directed self-assembly of azobenzene-bridged alkoxysilanes containing propylene linkers. Three types of precursors with tri-, di- and mono-ethoxysilyl groups (1, 2 and 3 in Scheme 1) were used to study the effects of (i) the number of Si–OH groups on self-assembly and (ii) the degree of Si–O–Si cross-linking on photo-isomerization of bridged azobenzene groups. Powder samples (P1, P2 and P3) were obtained as precipitates by acid-catalyzed hydrolysis and polycondensation in THF–H2O solutions. Furthermore, to ensure high-efficiency light absorption,13,16–21 thin films were prepared on glass substrates by either an evaporation induced self-assembly (EISA) technique (F2 and F3) or solid-state reactions (F1′, F2′ and F3′). The trans–cis isomerization of azobenzene groups in hybrid thin films upon UV/Vis irradiation was investigated.
 | ||
| Scheme 1 Structures of bridged-type 1, 2 and 3 precursors and the preparation pathways of hybrid powders and films. | ||
2 Experimental
Materials
Allyl bromide (>98.0%), 4-aminophenol (>98.0%), phenol (>99.0%), sodium nitrite (>98.5%), hydrochloric acid (HCl, 1 M), sodium hydroxide (NaOH, >97.0%), N,N-dimethylformamide (DMF, dehydrated, >99.5%), ethyl acetate (>99.0%) and dimethylethoxysilane were purchased from Wako Pure Chemical Industry. Sodium hydride (NaH) (60% dispersion in paraffin liquid), triethoxysilane (>97.0%) and diethoxymethysilane (>95%) were purchased from Tokyo Chemical Industry. Platinum (0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complexes in xylene (Pt ∼2%), sulfamic acid (≥99.5%) and acetic acid (≥99.5%) were purchased from Sigma-Aldrich. All chemicals were used as received, without further purification.Synthesis of 4,4′-dihydroxyazobenzene
The procedure was conducted as per a previous report,22 with slight modification. 4-Aminophenol (2 g, 18.3 mmol) and 16% HCl (8.4 mL) were added to a 20 mL flask (flask A). After being stirred for 45 min at room temperature, the mixture was cooled to 0 °C. Then, a 2 M NaNO2 solution was slowly added while stirring at 0 °C. The mixture was stirred at 0 °C for another 2 h, followed by the addition of sulfamic acid to remove the excess NaNO2. In a 50 mL flask (flask B), phenol (1.72 g, 18.3 mmol) was mixed with a 2 M NaOH solution (1.47 g, 36.6 mmol in 17 mL H2O). The solution in flask A was then added to flask B, and subsequently stirred at room temperature for 10 h. The crude product was recrystallized in an EtOH–H2O solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v), yielding a 1.56 g dark red product (40% yield). 1H NMR (δ, 270 MHz, DMSO-d6): 6.89–6.93 (d, 4H, ArH), 7.70–7.80 (d, 4H, ArH), 10.13 (s, 2H, Ar–OH). 13C NMR (δ, 67.8 MHz, DMSO-d6): 115.76, 124.12, 145.25, 159.96.
:5 v/v), yielding a 1.56 g dark red product (40% yield). 1H NMR (δ, 270 MHz, DMSO-d6): 6.89–6.93 (d, 4H, ArH), 7.70–7.80 (d, 4H, ArH), 10.13 (s, 2H, Ar–OH). 13C NMR (δ, 67.8 MHz, DMSO-d6): 115.76, 124.12, 145.25, 159.96.
Synthesis of 4,4′-diallyloxyazobenzene
In a 100 mL Schlenk flask, 4,4′-dihydroxyazobenzene (1.00 g, 4.66 mmol) was dissolved in 10 mL of DMF. Activated NaH (0.56 g, 14.0 mmol), dispersed in DMF (20 mL), was added to the flask. After stirring at room temperature for 2 h, allyl bromide (3.49 g, 11.7 mmol) was added, and the mixture was stirred at 60 °C for 1 day under nitrogen atmosphere. The product was extracted with ethyl acetate, washed with cold water and dried over MgSO4. Evaporation of ethyl acetate gave a dark red, crude solid product. Yellow crystals (0.64 g; yield of 47%) were obtained after recrystallization from ethyl acetate. 1H NMR (δ, 270 MHz, CDCl3): 4.60–4.61 (d, 4H, ArOCH2), 5.30–5.48 (m, 4H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.10–6.15 (m, 2H, ArCH2CH), 6.98–7.04 (d, 4H, ArH), 7.84–7.89 (d, 4H, ArH). 13C NMR (δ, 67.8 MHz, CDCl3): 69.04, 114.94, 118.00, 124.33, 132.87, 147.18, 160.60.
