Highly dispersed Cu(II), Co(II) and Ni(II) catalysts covalently immobilized on imine-modified silica for cyclohexane oxidation with hydrogen peroxide†
R. Antonya,
S. Theodore David Manickam*a,
Pratap Kollub,
P. V. Chandrasekarc,
K. Karuppasamya and
S. Balakumara
aCentre for Scientific and Applied Research, PSN College of Engineering and Technology, Tirunelveli-627 152, Tamil Nadu, India. E-mail: s.theodore.david@gmail.com
bDST-INSPIRE Faculty, Department of Metallurgical Engineering and Materials Science, Indian Institute of Technology Bombay, Mumbai-400076, India
cCollege of Physics and Information Engineering, Institute of Optoelectronic Display, Fuzhou University, Fuzhou-350002, PR China
First published on 7th May 2014
Abstract
This paper describes the synthesis of Cu(II), Co(II) and Ni(II) catalysts immobilized on imine-functionalized silica gel through a 3-aminopropyltriethoxysilane linker. The synthesized catalysts were characterized by spectroscopic techniques, namely EDS, FTIR, UV-Vis, 29Si MAS NMR, powder XRD and ESR spectroscopy. These analytical methods evidently confirmed the formation of silica-supported catalysts. Thermal properties of catalysts were studied between 30 and 800 °C by thermogravimetric-differential thermal gravimetric (TG-DTG) analysis. The surface roughness of the silica gel was increased upon modification but without losing its lumpy shape, as evidenced by SEM investigation. Magnified SEM and AFM images both suggested the high dispersive nature of the catalysts. Cyclohexane was successfully converted into cyclohexanol and cyclohexanone by the catalysts with the aid of hydrogen peroxide (oxidant). Comparatively, Cu(II) catalyst exhibited better cyclohexane conversion than the other two catalysts. The reusable nature of the catalysts was established by performing five consecutive catalytic runs with Cu(II) catalyst. Comparatively, the present reported catalytic systems were simple, reusable and effective models for higher cyclohexane conversion with better product selectivity.
1. Introduction
The oxidation of organic substrates containing inert C–H bonds is one of the desirable and challenging organic transformations1,2 for researchers. Specifically, the oxidation of C–H bonds in cyclohexane is an important area in synthetic chemistry as there is an upward attention on the use of its products, i.e. cyclohexanol and cyclohexanone (also known as KA oil). For example, its products are employed as starting materials for the manufacture of adipic acid, polyamide-6, acidulant in baking powder and caprolactam. Appreciably, caprolactam is an important precursor in the manufacture of commercial products such as nylon-6 and nylon-6,6′ (ref. 3 and 4). The current industrial procedure involves a temperature of 150 °C and pressure of 1–2 MPa over metal cobalt salt or metal-boric acid catalyst for just a 4% conversion of cyclohexane into cyclohexanol and cyclohexanone. Besides, this commercial technique yields more cyclohexanol, and some additional steps are required to get a better cyclohexanol/cyclohexanone ratio.5,6 Because of these limitations, the search of new catalysts for the effective oxidation of cyclohexane has become imperative. In this exploration, heterogeneous catalysts are often preferred over homogeneous catalysts due to easy catalyst separation, excellent reusability and chemical and thermal stability.7,8 These outstanding qualities establish heterogeneous catalysts as environmentally benign catalysts. Different types of heterogeneous catalysts have been attained for cyclohexane oxidation, and a few instances include Co(II) and Cr(VI) on poly(4-vinylpyridine-co-divinylbenzene) (4.64% conversion);9 scorpionate Fe(III) complex on carbon materials (20.8% conversion)10 and desilicated MOR zeolite (37.2% conversion)11 in the presence of co-catalyst and co-oxidant and gold nanoparticles on carbon materials (3.6% conversion).12 In spite of these examples, silica gel is a more desirable support than aforementioned supports due to its low price, excellent stabilities, good accessibilities as well as the fact that organic groups can be robustly anchored onto the surface of silica gels.13,14 With these salient features, silica-supported catalysts have exhibited superior cyclohexane conversion. A few examples are listed here. Urus et al. achieved a 99.99% cyclohexane conversion with silica-supported complexes. However, they employed microwave irradiation and attained very poor cyclohexanol and cyclohexanone selectivity.15 Mesoporous silica materials such as MCM-41 (45.5% conversion with 51.9% cyclohexanol and 48.1% cyclohexanone selectivity)16 and SBA-15 (71.1% conversion with 26% cyclohexanol and 74% cyclohexanone selectivity)17 have also been synthesised in attempts to attain better cyclohexane conversions. However, the synthesis of mesoporous silica materials involves tedious procedures, more chemicals and a relatively high cost. In this circumstance, aforesaid disadvantages exclude the use of such silica-supported catalysts. Therefore, it is important to find simple, inexpensive and effective silica-supported catalysts for oxidation of cyclohexane with enhanced selectivity.In the drive towards the preparation of heterogeneous catalysts using silica materials, various approaches have been followed. Among them, the incorporation of an organic entity (flexible spacer) onto silica is considered the best. The major benefit in anchoring organic groups onto silica is the easier formation of the catalytic centre.18,19 The method of organo-modification of inorganic materials is usually divided into two classes. In class I, organic and inorganic components are embedded, and only hydrogen bonds, van der Waals forces or ionic bonds provide cohesion to the whole structure. On the other hand, in class II, the two phases (organic and inorganic) are linked together through strong covalent or coordinative bonds.20–22 The class II type is more favoured than class I, since it affords additional chemical strength to the organo-modified inorganic materials. Furthermore, the covalent immobilization of metal complexes to solid support offers high catalytic efficiency and better recyclability without the inherent problems such as leaching of complex/ligand during the reaction.23,24 Silica-based materials have been frequently organo-modified by the class II method using 3-aminopropyltriethoxysilane (APTES) as the linker between silica materials and the organic entity.22,25,26 This linker may go through the formation of Schiff base ligands on the route to organo-modification of silica. The added applications along with the formation of Schiff bases are their chelating ability with transition metal ions, stability under a variety of oxidative and reductive conditions and borderline nature between hard and soft Lewis bases. Additionally, when Schiff base ligands are administered as their metal complexes, the range of their applications are enhanced.27,28 Isatin is the most reported endogenous indole due to its reaction with primary amines for the preparation of Schiff bases.29,30
Based on the facts above, we aspire to accomplish new recyclable silica-supported catalytic systems with sufficient catalytic ability and selectivity under environmentally tolerable conditions for cyclohexane oxidation. New silica-supported Cu(II), Co(II) and Ni(II) catalysts (catalysts 1, 2 and 3, respectively), derived through 3-amino modification and imine functionalization of silica gel, were synthesised and applied in catalytic cyclohexane oxidation using hydrogen peroxide as the oxidant. The reason for using hydrogen peroxide is its environmental benign character as it decomposes to give only water and oxygen. Thus, the use of hydrogen peroxide is a safe and promising approach for a variety of oxidation reactions.31 This work also comprises the characterization of catalysts by EDS, FT-IR, UV-Vis., 29Si NMR, powder XRD and ESR; surface studies by SEM and AFM; and thermal study by TG-DTG.
2. Experimental
2.1. Materials
3-Aminopropyl triethoxysilane (APTES) was procured from Himedia. Silica gel, metal salts, isatin, cyclohexane, acetonitrile, toluene and hydrogen peroxide were purchased from E-Merck. All the chemicals and solvents were employed without further purification.2.2. Synthesis of amino-modified silica gel (SiO2–NH2)
Pre-treatment of silica gel was done by refluxing 50 g silica gel with an excess amount of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrochloric acid for 6 h. It was filtered off and washed with a sufficient amount of deionised water until the pH became 7. The resultant product was dried at 120 °C for 12 h. Then, 20 g pre-treated silica was taken in 100 ml toluene followed by the addition of 20 ml APTES, and the resultant mixture was refluxed for 72 h. The final suspension was filtered off, washed with an excess amount of toluene, ethanol and diethyl ether and dried at 100 °C under vacuum.
