Formation of carbon-coated ZnFe2O4 nanowires and their highly reversible lithium storage properties†
Jong Guk Kimab,
Youngmin Kimab,
Yuseong Nohab and
Won Bae Kim*ab
aSchool of Materials Science and Engineering, Gwangju Institute of Science and Technology (GIST), 261 Cheomdan-gwagiro, Buk-gu, Gwangju 500-712, South Korea. E-mail: wbkim@gist.ac.kr; Fax: +82 62 7152304; Tel: +82 62 7152317
bResearch Institute for Solar and Sustainable Energies (RISE), Gwangju Institute of Science and Technology (GIST), 261 Cheomdan-gwagiro, Buk-gu, Gwangju 500-712, South Korea
First published on 13th June 2014
Abstract
In this paper, carbon-decorated ZnFe2O4 nanowires, having one-dimensional geometry with diameters of 70–150 nm and lengths of several micrometers, were prepared and used as a highly reversible lithium ion anode material. They can be obtained from calcination of glucose-coated ZnFe2(C2O4)3 nanowires, which were prepared in glucose containing microemulsion solutions. The physicochemical properties of carbon-coated ZnFe2O4 nanowires were investigated by scanning electron microscopy, X-ray diffraction, Raman spectroscopy, transmission electron microscopy, and X-ray photoelectron spectroscopy. The carbon-coated ZnFe2O4 nanowires showed a substantially increased discharge capacity of ca. 1285.1 mA h g−1 at the first cycle as compared with non-carbon-coated ZnFe2O4 nanowires (ca. 1024.3 mA h g−1) and ZnFe2O4 nanoparticles (ca. 1148.7 mA h g−1). Moreover, the discharge capacity of the carbon-coated ZnFe2O4 nanowires was maintained with no degradation even after 100 charge/discharge cycles. The high cycling durability, rate capability, and coulombic efficiency suggest that the carbon-coated ZnFe2O4 nanowires prepared here can be promising anode candidates for a highly reversible lithium storage electrode.
Introduction
The increasing demand for high performance lithium ion batteries (LIBs) has triggered tremendous research efforts for the development of electrode materials with high lithium storage capacity, long-term cycling stability, and excellent rate capability.1–3 In particular, 3d transition-metal oxides have attracted special attention as an alternative anode material to current commercial graphite due to their high lithium storage capacity, safety and relatively low cost.1–5 Among them, iron oxides have been explored as potential anode materials because of their high specific capacity, low cost, and environmental friendliness.6,7 However, long-term cycling stability of iron oxides still remains a challenge for the practical LIB application due to their poor electronic conductivity and large volume variations during repeated charge/discharge processes, which could lead to eventual cracking of electrode and rapid capacity fade.8–10 Recently, several strategies were proposed to improve the electronic conductivity and cycling stability of iron oxide electrodes. For instance, to minimize Li+ diffusion pathways and mechanical stress during charge/discharge, various nanostructured iron oxides have been proposed, such as urchin-like spheres,6 nanorods,8 nanotubes,9 hollow spheres,11 etc. Especially, one-dimensional (1D) nanostructured iron oxides can offer high specific capacity, improved cycling stability and rate capability, because they can make electron and Li+ transport faster by offering large electrode/electrolyte contact areas and efficient accommodation of strain energy without cracking or crumbling.8,9,12 In addition, to enhance charge transfer rate and buffer the rapid volume changes during charge/discharge, hybridizations with another materials such as amorphous carbon,13 graphene,14 and carbon nanotube15 were also proposed. Among these hybridization