CH), 6.10–6.15 (m, 2H, ArCH2CH), 6.98–7.04 (d, 4H, ArH), 7.84–7.89 (d, 4H, ArH). 13C NMR (δ, 67.8 MHz, CDCl3): 69.04, 114.94, 118.00, 124.33, 132.87, 147.18, 160.60.
Synthesis of 4,4′-[3-(triethoxysilyl)propoxy]azobenzene (1)
Hydrosilylation of 4,4′-diallyloxy-azobenzene (0.294 g, 1.00 mmol) in an excess amount of triethoxysilane (3.28 g, 20.0 mmol) was performed in toluene (20 mL) in the presence of a Pt catalyst (0.027 g, 2 × 10−5 mol). The mixture was stirred at 70 °C for 24 h under nitrogen atmosphere, and the solvent and unreacted triethoxysilane were removed in vacuo. 1 was obtained as red crystals (0.529 g, yield of 85%) after purification using gel permeation chromatography (GPC), with chloroform as the eluent. 1H NMR (δ, 270 MHz, CDCl3): 0.76–0.82 (m, 2H, OCH2CH2CH2Si), 1.21–1.26 (m, 9H, Si(OCH2CH3)3), 1.78–2.00 (m, 2H, OCH2CH2CH2Si), 3.81–3.89 (m, 6H, Si(OCH2CH3)3), 4.00–4.05 (m, 2H, OCH2CH2CH2Si), 6.96–7.00 (d, 2H, ArH), 7.83–7.88 (d, 2H, ArH). 13C NMR (δ, 67.8 MHz, CDCl3): 6.50, 18.32, 22.77, 58.46, 70.13, 114.67, 124.30, 146.95, 161.11. 29Si NMR (δ, 53.45 MHz, CDCl3): −45.53. ESI-MS: m/z: 645.2994 [M + Na]+.Synthesis of 4,4′-[3-(diethoxymethylsilyl)propoxy]azobenzene (2)
The chemical 4,4′-diallyloxyazobenzene (0.294 g, 1.00 mmol) dissolved in toluene (15 mL) was mixed with diethoxymethylsilane (2.68 g, 20.0 mmol) and 0.02 g (2 × 10−5 mol) of a Pt catalyst. The mixture was stirred under nitrogen atmosphere at 70 °C for 24 h. Yellow crystals (0.51 g, yield of 90%) were obtained after solvent evaporation, followed by purification using GPC. 1H NMR (δ, 270 MHz, CDCl3): 0.163 (s, 3H, SiCH3), 0.74–0.80 (m, 2H, OCH2CH2CH2Si), 1.20–1.28 (m, 6H, Si(OCH2CH3)2), 1.75–1.95 (m, 2H, OCH2CH2CH2Si), 3.75–3.83 (m, 4H, Si(OCH2CH3)2), 3.98–4.03 (m, 2H, OCH2CH2CH2Si), 6.95–7.01 (d, 2H, ArH), 7.83–7.89 (d, 2H, ArH). 13C NMR (δ, 67.8 MHz, CDCl3): −4.87, 9.96, 18.42, 22.82, 58.19, 70.36, 114.67, 124.30, 146.96, 161.08. 29Si NMR (δ, 53.45 MHz, CDCl3): −5.08. ESI-MS: m/z: 585.2786 [M + Na]+.Synthesis of 4,4′-[3-(ethoxydimethylsilyl)propoxy]azobenzene (3)
4,4′-Diallyloxy-azobenzene (0.294 g, 1.00 mmol) dissolved in toluene (15 mL) was mixed with diethoxymethylsilane (2.08 g, 20.0 mmol) and 0.02 g (2 × 10−5 mol) of a Pt catalyst. The mixture was stirred under nitrogen atmosphere at 70 °C for 24 h. Yellow crystals (0.43 g, yield of 85%) were obtained after solvent evaporation, followed by purification using GPC. 1H NMR (δ, 270 MHz, CDCl3): 0.151 (s, 6H, Si(CH3)2), 0.71–0.77 (m, 2H, OCH2CH2CH2Si), 1.18–1.23 (m, 3H, SiOCH2CH3), 1.82–1.93 (m, 2H, OCH2CH2CH2Si), 3.65–3.68 (m, 2H, SiOCH2CH3), 3.99–4.04 (m, 2H, OCH2CH2CH2Si), 6.97–7.01 (d, 2H, ArH), 7.84–7.88 (d, 2H, ArH). 13C NMR (δ, 67.8 MHz, CDCl3): −2.10, 12.39, 18.57, 23.15, 58.34, 70.64, 114.67, 124.31, 146.96, 161.10. 29Si NMR (δ, 53.45 MHz, CDCl3): 17.18. ESI-MS: m/z: 525.2576 [M + Na]+.Preparation of hybrid powders (P1, P2 and P3)
An HCl aqueous solution was added to the THF solutions of 1, 2 and 3, yielding molar compositions of 1:THF:H2O:HCl = 1:50:15:0.03, 2:THF:H2O:HCl = 1:50:10:0.02, and 3:THF:H2O:HCl = 1:50:10:0.02, respectively. The mixtures were stirred at room temperature for several hours. The resulting precipitates were collected by filtration and subsequently heated overnight at 120 °C, giving P1, P2 and P3.
Preparation of hybrid films (F2 and F3) by EISA processes