:1 hydrochloric acid for 6 h. It was filtered off and washed with a sufficient amount of deionised water until the pH became 7. The resultant product was dried at 120 °C for 12 h. Then, 20 g pre-treated silica was taken in 100 ml toluene followed by the addition of 20 ml APTES, and the resultant mixture was refluxed for 72 h. The final suspension was filtered off, washed with an excess amount of toluene, ethanol and diethyl ether and dried at 100 °C under vacuum.
2.3. Synthesis of imine-modified silica gel (L)
For the preparation of L, 5 mmol isatin and 5 g SiO2–NH2 were taken in 100 ml deionised water. The ensuing mixture was then magnetically stirred at 60 °C for 24 h in the reaction flask. The brown solid product was filtered, washed repeatedly with water and dried at 100 °C.2.4. Synthesis of catalysts
2 g L and 50 ml deionised water were mixed in a reaction flask for the synthesis of catalyst 1. To this mixture, 5 mmol copper chloride was added and then magnetically stirred at ambient temperature for 24 h. The product was filtered and washed thoroughly with water and dried at 90 °C under vacuum. Likewise, catalysts 2 and 3 were synthesised from their corresponding metal chlorides. The schematic overview for the synthesis of catalysts is shown in Fig. 1.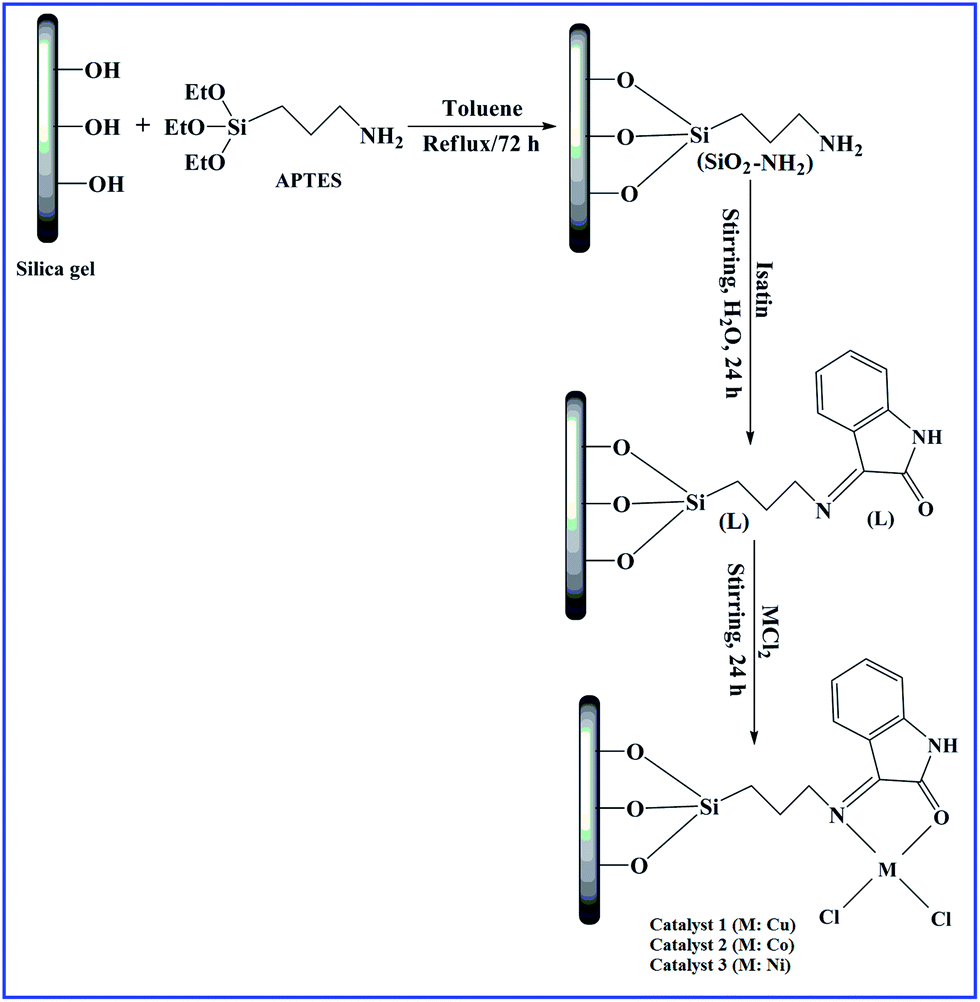 | ||
| Fig. 1 Schematic of the synthetic route of imine-modified silica gel (L) and catalysts. | ||
2.5. Characterization
FTIR spectra were recorded on a Jasco FTIR 4100 spectrophotometer from KBr pellets containing 1% of the sample in 4000–400 cm−1 wave number region with 2 cm−1 resolution. Electronic spectral studies were performed on a Shimadzu UV-2600 spectrophotometer in 200–800 nm wavelength range using the diffuse reflectance (DR) technique. 29Si cross-polarized magic-angle spinning nuclear magnetic resonance (29Si CP MAS NMR) spectral analysis of L and catalyst 1 was conducted on a Mercury Plus 300 MHz NMR spectrometer (VARIAN, USA) equipped with a 5 mm dual broad band probe. Thermal properties of the compounds were investigated by the TG-DTG technique using a Mettler Toledo star system under dynamic N2 atmosphere within the 30–800 °C temperature range at a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) determination was executed on an X-ray diffractometer (XPERT PRO PANalytical, Netherland) for phase identification. The patterns were run with CuKα radiation with a secondary monochromator (λ = 0.1545 nm) at 40 kV and 30 mA. The morphologies, dimensions and elemental compositions of catalysts were explored using a scanning electron microscope of the Hitachi S-3400N model attached with an energy dispersive spectroscope. SEM images were taken at 15 kV accelerating voltage with 2k× magnification range under liquid N2 atmosphere. AFM analysis was accomplished on catalyst 1 using AFM XE 70, Park Systems for the study on particle distribution.2.6. Oxidation of cyclohexane
Oxidation reactions were carried out in a 25 ml flask equipped with a magnetic stirrer. The flask was charged with 0.05 g of the catalyst (1/2/3) in 10 ml acetonitrile. Then, 10 mmol of a 30% H2O2 solution and 5 mmol of cyclohexane were added successively. This reaction mixture was stirred in the reaction flask at 70 °C under atmospheric pressure conditions for 12 h. Aliquots from the reaction mixture were taken every 2 h for product analysis to examine the effect of reaction time on cyclohexane conversion percentage. Two other separate catalytic experiments (one without catalyst and another without oxidant) were also performed using a similar reaction procedure. The product samples were investigated using a Hewlett–Packard gas chromatograph (HP 6890) equipped with an FID detector, a capillary column (HP-5), and a programmed oven with a temperature range from 50 to 200 °C and N2 carrier gas at a flow rate of 0.5 cm3 min−1. The conversion percentage of cyclohexane and selectivity for cyclohexanol and/or cyclohexanone were calculated using the equations given below:| Conversion% of cyclohexane = 100 × [initial% − final%]/initial% |
| Selectivity (%) = 100 × [GC peak area% of cyclohexanol and/or cyclohexanone]/∑peak area of total products. |
3. Results and discussion
3.1. Characterization of catalysts
The presence of expected elements in the respective prepared compounds was confirmed by EDS analysis. The EDS spectra of silica gel, SiO2–NH2, L and catalysts (1, 2 and 3) are illustrated in ESI.† The formation of catalysts was identified by the peaks indicative of their corresponding metal ions in their EDS spectra.The FTIR spectra of silica gel, SiO2–NH2 and L are compared, as shown in Fig. 2. In the FTIR spectrum of silica gel (Fig. 2a), the bands at 3548 (broad) and 1632 cm−1 are assigned to the stretching and bending vibrations of silanol groups (Si–OH), respectively. The bands found at 1094 and 798 cm−1 are the characteristic anti-symmetric and symmetric stretching modes (Si–O–Si) of [SiO4] units, respectively. A small shoulder observed at 974 cm−1 is allocated to the Si–OH stretching mode, and the band at 462 cm−1 is assigned to Si–O–Si bending vibrations.32,33 As shown in Fig. 2b, the appearance of a new continuing band in the region of 3290–3000 cm−1, characteristic of the stretching vibration of the –NH2 group, confirms the aminopropylation of the silica support. It is further supported by the stretching and bending vibration modes of aliphatic CH2 groups appearing at 2938 and 1495 cm−1, respectively. The NH2 bending vibration of SiO2–NH2 emerges at 1572 cm−1. In the FTIR spectrum of L (Fig. 2c), the stretching and bending vibrations of –NH2 group disappear because of imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) group formation by the reaction of isatin with SiO2–NH2. The occurrence of this reaction is also proved by the presence of new characteristic bands at 1714 and 1629 cm−1, which are peaks indicative of the stretching modes of CO and CN groups, respectively. In addition to these changes, a new band is found at 1467 cm−1 caused by an aromatic –CC– stretch. The anticipated amide –NH stretch is masked by the broad Si–OH stretching vibration. In the FTIR spectra of catalysts, it is expected that the coordination of the nitrogen centre to the metal ion would reduce the electron density in the imine link and shift the CN stretching frequency to lower wave numbers. In Fig. 3, the downward shift in CN stretch is found in FT-IR spectra of all catalysts, which implies the successful coordination of azomethine nitrogen to the metal centre.34 Furthermore, the CO group of L is also expected to coordinate with the metal ion through its oxygen atom, and this is confirmed by the shift in its stretching frequency to a lower region. The stretching bands for M–N and M–O could be superimposed with the sharp Si–O–Si bending vibration modes noted at 460 cm−1. Besides the appearance of characteristic bands of the silica support in the FTIR spectra of SiO2–NH2, L and all catalysts show that the fundamental structure of the parent silica support is not disturbed even after organo-modification and catalyst formation.