methods, carbon decoration technique has been widely adopted for highly stable electrode materials because of its simple synthetic route.13,16Recently, besides simple iron oxides, spinel structured ternary iron oxides, such as CoFe2O4,17–19 CuFe2O4,20,21 NiFe2O4,22,23 and ZnFe2O424–27 have attracted great interest for improving the capacity retention of iron-based oxide. ZnFe2O4 has been proposed as a promising candidate for a LIB anode because of its high theoretical specific capacity (1072 mA h g−1), low toxicity, and abundance.28–32 However, there are still some issues to be solved in ZnFe2O4 materials such as low electronic conductivity, and large volume variations during repeated cyclings.33,34 In this regard, carbon-decorated 1D ZnFe2O4 nanowires (C@ZFO NWs) are prepared for the LIB electrodes, whose configuration could improve the charge transfer rate and cycling reversibility with following features: (i) carbon coated layers and 1D structure could improve the conductivity of the electrode system, which could enhance electron transfer and decrease ohmic losses.35 (ii) Large surface-to-volume ratio of 1D C@ZFO NWs could enable electrolyte permeability to increase for faster Li+ diffusion.36
Herein, we report a facile approach for the synthesis of C@ZFO NWs by a calcination of glucose-coated ZnFe2(C2O4)3 nanowires, which were prepared in glucose containing microemulsion system. To the best of our knowledge, this is the first time that 1D C@ZFO NWs have been applied to the LIB electrodes with highly reversible charge/discharge and high rate capability. The prepared C@ZFO NWs electrode shows an excellent initial discharge capacity (ca. 1285.1 mA h g−1), long-term cycling stability, and high coulombic efficiency, as compared with non-carbon-coated ZnFe2O4 NWs (ZFO NWs) and ZnFe2O4 nanoparticles (ZFO NPs). From their unique architecture and good electrochemical performance, these C@ZFO NWs could be considered as promising electrode materials for next-generation lithium storage platforms.
Experimental
Synthesis of carbon-coated ZnFe2O4 nanowires
For the fabrication of C@ZFO NWs, 1.0 g of cetyltrimethylammonium bromide (≥99%, Aldrich) was dissolved in mixed solvent of 35 mL of cyclohexane (Kanto Chemical) and 1.5 mL of n-pentanol (≥99%, Aldrich), which was stirred for 30 min. After then, 2.0 mL of 1.0 M aqueous solution of oxalic acid (Junsei Chemical) was added into above solution which was stirred additionally for 1 h until it became transparent. The 1.25 mL of an aqueous solution containing 0.05 M of Zn(NO3)2·6H2O (98%, Aldrich) and 0.1 M of FeSO4·7H2O (99.9%, Aldrich) was added to the above solution and was stirred for 6 h. Finally, 2.0 mL of 0.1 M glucose aqueous solution was added and stirred for 6 h. The final product was rinsed thoroughly with absolute acetone to remove residual salts, and subsequently dried in a convection oven at 70 °C. The resulting material was heat-treated in an Ar atmosphere at 450 °C for 2 h with a heating rate of 2 °C min−1.For comparison, non-carbon-coated ZFO NWs were prepared via a same procedure without addition of glucose aqueous solution. The ZnFe2(C2O4)3 NWs and ZFO NWs were obtained with different combinations of Zn:Fe precursors (see Fig. S1 in the ESI†). ZFO NPs were synthesized via a hydrothermal route. In a typical synthesis, 0.22 g of (CH3COO)2Zn·2H2O (≥99%, Aldrich) and 0.35 g of (CH3COO)2Fe (≥99.99%, Aldrich) were dissolved in 50 mL of ethylene glycol. This solution was stirred for 30 min under atmospheric conditions and maintained at 160 °C for 12 h, followed by cooling to room temperature. Obtained material was rinsed thoroughly with distilled water and absolute ethanol and then dried in a convection oven at 70 °C.