An HCl aqueous solution was added to THF solutions of 2 and 3 (molar ratios of 2:THF:H2O:HCl = 1:100:10:0.02 and 3:THF:H2O:HCl = 1:100:10:0.005). The mixtures were stirred at room temperature for 2 h and 1 h, respectively. A portion of the mixtures was spin-coated on glass substrates to produce thin films. To induce further polymerization, the film prepared from 2 was exposed to 5 M HCl vapour for 30 min, and the film prepared from 3 was heated at 120 °C for 3 h to give F2 and F3, respectively. Note that film samples cannot be obtained from 1 by this procedure due to precipitation upon hydrolysis.
Preparation of hybrid films (F1′, F2′ and F3′) by solid-state reactions
First, crystalline thin films of precursors 1, 2 and 3 were prepared by spin-coating (3000 rpm) their THF solutions (ca. 0.25 M) on glass substrates. Subsequently, F1′, F2′ and F3′ films were obtained by solid–liquid reactions, that is, by treating the precursor films in a 0.1 M HCl solution at room temperature for 15 days (F1′) and 9 days (F2′ and F3′), followed by air drying.Characterizations
Solution-state 1H-, 13C- and 29Si-NMR spectra were recorded on a JEOL JNM-270 spectrometer at 270, 67.8 and 53.45 MHz, respectively. DMSO-d6 or CDCl3 was used as the solvent and tetramethylsilane as the internal reference. High resolution ESI mass analysis was carried out with a JEOL JMS-T100 CS instrument. Samples were dissolved in methanol. X-ray diffraction (XRD) patterns were obtained using a Rigaku Ultima IV diffractometer operated at 40 kV and 40 mA, with CuKα radiation (0.15406 nm). The measurements were performed over a 2θ range of 2–30° at scanning speeds of 2° and 1° min−1 with a step width of 0.02° for precursors and hybrid materials, respectively. Fourier transform infrared (FT-IR) spectra were recorded using a JASCO FT/IR-6100 spectrometer by the KBr pellet technique. UV-Vis absorption spectra were recorded on a JASCO V-670 instrument. Morphologies of the samples were observed on a field-emission scanning electron microscope (FE-SEM, Hitachi S-900) with an accelerating voltage of 6 kV. Before observation, samples were sputter-coated with Pt. A SUPERCURE-204S UV light source (San-ei electric) with an intensity of 67–76 mW cm−2 was employed for UV and visible light irradiation of the samples. UV cut (HOYA L-420 nm) and UV pass (HOYA U-340 nm) filters were used. Thermogravimetry (TG)-differential thermal analysis (DTA) measurements were performed on Rigaku 8120 in helium atmosphere with a heating rate of 5 K min−1.3 Results and discussion
Characterizations of precursors 1, 2 and 3
The bridged-type precursors 1, 2 and 3 have similar structures. They possess the same ‘bridge’ (organic fragment between the two Si atoms, having a length of ca. 1.9 nm) while differ in the number of ethoxy groups attached to each Si atom (3, 2 and 1 for 1, 2 and 3, respectively).Powder XRD patterns of the precursors 1, 2 and 3 are shown in Fig. 1. The first strong peaks are attributed to the diffractions from crystals in one orientation, corresponding to the d-spacings of 1.42, 1.70 and 1.93 nm, respectively. Differences in the d-spacings indicate the varying arrangement and/or orientation of the organic bridges. TG-DTA curves of 1, 2 and 3 show endothermic peaks at ca. 85 °C, 60 °C and 79 °C, respectively, without weight losses (Fig. S1 in ESI†), which is attributed to the melting of the molecular crystals.