N) group formation by the reaction of isatin with SiO2–NH2. The occurrence of this reaction is also proved by the presence of new characteristic bands at 1714 and 1629 cm−1, which are peaks indicative of the stretching modes of CO and CN groups, respectively. In addition to these changes, a new band is found at 1467 cm−1 caused by an aromatic –CC– stretch. The anticipated amide –NH stretch is masked by the broad Si–OH stretching vibration. In the FTIR spectra of catalysts, it is expected that the coordination of the nitrogen centre to the metal ion would reduce the electron density in the imine link and shift the CN stretching frequency to lower wave numbers. In Fig. 3, the downward shift in CN stretch is found in FT-IR spectra of all catalysts, which implies the successful coordination of azomethine nitrogen to the metal centre.34 Furthermore, the CO group of L is also expected to coordinate with the metal ion through its oxygen atom, and this is confirmed by the shift in its stretching frequency to a lower region. The stretching bands for M–N and M–O could be superimposed with the sharp Si–O–Si bending vibration modes noted at 460 cm−1. Besides the appearance of characteristic bands of the silica support in the FTIR spectra of SiO2–NH2, L and all catalysts show that the fundamental structure of the parent silica support is not disturbed even after organo-modification and catalyst formation.
 | ||
| Fig. 2 FTIR spectra of silica gel (a), amino-modified silica gel, SiO2–NH2 (b) and imine-modified silica gel, L (c). | ||
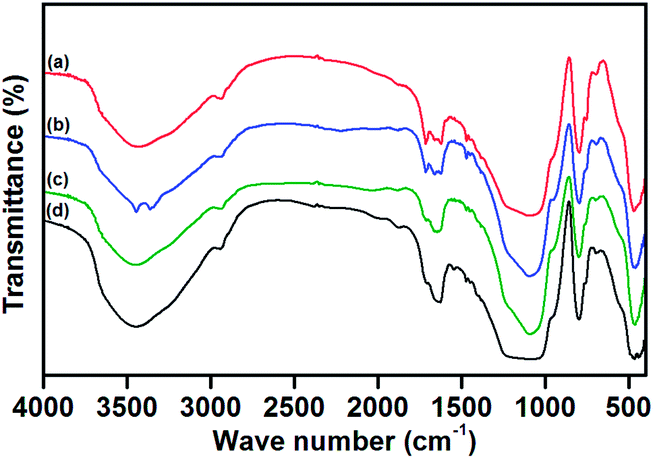 | ||
| Fig. 3 FTIR spectra of imine-modified silica gel, L (a) and catalysts 1 (b), 2 (c) and 3 (d). | ||
UV-Vis spectroscopy is the best method to assign the geometry of the metal complexes around the metal centre. The DR UV-Vis spectra of L and catalysts are illustrated in Fig. 4, while the same for silica gel and SiO2–NH2 are presented in supplementary file (ESI). In general, two types of electronic transitions are expected (π–π* and n–π* transitions) for an aromatic ring containing Schiff base ligands. The UV-Vis spectrum of L (Fig. 4a) displays two such characteristic absorption bands below 400 nm. The band at 220 nm is due to a π–π* transition (characteristic of π-bonds of aromatic ring, CN and CO groups). Another band at 340 nm is assigned to a n–π* transition, which was characteristic of non-bonded electrons available on nitrogen (CN) and oxygen (CO).
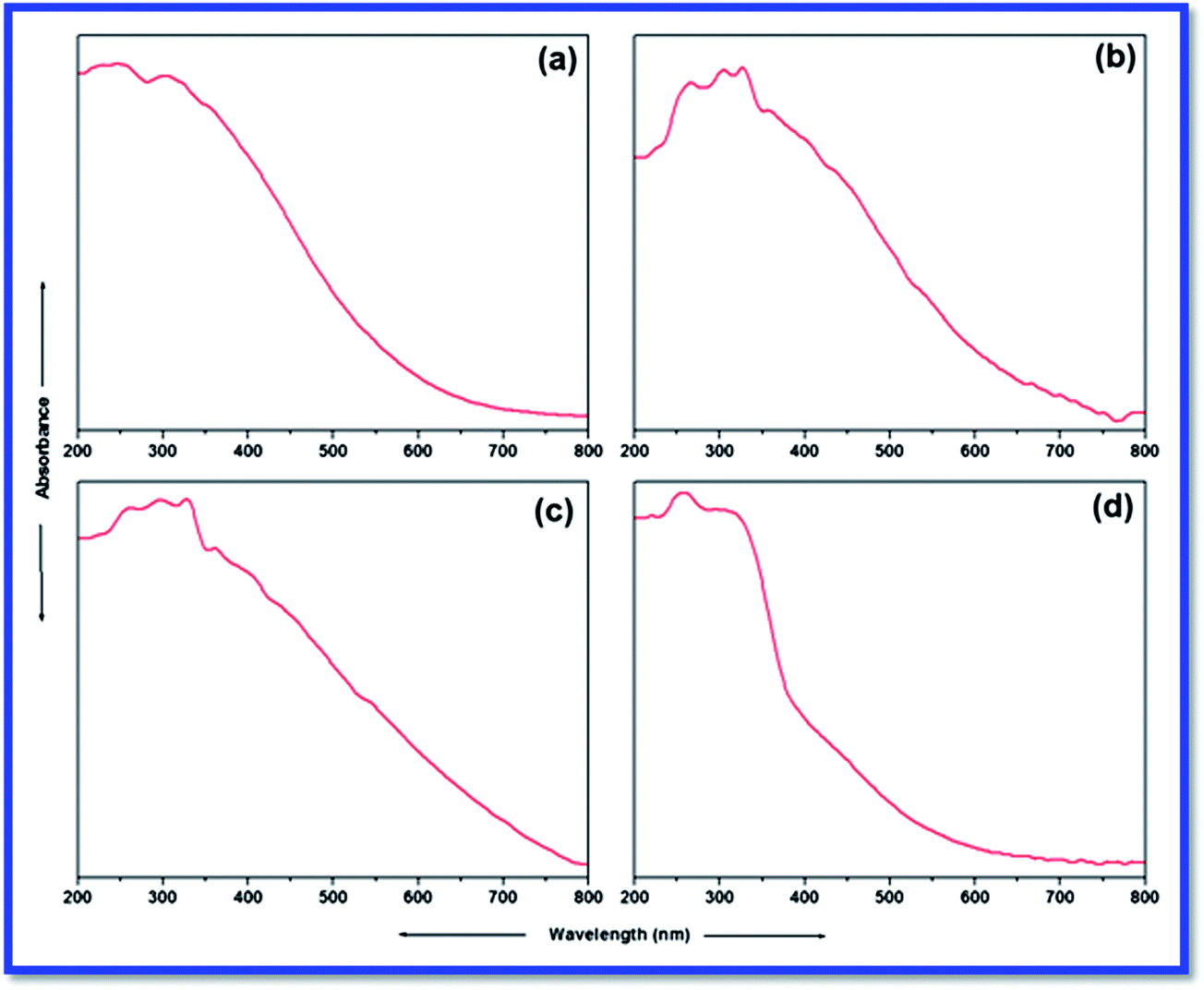 | ||
| Fig. 4 DR UV-Vis spectra of imine-modified silica gel, L (a) and catalysts 1 (b), 2 (c) and 3 (d). | ||
The UV-Vis spectra of catalysts 1 and 2 reveal a similar pattern; however they are found to be different from that of the catalyst 3. As found in the UV-Vis spectrum of L, characteristic bands for π–π* and n–π* transitions at around 220 and 340 nm, respectively, are also detected in the UV-Vis spectra of catalysts. A new band at 450 nm is attributed to the ligand to metal charge transfer (LMCT). This suggests the coordination of L with the metal ions. This coordination behaviour of L is further endorsed by the appearance of a new significant