Physicochemical characterization and electrochemical measurements
The shapes and morphology of the samples were investigated by using scanning electron microscopy (SEM; JEOL JSM-7500F). The X-ray diffraction (XRD) patterns were recorded with a Rigaku Rotalflex RU-200B diffractometer using a Cu-Kα (λ = 1.5418 Å) source with a Ni filter at 40 kV, 40 mA and a scan rate of 0.02° s−1. Raman spectra (Renishaw Invia) were obtained using 514 nm Ar+ laser excitation with a power of 8.5 mW. The spectra were recorded in wave number range between 2000 and 100 cm−1. The carbon content (wt%) in the obtained C@ZFO NWs was determined by thermogravimetric analysis (TGA; Shimadzu TGA-50) at a heating rate of 10 °C min−1 in air. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) observations coupled with energy-dispersive X-ray spectrometry (EDX) were carried out (Tecnai G2 F30 S-Twin) at 300 kV. The surface area and pore size distribution of the sample were characterized by Brunauer–Emmett–Teller (BET) analysis using a N2 adsorption on an ASAP 2020 (Micromeritics, USA). X-ray photoelectron spectroscopy (XPS; MultiLab 2000) analysis was conducted using a monochromic Al-Kα source (E = 1486.6 eV). Data processing of the XPS measurements was conducted with the XPSPEAK software program.The C@ZFO NWs, carboxylmethyl cellulose binder, and carbon black were mixed in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The prepared slurry was pasted onto a copper foil using a doctor blade method. The specific capacity of C@ZFO NWs was calculated based on their total mass. Prior to the battery cycling tests, the electrode was dried in a vacuum oven overnight at 120 °C. The electrolyte consisted of 1 M LiPF6 in a 1:1 v/v mixture of ethylene carbonate and diethyl carbonate (Cheil Industries). Pure lithium foil was used as counter and reference electrode, and Cellgard 2400 was used as a separator film. The cell (CR2032 coin type) was assembled in an argon-filled glove box in which the moisture and oxygen concentrations were maintained below 1 ppm. The cells were aged for 24 h before the electrochemical measurements, and the cyclic voltammograms (CVs) were recorded at a scan rate of 0.05 mV s−1 from 3.0 to 0.01 V with an AMETEK Solartron Analytical 1400. The assembled cells were also galvanostatically cycled at a current rate of 100 mA g−1 between 0.01 and 3.0 V on a WBCS 3000 battery tester (WonA Tech). Galvanostatic intermittent titration technique (GITT) experiment was performed by charging/discharging the cells for 30 min at a current density of 100 mA g−1 and relaxing for 1 h to reach quasi-equilibrium state and then repeating the process to the end of voltage limit after 10 cycles. The impedance response at the AC amplitude of 5 mV in the frequency range of 100 kHz to 10 mHz was measured using a Solartron 1260 frequency response analyzer after the first cycle.
:10:10. The prepared slurry was pasted onto a copper foil using a doctor blade method. The specific capacity of C@ZFO NWs was calculated based on their total mass. Prior to the battery cycling tests, the electrode was dried in a vacuum oven overnight at 120 °C. The electrolyte consisted of 1 M LiPF6 in a 1:1 v/v mixture of ethylene carbonate and diethyl carbonate (Cheil Industries). Pure lithium foil was used as counter and reference electrode, and Cellgard 2400 was used as a separator film. The cell (CR2032 coin type) was assembled in an argon-filled glove box in which the moisture and oxygen concentrations were maintained below 1 ppm. The cells were aged for 24 h before the electrochemical measurements, and the cyclic voltammograms (CVs) were recorded at a scan rate of 0.05 mV s−1 from 3.0 to 0.01 V with an AMETEK Solartron Analytical 1400. The assembled cells were also galvanostatically cycled at a current rate of 100 mA g−1 between 0.01 and 3.0 V on a WBCS 3000 battery tester (WonA Tech). Galvanostatic intermittent titration technique (GITT) experiment was performed by charging/discharging the cells for 30 min at a current density of 100 mA g−1 and relaxing for 1 h to reach quasi-equilibrium state and then repeating the process to the end of voltage limit after 10 cycles. The impedance response at the AC amplitude of 5 mV in the frequency range of 100 kHz to 10 mHz was measured using a Solartron 1260 frequency response analyzer after the first cycle.