 | ||
| Fig. 1 Powder XRD patterns of the precursors: (a) 1, (b) 2 and (c) 3. | ||
These precursors can also be obtained as thin films by spin-coating their THF solutions on glass substrates. In these films, lamellar structures are highly orientated relative to the surface, as suggested by the disappearance of diffraction peaks at high angles (Fig. S2 in ESI†). Polarized optical microscopic images of 1, 2 and 3 films (Fig. S3 in ESI†) depict birefringent texture of crystals, indicating submicroscopic ordered structures.
The trans–cis photo-isomerization properties of precursors 1–3 were investigated by UV-Vis spectra of their dilute EtOH solutions (ca. 5 × 10−5 M) upon UV/Vis irradiation. As depicted in Fig. 2, all precursors before irradiation exhibited large absorption peaks at ca. 360 nm, due to π–π* transitions of thermally stable trans isomers. After 1 min of UV irradiation, the peaks significantly decreased, while the broad absorption peaks at ca. 440 nm due to n–π* transitions of cis isomers increased. A further 1 min of Vis irradiation almost recovered the spectra to the original states, in which trans isomers are dominant. In all cases, 1 min of UV or Vis irradiation was enough to drive azobenzenes to reach a stable isomerization state, suggesting that reversible, fast trans–cis photo-isomerization can be achieved in dilute EtOH solutions of 1, 2 and 3 precursors.
 | ||
| Fig. 2 UV-Vis spectra of EtOH solutions of precursors (a) 1, (b) 2 and (c) 3: before irradiation (red line), after 1 min of UV irradiation (blue line), and after a further 1 min of Vis irradiation (green line). | ||
Hybrid materials prepared by hydrolysis and polycondensation of precursors 1, 2 and 3
 | ||
| Fig. 3 FE-SEM images of (a) P1, (b) P2 and (c) P3 (scale bar: 1 μm). | ||
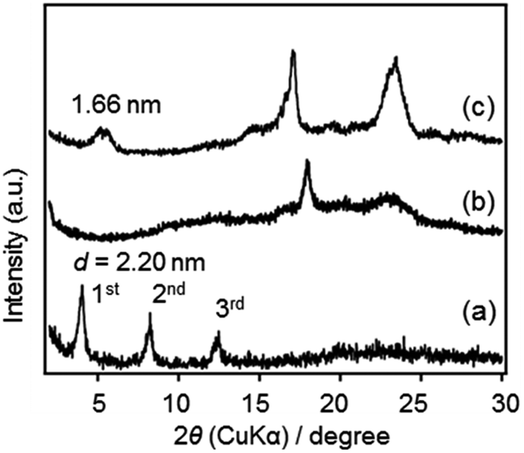 | ||
| Fig. 4 Powder XRD patterns of (a) P1, (b) P2 and (c) P3. | ||
To obtain information about the progress of hydrolysis and polycondensation, FT-IR spectra of P1, P2 and P3 were compared with those of the precursors and as-formed precipitates (i.e. P1, P2 and P3 before heating). The as-formed precipitates derived from 1 (Fig. 5 (a, middle line)) exhibit no OCH2CH3 absorption peaks (2995 cm−1, cf. Fig. 5 (a, bottom line) for 1), suggesting that the ethoxysilyl (SiOEt) groups of 1 were fully hydrolyzed. The new peak at 896 cm−1 was ascribed to the silanol (SiOH) groups. The large absorption band centred at ca. 3245 cm−1 can be assigned to the O–H stretching modes of SiOH and physisorbed water. No peak of isolated, free silanol groups (∼3750 cm−1)25 was observed. The as-formed precipitates have a highly ordered lamellar structure (Fig. S4(a), in ESI†), which is probably stabilized by H-bondings of SiOH groups.26 After heating (Fig. 5 (a, top line)), the absorption bands of SiOH decreased greatly, accompanied by an increase in Si–O–Si absorption bands (e.g. an asymmetric stretching band of Si–O–Si at 1023 cm−1). On the other hand, for P2 and P3, both hydrolysis and polycondensation proceeded to a high extent even prior to heating, as judged from the intense Si–O–Si bands and relatively small SiOH bands (Fig. 5 (middle lines of (b) and (c), respectively). No significant changes were observed after heating (Fig. 5 (top lines of (b) and (c)).