d–d transition band of the metal ions. Catalyst 1 shows a broad band centred at 600 nm, which could be attributed to a d–d transition of Cu(II) (Fig. 4b). The d–d transitions of very close energies may overlap and lead to the emergence of a very broad single d–d band. Similarly, catalyst 2 shows d–d transition as a broad band centred at 600 nm (Fig. 4c). For catalyst 3, this characteristic d–d band is detected at 520 nm (Fig. 4d), and it is not broad like those of catalysts 1 and 2. The appearance of this characteristic d–d transition band in the UV-Vis spectra of all catalysts confirms the FTIR investigation in assuring the formation of catalysts and suggests approximate square planar geometry.35–37
The changes in the silicon environment of silica gel after modification is studied by the 29Si CP MAS NMR technique. For the unmodified silica gel, the anticipated characteristic peaks regularly appear at ∼110, ∼105 and ∼90 ppm, which are allocated to ((SiO)4Si) silica sites, ((SiO)3SiOH) terminal silanol sites and ((SiO)2Si(OH)2) geminal silanol groups, respectively. Among them, the dominant peak at 110 ppm is due to the abundance of ((SiO)4Si) silica sites.38–41 Fig. 5a and b show 29Si NMR spectra of L and catalyst 1, respectively. In these spectra, the intense peak at 109 ppm corresponds to ((SiO)4Si) silica sites. In contrast, the other two peaks are less intense. Importantly, in the 29Si NMR spectra of both L and catalyst 1, an additional broad peak is seen at ∼60 ppm, caused by the formation of new ((SiO)3SiC) sites.42 These observations are indications of the covalent modification of the silica gel backbone through the reaction between the terminal and geminal surface silanol groups. Although these peaks are found in 29Si NMR spectra of both L and catalyst 1, their position and intensity are slightly different, which may be due to the coordination of Cu(II) with L.
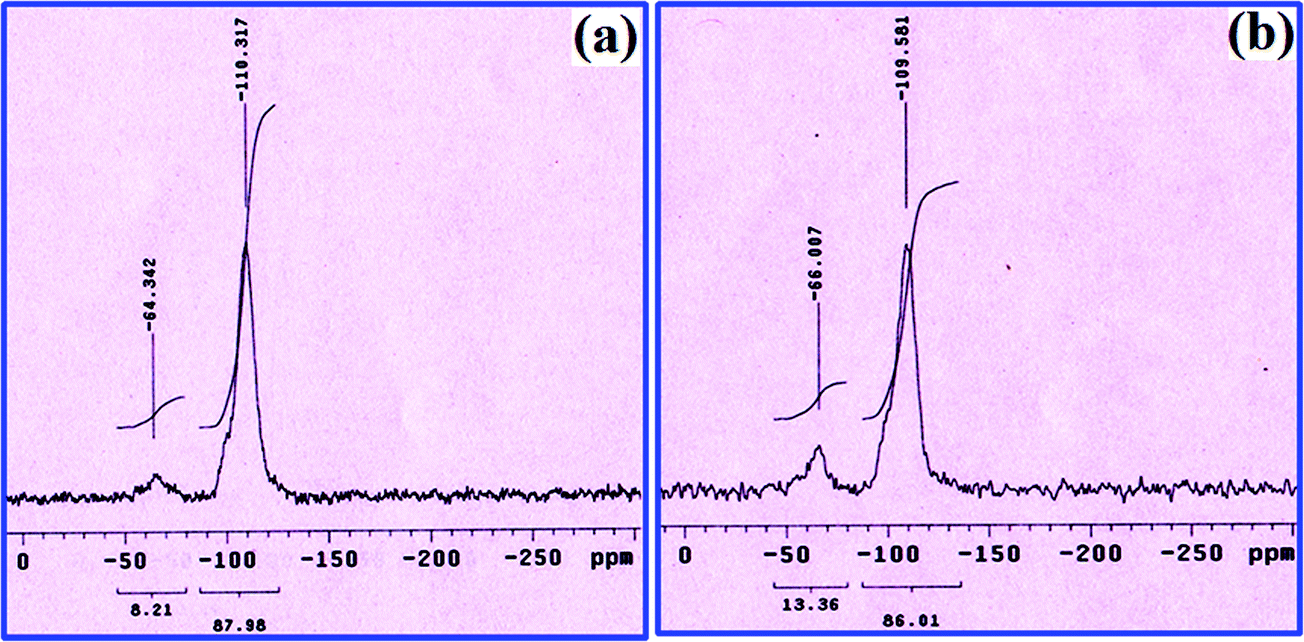 | ||
| Fig. 5 29Si CP MAS NMR spectra of imine-modified silica gel, L (a) and catalyst 1 (b). | ||
The wide angle PXRD patterns of the unmodified silica gel, SiO2–NH2 and all catalysts are shown in Fig. 6. The obtained patterns of all the compounds display a similar broad band centred at 22° as the substantiation to amorphous nature and topological structure of the SiO2 support.38,43,44 On comparison to the unmodified silica gel (Fig. 6a), the intensity of this characteristic peak in PXRD patterns of all modified silica gels, is found to decrease along with line broadening. This decrease in intensity is probably caused by the filling of pores of the silica surface by metal ions or a reduction in X-ray scattering contrast between the channel wall of the silicate framework and L. Furthermore, no new peaks are acquired after amino modification, imine modification and catalyst formation. It can be said that the amorphous nature of silica is maintained even after modification reactions. However, this amorphous nature is not maintained in the case of catalyst 1 as evidenced by the new peaks appearing in Fig. 6d, which means that the crystallinity of L is enhanced after metalation with Cu(II) ions.