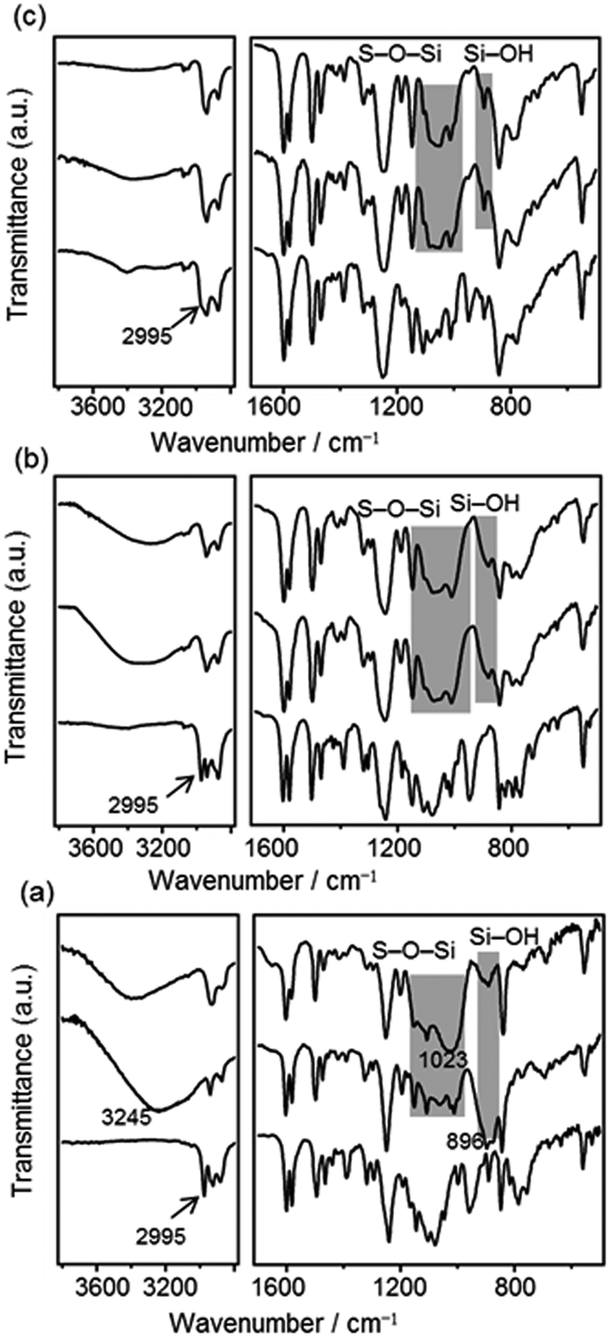 | ||
| Fig. 5 FT-IR spectra of (a) precursor 1 (bottom), as-formed precipitates (middle) and P1 (top), (b) precursor 2 (bottom), as-formed precipitates (middle) and P2 (top) and (c) precursor 3 (bottom), as-formed precipitates (middle) and P3 (top). | ||
The ordered structure of P1 was considered to be formed by self-assembly of amphiphilic hydrolyzed species through hydrophobic (and/or π–π) interactions between organic bridges and H-bondings between silanol groups. Three steps, i.e. hydrolysis, self-assembly (precipitation) and polycondensation, should proceed sequentially, as supported by the above results. In the cases of less-ordered P2 and P3, polycondensation likely occurred in the solutions, forming precipitates without self-assembly. The difference is attributed to the number of SiOH groups; precursors 1, 2 and 3 can form 6, 4 and 2 SiOH groups, respectively, after complete hydrolysis. The lack of self-assembly ability of 2 and 3 may be due to the absence of adequate H-bonding interactions to stabilize lamellar structures, leading to the formation of less ordered structures.