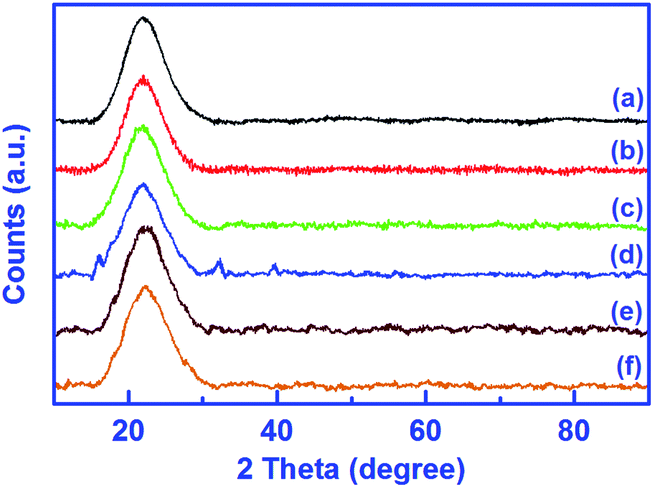 | ||
| Fig. 6 XRD patterns of silica gel (a), amino-modified silica gel, SiO2–NH2 (b), imine-modified silica gel, L (c) and catalysts 1 (d), 2 (e) and 3 (f). | ||
ESR spectra of catalysts 1 and 2 (given in Fig. 7a and b, respectively) were recorded at 77 K to verify the geometry around central metal ions of the catalysts. The resultant spectra are typical for the geometry of axial nature, and the detected g factors are in the order of g‖ > g⊥ > 2.0023 for both catalysts. In the case of catalyst 1, this order indicates that the unpaired electron most probably dwells in the dx2 – dy2 orbital and it is likely to be square planar geometry as suggested by UV-Vis spectroscopy.45 The g‖ value of less than 2.3 is a clear indication of the covalent character of Cu–L bond as suggested by Kivelson and Neiman.46 The exchange coupling factor (G) is expressed by the subsequent equation:
| G = (g‖ − 2)/(g⊥ − 2) |
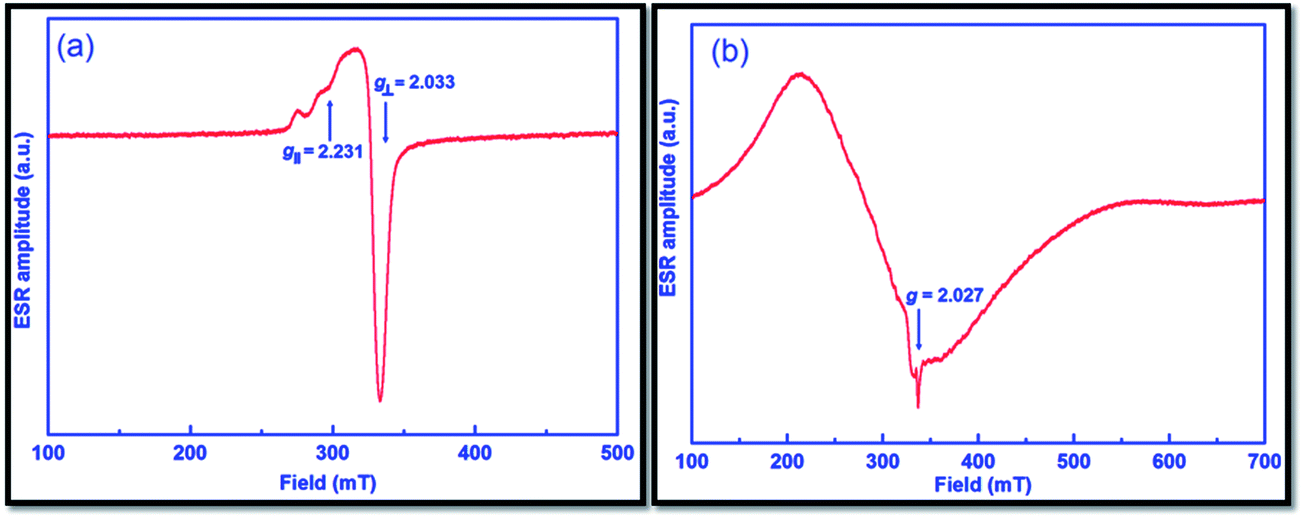 | ||
| Fig. 7 ESR spectra of catalyst 1 (a) and catalyst 2 (b). | ||
As reported earlier, the G value for the parallel-aligned or slightly misaligned local axes is greater than 4.0, whereas it is less than 4.0 for appreciably misaligned local axes with considerable exchange coupling. From the ESR spectrum of catalyst 1, the calculated G value is 7.0, and hence implied negligible exchange coupling.47,48 On the other hand, catalyst 2 displays a very broad ESR spectrum, perhaps due to differences in the relaxation parameters and the spin–spin interaction between Co(II) ions.49
Fig. 8 depicts the TG-DTG patterns of L and all the catalysts studied. In Fig. 8a, L exhibits three mass loss stages, corresponding to the desorption of physically adsorbed water molecules (0–110 °C), decay of organic contents (160–370 °C) and dehydroxylation of surface silanol groups (370–700 °C). Similar thermal decomposition steps are also observed in TG-DTG curves of all catalysts (Fig. 8b–d). However, the calculated% weight loss values are not comparable as in the case of L, and most probably, it is due to the coordination of metal ions with L. Contrary to L, metal complexes exhibit a new weight gain step above 600 °C. This is characteristic of the formation of metal oxides through oxidation reactions. The TG-DTG studies also support the formation of catalysts from L. The details of weight loss and weight gain values are shown separately as a table in ESI.†
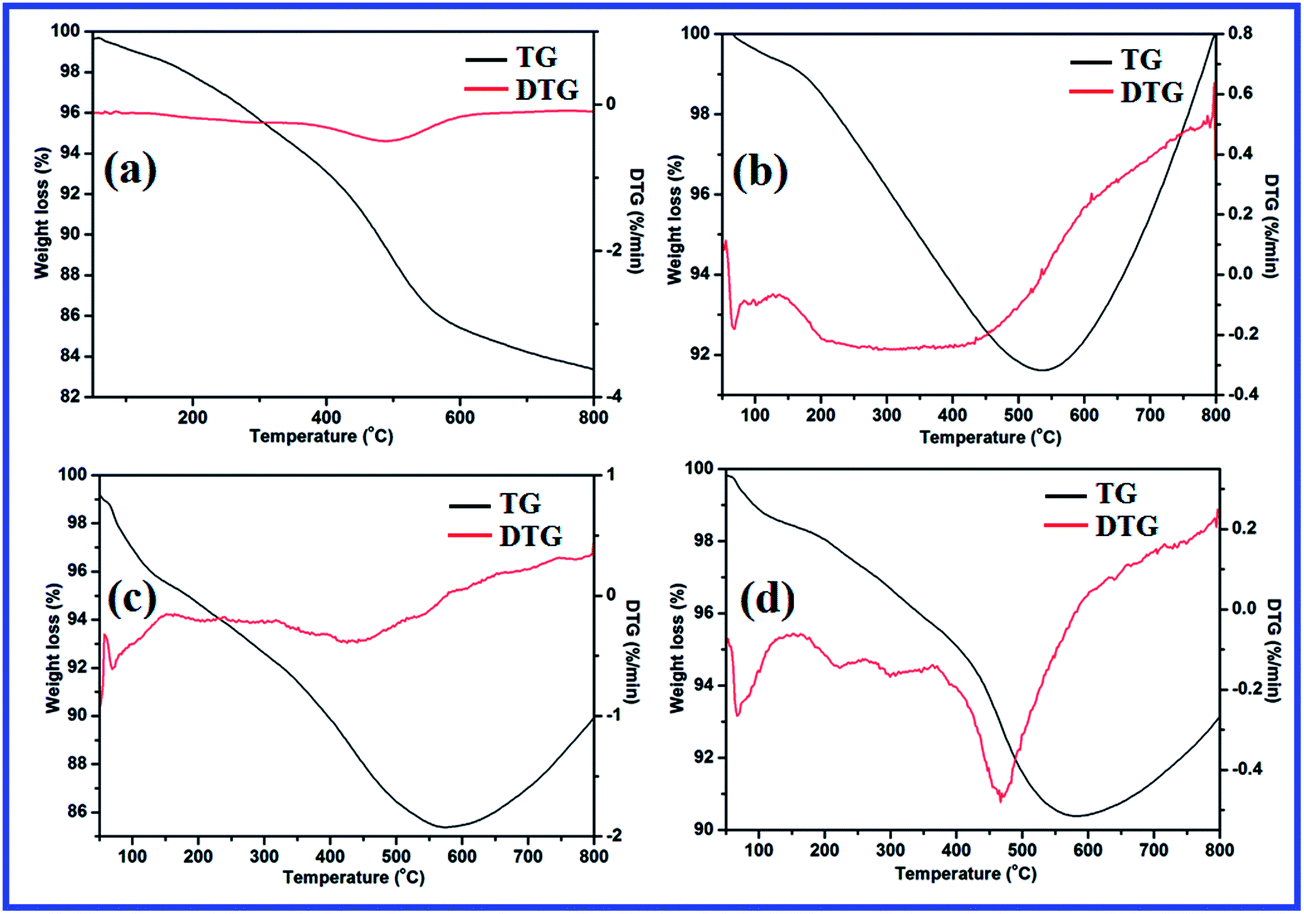 | ||
| Fig. 8 TG-DTG patterns of imine-modified silica gel, L (a) and catalysts 1 (b), 2 (c) and 3 (d). | ||
In order to find out the difference between the size and surface morphology of SiO2–NH2 and its modified forms (L and catalysts), SEM study was employed. The SEM photographs are given in Fig. 9. For comparison, an SEM photograph of the unmodified silica gel is given in supplementary file (ESI). Silica gel displays a clear surface, and there is no evidence of the presence of any adsorbed particles on its surface. After modification with APTES, the surface shows the presence of adsorbed particles as seen in the SEM image of SiO2–NH2 (Fig. 9a). The number of particles on the surface is found to be enhanced upon organo-modification of SiO2–NH2 with isatin for the formation of L. This noteworthy change in surface (Fig. 9b) further confirms the formation of L. In general, the modification of porous silica gel with organic functionalities may assist in the effective adsorption of metal ions.50,51 In fact, as evidenced by the SEM images of catalysts, the metal ions are adsorbed via complex formation throughout the surface of L in a dispersed manner. To confirm the dispersion of metal ions, a magnified SEM image of catalyst 1 is also illustrated in Fig. 9f. This uniform scattering of metal ions on L probably provides a greater surface area for better catalytic activity of catalysts. Throughout the modification process, silica gel particles do not lose their lumpy shape.
 | ||
| Fig. 9 SEM photographs of amino-modified silica gel, SiO2–NH2 (a), imine-modified silica gel, L (b) and catalysts 1 (c), 2 (d), 3 (e) and magnified SEM image of catalyst 1 (f). | ||
Fig. 10 demonstrates the 2D and 3D AFM images of catalyst 1 in two different magnifications for the further investigation of surface properties, and particularly particle dispersion. From the AFM images, it is obvious that the particles are found to emerge as cone-shaped heaps. They are not agglomerated and are well strewn from each other. These observations supplement surface information obtained from the SEM images.
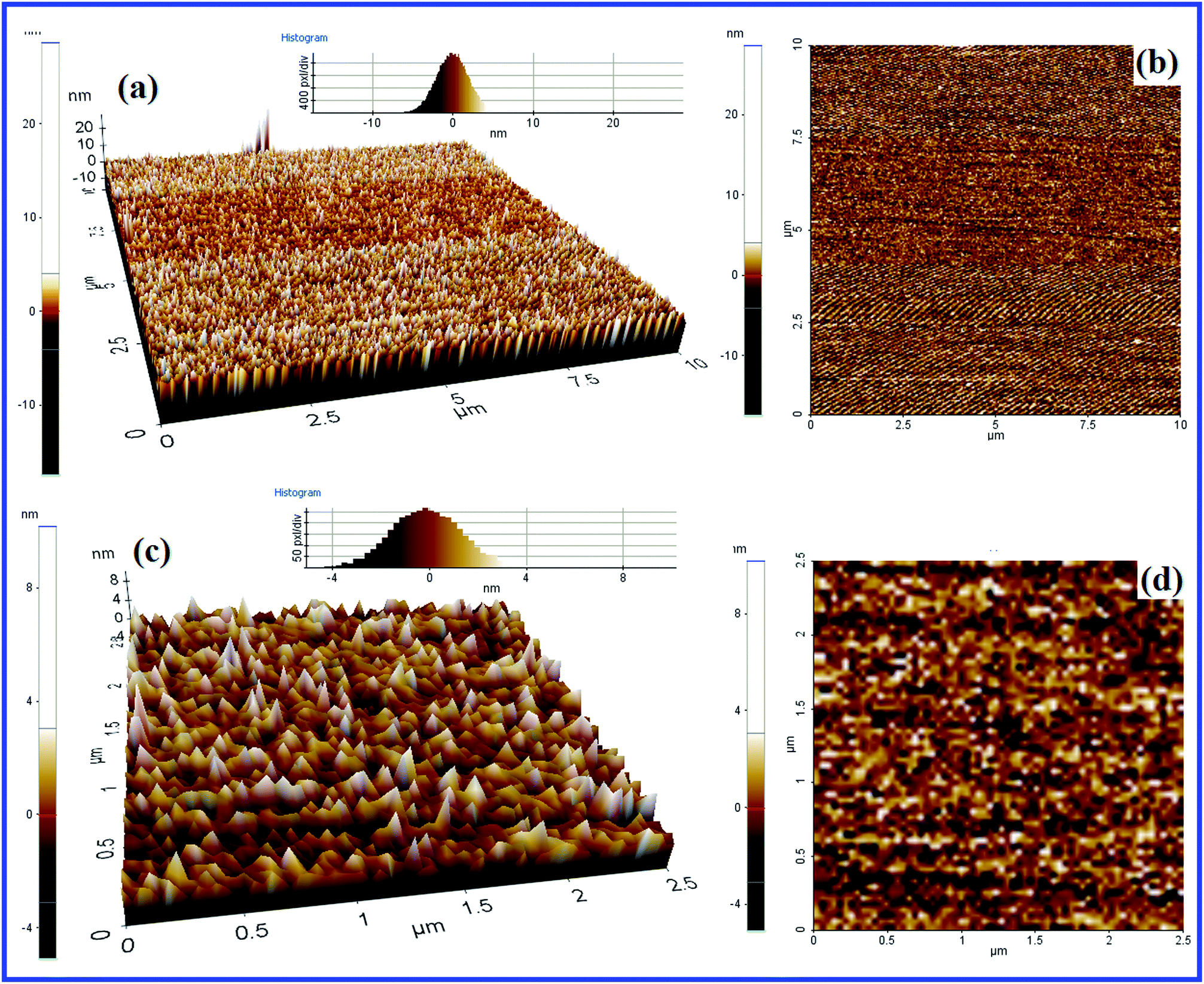 | ||
| Fig. 10 2D (a) and 3D (b) AFM images (10 × 10 μm); 2D (c) and 3D (d) AFM images (2.5 × 2.5 μm) of catalyst 1. | ||
3.2. Catalytic study
Owing to the commercial importance of cyclohexane oxidation, it has been selected as the model reaction in the study of the catalytic properties of catalysts 1, 2 and 3. As it leaves only water as the by-product, H2O2 was employed as the oxidant. According to Corma et al., the role of solvent in catalytic reactions is sometimes crucial as it merges different phases into a uniform one and thus upholds mass transportation. Solvents can affect intermediate species, which further alter reaction mechanisms and also take part in the tuning of surface properties of the catalysts and reaction pathways.52 By comparison, acetonitrile was found to be a more effective solvent than others as evidenced by the literature.17,53 Hence, in this study, the catalytic cyclohexane oxidation was performed in acetonitrile.Catalyst 1/2/3-catalysed oxidation of cyclohexane mainly yields cyclohexanone and cyclohexanol. There is no evidence of other products from gas chromatograph analysis. After the completion of catalytic reactions, acid–base titration was carried out on the product mixture to check if the acidic product, adipic acid, was formed. On cooling the product mixture, no white precipitate was developed. This observation and acid–base titration results suggested that no adipic acid was formed. A pictorial representation of cyclohexane oxidation to cyclohexanol and cyclohexanone is shown in Fig. 11.