 | ||
| Fig. 6 XRD patterns of (a) F2 and (b) F3. | ||
The XRD patterns of F1′–F3′ films are shown in Fig. 7. All films showed lamellar structures, with d = 2.28, 1.85 and 1.99 nm for F1′, F2′ and F3′, respectively. Relatively weak peaks in F1′ and F2′ indicated less ordered structures, likely due to a partial loss of the crystalline arrangement of precursor molecules during the solid-state reaction. The d-spacings of F2′ and F3′ were similar to those of the precursor films (1.89 nm and 1.93 nm, Fig. S2 in ESI†) indicating the almost unchanged molecular arrangements, despite the conversion of SiOEt to Si–O–Si.
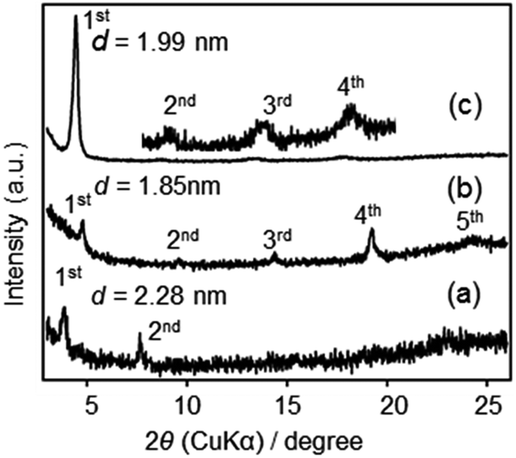 | ||
| Fig. 7 XRD patterns of (a) F1′, (b) F2′ and (c) F3′. | ||
Photo-responsive properties of the hybrid films
The photo-responsive properties of the hybrid films (F2, F3, F1′, F2′ and F3′) were investigated by measuring their UV-Vis spectra upon light irradiation. We have recently reported that azobenzene–siloxane hybrid films prepared from pendant-type precursors showed partial but reversible trans–cis isomerizations.16 Bridged-type hybrids may suffer from a low mobility of bridging azobenzene groups due to greater constraints at both ends, thereby limiting trans–cis isomerization. It is expected that a decrease in the degree of Si–O–Si cross-linking by utilizing precursors with less SiOEt groups will increase the flexibility of siloxane networks so that the mobility of azobenzene groups can be improved.As azobenzenes are less mobile in films than in solutions, a longer time (5 min) of irradiation (1 min for solutions) was conducted. As depicted in Fig. 8(a), F2 shows an apparent increase in absorption at 450 nm upon UV irradiation, indicating a partial of trans to cis isomerization. However, absorption at 350 nm also increased after UV irradiation, which is possibly due to an orientation change of azobenzene moieties relative to incident light caused by order–disorder transition upon irradiation, as described later. In contrast, F3 (Fig. 8(b)) showed obviously reversible trans–cis isomerization upon UV/Vis irradiation, with the isomerization degree comparable to that of pendant-type hybrids.16
 | ||
| Fig. 8 UV-Vis spectra of (a) F2 and (b) F3 films before irradiation (red line), after 5 min of UV irradiation (blue line), and after a further 5 min of visible light irradiation (green line). | ||
XRD patterns of F2 and F3 before and after UV/Vis irradiations are shown in Fig. 9. For F2, UV irradiation caused a disappearance of the XRD peaks and a further Vis irradiation did not recover the lamellar structure (Fig. 9(a)). This order-to-disorder transition upon UV irradiation was considered to be caused by the partial trans-to-cis transition of azobenzene (described as above). Further polycondensation may occur after UV irradiation, fixing the disordered structure, and thereby inhibiting a reversible change into the initial ordered structure even after Vis irradiation (Scheme S1 in ESI†). Further polycondensation after UV irradiation was confirmed by FT-IR spectra (Fig. S7 in ESI†). On the other hand, neither a change in the diffraction peak intensity nor position was observed for F3 upon UV/Vis irradiation (Fig. 9(b)). One possible explanation is that F3 contains some disordered domains, where azobenzene groups can more easily undergo photoisomerization.