 | ||
| Fig. 11 Schematic of catalytic cyclohexane oxidation into cyclohexanol and cyclohexanone. | ||
 | ||
| Fig. 12 Effect of temperature on cyclohexane oxidation catalysed by Cu(II), Co(II) and Ni(II) catalysts containing imine-modified silica gel, L. | ||
| Catalyst | Time (h) | Conversion (%) | Selectivity (%) | |
|---|---|---|---|---|
| Cyclohexanol | Cyclohexanone | |||
| a Reaction conditions: 5 mmol cyclohexane, 10 mmol 30% H2O2, 0.05 g catalyst, 10 ml CH3CN and 70 °C. | ||||
| 1 | 2 | 8 | 71 | 29 |
| 4 | 16 | 62 | 38 | |
| 6 | 30 | 50 | 50 | |
| 8 | 41 | 41 | 59 | |
| 10 | 46 | 35 | 65 | |
| 12 | 49 | 27 | 73 | |
| 2 | 2 | 6 | 67 | 33 |
| 4 | 13 | 54 | 46 | |
| 6 | 24 | 45 | 55 | |
| 8 | 33 | 31 | 69 | |
| 10 | 39 | 22 | 78 | |
| 12 | 40 | 14 | 86 | |
| 3 | 2 | 5 | 69 | 31 |
| 4 | 11 | 59 | 41 | |
| 6 | 20 | 52 | 48 | |
| 8 | 31 | 40 | 60 | |
| 10 | 35 | 28 | 72 | |
| 12 | 37 | 21 | 79 | |
Catalyst 1 showed a better catalytic efficiency compared to catalysts 2 and 3. In detail, it demonstrated 49% cyclohexane conversion at 12 h, with 27% cyclohexanol and 73% cyclohexanone product selectivity. Catalysts 2 and 3 also revealed comparatively adequate cyclohexane conversion (40 and 37%, respectively). The higher catalytic efficiency of catalyst 1 could be attributed to its effectiveness in decomposing hydrogen peroxide. Although the catalytic efficacy of catalysts 2 and 3 was similar, catalyst 2 yielded better cyclohexanone selectivity (14% cyclohexanol and 86% cyclohexanone). The variation in product selectivity may be due to the nature of the metal ions in the catalysts. The coordinatively unsaturated metal ion species such as Co(II), Ni(II) and Cu(II) present in its corresponding catalysts play a crucial role on promoting catalytic performance. The adequate catalytic activity of all studied catalysts (1, 2 and 3) may be due to their coordination of metal ions with the dispersed imine sites of imine-modified silica gel (L). Two blank experiments were also accomplished separately; one without oxidant and another without catalyst with other conditions unchanged. The product yields from these blank experiment were very meagre. It supported the extensive role of catalysts (metal ion species) in cyclohexane oxidation reaction.
The results of this reusability study are illustrated in Fig. 13. Cyclohexane conversion efficiency of the catalyst was comparable throughout the catalytic cycle. Comprehensively, the catalyst lost only 3% of its original catalytic activity in the fifth catalytic run, and the selectivity of the products was also comparable in each catalytic run. It proves the excellent stability of catalysts covalently immobilized on silica gel. This catalytic study suggests that no copper ion was leached out of the surface of silica gel into the catalytic solution. This implies good stability of the silica-supported complex. Further, the FTIR spectra of the fresh catalyst and the used catalyst were compared, and both of them were found to be similar. These experiments substantiate that the reported catalysts are heterogeneous in nature due to the silica support afforded via the covalent approach.
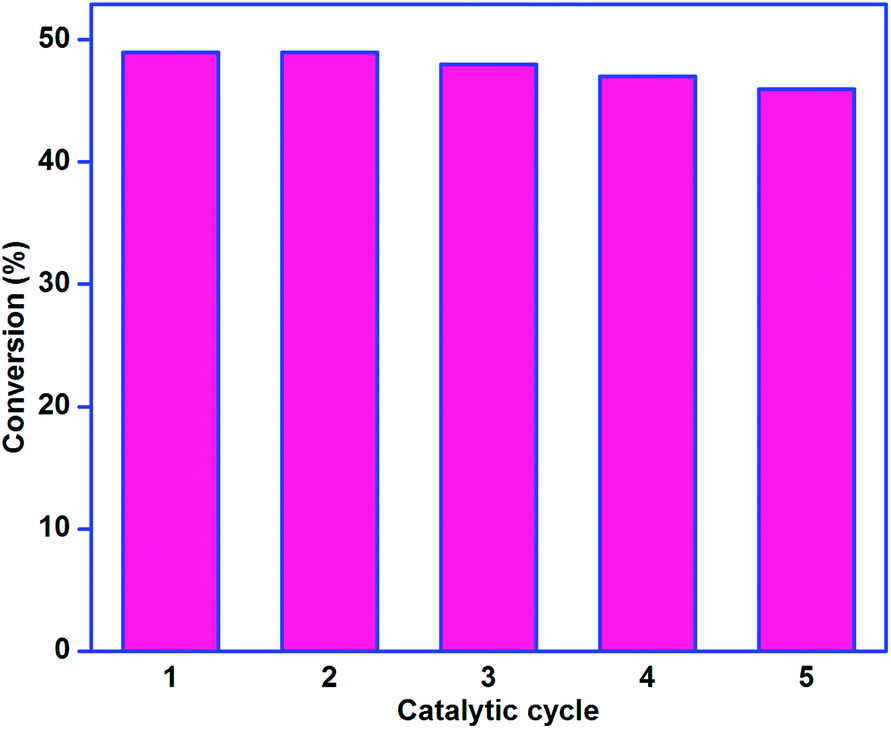 | ||
| Fig. 13 Recyclability of catalyst 1. Reaction conditions: 5 mmol cyclohexane, 10 mmol 30% H2O2, 0.05 g catalyst, 10 ml CH3CN, 70 °C and 12 h. | ||
In comparison, the silica-supported catalysts depict superior catalytic performance in cyclohexane conversion than our previously reported chitosan-supported catalysts.54 This is probably because of the well-dispersed surface of silica-supported catalysts than that of chitosan-supported analogues. Also, these silica-supported catalysts have better recyclability than their corresponding chitosan-supported catalysts.
4. Conclusion
In summary, Cu(II), Co(II) and Ni(II) catalysts derived by the coordination reaction with imine-modified silica gel were highly dispersed as examined by SEM and AFM studies. This highly dispersive nature of reported catalysts compelled us to apply them in cyclohexane oxidation using hydrogen peroxide as the oxidant. The formation of catalysts and the structure of ligand (L) were confirmed by spectral techniques such as EDS, FT-IR, DR UV-Vis, PXRD and 29Si CP MAS NMR. UV-Vis and ESR spectroscopic methods both suggested square planar geometry of the catalysts irrespective of metal ions. The weight gain detected in TG-DTG curves of the catalysts further confirmed the coordination of metal ions to the silica support. Among the catalysts studied, Cu(II) catalyst (catalyst 1) displayed superior catalytic behaviour (49% cyclohexane conversion) than the other two catalysts. However, in terms of selectivity, Co(II) catalyst (catalyst 2) was the better catalyst as it yielded 86% cyclohexanone. Regardless of catalysts, the increase in reaction time led to a higher cyclohexanone selectivity due to the subsequent oxidation of cyclohexanol. The reported covalently bound silica-supported catalysts were stable, simple, inexpensive, easily prepared and are good heterogeneous catalysts as they maintained their original catalytic behaviour in repeated catalytic runs. Therefore, in terms of green chemistry, these catalysts may open a new avenue for cyclohexane oxidation with hydrogen peroxide.Acknowledgements
STDM expresses his gratitude for financial aid (Sanction no: (2010/34/8/BRNS/772)) sanctioned by Department of Atomic Energy-Board of Research in Nuclear Sciences (DAE-BRNS), Government of India, Mumbai to carry out this work. RA thanks DAE-BRNS for the financial support in the form of Junior and Senior Research Fellowships. PK acknowledges Department of Science and Technology (DST) for DST-INSPIRE faculty fellowship and Sophisticated Analytical and Instrumentation Facility, Indian Institute of Technology Bombay, Mumbai, India for helping with characterization. All the authors are deeply grateful to Mr K. Saravanan, CSIR-CSMCRI, Bhavnagar, India for his valuable contribution in discussing the catalytic results.References
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- R. Sheldon, Metal-catalyzed oxidations of organic compounds: mechanistic principles and synthetic methodology including biochemical processes, Elsevier, 2012 Search PubMed.