 | ||
| Fig. 9 XRD patterns of (a) F2 and (b) F3: (bottom) before irradiation, (middle) after 5 min of UV irradiation, (top) after a further 5 min of visible light irradiation. | ||
UV-Vis spectra of F1′, F2′ and F3′ (Fig. S8 in ESI†) show large absorption peaks for the trans isomers of azobenzene even after UV irradiation. There were no changes upon UV/Vis irradiation for F1′, which is also in clear contrast to pendant-type hybrids.16 The absorption peak of F1′ at 330 nm was obviously blue shifted in comparison to the ethanol solution of 1 (Fig. 2), as well as to the F2′ and F3′ films. This suggested an enhanced H-aggregation of azobenzene moieties.32,33 In F1′, the closer packing of azobenzene groups and the more rigid siloxane networks should hinder photo-isomerization. In contrast, F2′ and F3′ films showed slightly reversible trans–cis isomerizations after UV/Vis irradiation, likely due to the lower cross-linking degree of siloxane networks. However, these changes were smaller than those observed for F2 and F3, suggesting that solid-state reactions gave siloxane networks which limited the mobility of azobenzene moieties.
4 Conclusions
We have demonstrated the formation of lamellar hybrid materials from azobenzene-bridged ethoxysilane precursors. Three types of photo-responsive precursors differing in the number of ethoxy groups were used. The triethoxysilylated precursor, which generates the largest number of SiOH groups, had a strong tendency to form self-assembled, lamellar structures. By employing an evaporation induced self-assembly process or a solid-state reaction process, ordered lamellar structures were also prepared from di- and mono-alkoxysilyl precursors. The photo-responsive properties of the hybrid films were influenced by cross-linking degree of siloxane networks: trans–cis isomerization of azobenzene moieties was improved by decreasing of Si–O–Si bonds via utilizing precursors with less condensable ethoxy groups. Further structural design of such self-assembled hybrid materials will lead to the creation of novel photo-responsive materials.Acknowledgements
The authors thank Mr Masashi Yoshikawa (Waseda University) for his help in the ESI-MS analysis. This work was supported in part by a Grant-in-Aid for Scientific Research (C) from Japan Society for the Promotion of Science (JSPS) and by a Grant-in-Aid for Scientific Research on Innovative Areas ‘New Polymeric Materials Based on Element-Blocks (no. 2401)’ provided by The Ministry of Education, Culture, Sports, Science and Technology, Japan. S.G. acknowledges the financial support offered by the Monbukagakusho Scholarship and by the Global Centre for Excellence for Mechanical Systems Innovation (GMSI, The University of Tokyo).Notes and references
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed.
- (a) A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563 RSC; (b) A. Chemtob, L. Ni, C. Croutxé-Barghorn and B. Boury, Chem.–Eur. J., 2014, 20, 1790 CrossRef PubMed.
- W. Chaikittisilp, M. Kubo, T. Moteki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2011, 133, 13832 CrossRef CAS PubMed.
- K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26, 211 CrossRef CAS.
- A. Shimojima, Y. Sugahara and K. Kuroda, Bull. Chem. Soc. Jpn., 1997, 70, 2847 CrossRef CAS.
- L. D. Carlos, V. D. Bermudez, V. S. Amaral, S. C. Nunes, N. J. O. Silva, R. A. S. Ferreira, J. Rocha, C. V. Santilli and D. Ostrovskii, Adv. Mater., 2007, 19, 341 CrossRef CAS.
- H. Peng, J. Tang, J. Pang, D. Chen, L. Yang, H. S. Ashbaugh, C. J. Brinker, Z. Yang and Y. Lu, J. Am. Chem. Soc., 2005, 127, 12782 CrossRef CAS PubMed.