- R. Zhao, D. Ji, G. Lv, G. Qian, L. Yan, X. Wang and J. Suo, Chem. Commun., 2004, 904–905 RSC.
- N. V. Maksimchuk, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Chem. Commun., 2012, 48, 6812–6814 RSC.
- T. F. Silva, G. S. Mishra, M. F. G. da Silva, R. Wanke, L. M. Martins and A. J. Pombeiro, Dalton Trans., 2009, 9207–9215 RSC.
- R. Whyman, Applied organometallic chemistry and catalysis, Oxford University Press, 2001 Search PubMed.
- C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Adv. Synth. Catal., 2004, 346, 1316–1328 CrossRef.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- D. Lončarević, J. Krstić, J. Dostanić, D. Manojlović, Ž. Čupić and D. Jovanović, Chem. Eng. J., 2010, 157, 181–188 CrossRef PubMed.
- L. Martins, M. P. de Almeida, S. Carabineiro, J. Figueiredo and A. Pombeiro, ChemCatChem, 2013, 5, 3847–3856 CrossRef CAS.
- L. Martins, A. Martins, E. Alegria, A. Carvalho and A. Pombeiro, Appl. Catal., A, 2013, 464, 43–50 CrossRef PubMed.
- S. Carabineiro, L. Martins, M. Avalos-Borja, J. Buijnsters, A. Pombeiro and J. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS PubMed.
- G. Fink, B. Steinmetz, J. Zechlin, C. Przybyla and B. Tesche, Chem. Rev., 2000, 100, 1377–1390 CrossRef CAS PubMed.
- H. Guo, X. Liu, Q. Xie, L. Wang, D.-L. Peng, P. S. Branco and M. B. Gawande, RSC Adv., 2013, 3, 19812–19815 RSC.
- S. Uruş, M. Dolaz and M. Tümer, J. Inorg. Organomet. Polym., 2010, 20, 706–713 CrossRef.
- J. Zhao, W. Wang and Y. Zhang, J. Inorg. Organomet. Polym., 2008, 18, 441–447 CrossRef CAS.
- S. Alavi, H. Hosseini-Monfared and M. Siczek, J. Mol. Catal. A: Chem., 2013, 377, 16–28 CrossRef CAS PubMed.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 386, 321 CrossRef PubMed.
- A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403–1419 CrossRef CAS.
- S. Attia, A. Shames, I. Zilbermann, G. Goobes, E. Maimon and D. Meyerstein, Dalton Trans., 2014, 43, 103–110 RSC.
- C. Freire, C. Pereira and S. Rebelo, Catalysis, 2012, 24, 116 CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- C. E. Song and S.-g. Lee, Chem. Rev., 2002, 102, 3495–3524 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS.
- J. P. Reynhardt, Y. Yang, A. Sayari and H. Alper, Chem. Mater., 2004, 16, 4095–4102 CrossRef CAS.
- A. Schätz, M. Hager and O. Reiser, Adv. Funct. Mater., 2009, 19, 2109–2115 CrossRef.
- A. K. Singh, V. Gupta and B. Gupta, Anal. Chim. Acta, 2007, 585, 171–178 CrossRef CAS PubMed.
- S. Chang, L. Jones, C. Wang, L. M. Henling and R. H. Grubbs, Organometallics, 1998, 17, 3460–3465 CrossRef CAS.
- M. A. Ali, H. J. Hj Abu Bakar, A. Mirza, S. Smith, L. Gahan and P. V. Bernhardt, Polyhedron, 2008, 27, 71–79 CrossRef CAS PubMed.
- G. F. Caramori, R. L. Parreira and A. M. D. C. Ferreira, Int. J. Quantum Chem., 2012, 112, 625–646 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- G. Dıaz, R. Perez-Hernandez, A. Gomez-Cortes, M. Benaissa, R. Mariscal and J. Fierro, J. Catal., 1999, 187, 1–14 CrossRef.
- Z. Huang, F. Cui, H. Kang, J. Chen, X. Zhang and C. Xia, Chem. Mater., 2008, 20, 5090–5099 CrossRef CAS.
- S. Jana, B. Dutta, R. Bera and S. Koner, Langmuir, 2007, 23, 2492–2496 CrossRef CAS PubMed.
- W.-J. Zhou, B. Albela, P. Perriat, M.-Y. He and L. Bonneviot, Langmuir, 2010, 26, 13493–13501 CrossRef CAS PubMed.
- C. Brückner, V. Karunaratne, S. J. Rettig and D. Dolphin, Can. J. Chem., 1996, 74, 2182–2193 CrossRef.
- F. Feigl, Chemistry of specific, selective and sensitive reactions, Academic Press, 1949 Search PubMed.
- P. Yin, Y. Tian, Z. Wang, R. Qu, X. Liu, Q. Xu and Q. Tang, Mater. Chem. Phys., 2011, 129, 168–175 CrossRef CAS PubMed.
- C. Pereira, J. F. Silva, A. M. Pereira, J. P. Araújo, G. Blanco, J. M. Pintado and C. Freire, Catal. Sci. Technol., 2011, 1, 784–793 Search PubMed.
- T. Yokoi, H. Yoshitake, T. Yamada, Y. Kubota and T. Tatsumi, J. Mater. Chem., 2006, 16, 1125–1135 RSC.
- M. A. B. Meador, E. F. Fabrizio, F. Ilhan, A. Dass, G. Zhang, P. Vassilaras, J. C. Johnston and N. Leventis, Chem. Mater., 2005, 17, 1085–1098 CrossRef CAS.
- X. Wang, G. Wu, W. Wei and Y. Sun, Catal. Lett., 2010, 136, 96–105 CrossRef CAS PubMed.
- N. An, W. Zhang, X. Yuan, B. Pan, G. Liu, M. Jia, W. Yan and W. Zhang, Chem. Eng. J., 2013, 215, 1–6 CrossRef PubMed.
- J. Wang, J. Sun, Y. Li and F. Wang, Luminescence, 2014, 29, 188–194 CrossRef CAS PubMed.
- S. Deshpande, D. Srinivas and P. Ratnasamy, J. Catal., 1999, 188, 261–269 CrossRef CAS.
- D. Kivelson and R. Neiman, J. Chem. Phys., 1961, 35, 149–155 CrossRef CAS PubMed.
- B. Hathaway and D. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- R. Dudley and B. Hathaway, J. Chem. Soc. A, 1970, 1725–1728 RSC.
- J. Evans, A. Zaki, M. El-Sheikh and S. El-Safty, J. Phys. Chem. B, 2000, 104, 10271–10281 CrossRef CAS.
- S. Uruş, S. Purtaş, G. Ceyhan and T. Erkenez, Sep. Purif. Technol., 2013, 118, 432–447 CrossRef PubMed.
- S. Uruş, S. Purtaş, G. Ceyhan and F. Tümer, Chem. Eng. J., 2013, 220, 420–430 CrossRef PubMed.
- A. Corma, M. Camblor, P. Esteve, A. Martinez and J. Perezpariente, J. Catal., 1994, 145, 151–158 CrossRef CAS.
- C. K. Modi and P. M. Trivedi, Arabian. J. Chem., 2013 DOI:10.1016/j.arabjc.2013.03.016.
- R. Antony, S. Theodore David, K. Saravanan, K. Karuppasamy and S. Balakumar, Spectrochim. Acta, Part A, 2013, 103, 423–430 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01960a |
| This journal is © The Royal Society of Chemistry 2014 |