- J. J. Moreau, L. Vellutini, M. Wong Chi Man, C. Bied, J.-L. Bantignies, P. Dieudonne and J.-L. Sauvajol, J. Am. Chem. Soc., 2001, 123, 7957 CrossRef CAS.
- J. J. E. Moreau, B. P. Pichon, M. Wong Chi Man, C. Bied, H. Pritzkow, J.-L. Bantignies, P. Dieudonne and J.-L. Sauvajol, Angew. Chem., Int. Ed., 2004, 43, 203 CrossRef CAS PubMed.
- J. J. E. Moreau, B. P. Pichon, G. Arrachart, M. Wong Chi Man and C. Bied, New J. Chem., 2005, 29, 653 RSC.
- J. J. E. Moreau, L. Vellutini, M. Wong Chi Man, C. Bied, P. Dieudonne, J.-L. Bantignies and J.-L. Sauvajol, Chem.–Eur. J., 2005, 11, 1527 CrossRef CAS PubMed.
- F. Ben, B. Boury and R. J. P. Corriu, Adv. Mater., 2002, 14, 1081 CrossRef CAS.
- N. Liu, K. Yu, B. Smarsly, D. R. Dunphy, Y.-B. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2002, 124, 14540 CrossRef CAS PubMed.
- M. Alvaro, M. Benitez, D. Das, H. Garcia and E. Peris, Chem. Mater., 2005, 17, 4958 CrossRef CAS.
- E. Besson, A. Mehdi, D. A. Lerner, C. Reye and R. J. P. Corriu, J. Mater. Chem., 2005, 15, 803 RSC.
- S. Guo, A. Sugawara-Narutaki, T. Okubo and A. Shimojima, J. Mater. Chem. C, 2013, 1, 6989 RSC.
- M.-H. Li, P. Keller, B. Li, X. Wang and M. Brunet, Adv. Mater., 2003, 15, 569 CrossRef CAS.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201 CrossRef CAS.
- T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796 RSC.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, R. Miyasato, Y. Naka, J. Mamiya, M. Kinoshita, A. Shishido, Y. Yu, C. J. Barrett and T. Ikeda, J. Mater. Chem., 2009, 19, 60 RSC.
- C. Kordel, C. Popeney and R. Haag, Chem. Commun., 2011, 47, 6584 RSC.
- S. S. Nobre, X. Cattoen, R. A. S. Ferreira, C. Carcel, V. Z. Bermudez, M. Wong Chi Man and L. D. Carlos, Chem. Mater., 2010, 22, 3599 CrossRef CAS.
- B. Menaa, M. Takahashi, P. Innocenzi and T. Yoko, Chem. Mater., 2007, 19, 1946 CrossRef CAS.
- X. Zhou, S. Yang, C. Yu, Z. Li, X. Yan, Y. Cao and D. Zhao, Chem.–Eur. J., 2006, 12, 8484 CrossRef CAS PubMed.
- G. Cerveau, S. Chappellet, R. J. P. Corriu, B. Dabiens and J. L. Bideau, Organometallics, 2002, 21, 1560 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- L. Nicole, C. Boissiere, D. Grosso, A. Quach and C. Sanchez, J. Mater. Chem., 2005, 15, 3598 RSC.
- A. Shimojima and K. Kuroda, Chem. Rec., 2006, 6, 53 CrossRef CAS PubMed.
- H. Muramatsu, R. Corriu and B. Boury, J. Am. Chem. Soc., 2003, 125, 854 CrossRef CAS PubMed.
- B. Boury, F. Ben and R. J. P. Corriu, Angew. Chem., Int. Ed., 2001, 40, 2853 CrossRef CAS.
- Z. T. Nagy, B. Heinrich, D. Guillon, J. Tomczyk, J. Stumpe and B. Donnio, J. Mater. Chem., 2012, 22, 18614 RSC.
- C. L. Yeung, S. Charlesworth, P. Iqbal, J. Bowen, J. A. Preeceb and P. M. Mendes, Phys. Chem. Chem. Phys., 2013, 15, 11014 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S8 and Scheme S1. See DOI: 10.1039/c4ra01709a |
| This journal is © The Royal Society of Chemistry 2014 |