Results and discussion
Fig. 1a shows schematic illustrations of the synthesis procedure for the C@ZFO NWs together with a hypothetical depiction of the Li+ storage process of as-prepared electrode system. In the first step, glucose molecule-coated ZnFe2(C2O4)3 NWs were prepared in the glucose-containing microemulsion solution. In the second step, the carbon layer was formed on the ZFO NWs through a calcination in Ar atmosphere, which allows both phase transformation of zinc iron oxalate and carbonization of glucose molecules for the formation of C@ZFO NWs. The conceptual Li+ storage process in the C@ZFO NWs electrode system is illustrated in Fig. 1a(iii), in which Li+ could migrate readily into the intervals between the C@ZFO NWs networks and then enter into the C@ZFO NWs material, which shortens the Li+ diffusion distance. Electrons could also be transported effectively in this architecture because both carbon layers and lD structure itself could allow easy electron transfer along the NWs axis direction.37–39 Fig. 1b shows a SEM image of the glucose-coated ZnFe2(C2O4)3 NWs, which were used as a precursor for the synthesis of the C@ZFO NWs in this work. | ||
| Fig. 1 (a) Illustration of the formation procedures for the C@ZFO NWs together with schematic description of charging process of as-prepared electrode system; (i) glucose-coated ZnFe2(C2O4)3 NWs, (ii) C@ZFO NWs, and (iii) electrochemical charging process. SEM images of (b) glucose-coated ZnFe2(C2O4)3 NWs and (c) C@ZFO NWs. | ||
The image shows the uniform 1D morphologies without agglomeration of other structures, indicating uniform formation of 1D glucose-coated ZnFe2(C2O4)3 NWs. The inset of Fig. 1b reveals the very smooth surface of glucose-coated ZnFe2(C2O4)3 NWs with diameters of 100–250 nm and lengths of several micrometers. The C@ZFO NWs were prepared through the calcination route at 450 °C in Ar atmosphere, as shown in Fig. 1c. As-prepared C@ZFO NWs maintained their original 1D structure from the precursor NWs after the calcination and had diameters of 70–150 nm and lengths of several micrometers. Note that the dimensions of C@ZFO NWs were reduced as compared to the glucose-coated ZnFe2(C2O4)3 NWs, because of the release of CO2 from ZnFe2(C2O4)3 NWs during the calcination process.40,41 Although the carbon layers cannot be recognized in Fig. 1c, the color change from brown of non-carbon-coated ZFO NWs to light-black of C@ZFO NWs could enable one to deduce the presence of carbon layers formed on the ZFO NWs.42
The crystal structure of the as-prepared materials was examined with XRD measurements in Fig. 2a. The diffraction peaks of glucose-coated ZnFe2(C2O4)3 NWs can be indexed to the standard patterns of zinc iron oxalate (JCPDS 49-1079), where the diffraction peaks at 2θ of 22.7, 24.9, 30.1, 33.7, 47.2, and 50.7° can be ascribed to the (002), (111), (40-2), (11-3), (023), and (312) planes of the zinc iron oxalate, respectively. These peak positions are also consistent with earlier reports.43–46 Note that the coated glucose did not affect the XRD patterns of ZnFe2(C2O4)3 NWs because the diffraction patterns of glucose-coated ZnFe2(C2O4)3 NWs are same as non-glucose-coated ZnFe2(C2O4)3 NWs (see Fig. S2a in the ESI†). After the calcination process, the XRD patterns indicate that the C@ZFO NWs exhibit the spinel phase of ZnFe2O4 from comparison with the standard material (JCPDS 22-1012). The diffraction peaks at 2θ of 30.0, 31.6, 35.3, 42.9, 56.6, 62.6, and 67.8° correspond to the planes (220), (200), (311), (400), (511), (440), and (442), respectively, of the polycrystalline ZnFe2O4 phase. The spinel ZnFe2O4 belongs to space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227) with lattice parameters of a = 8.441 Å.45 The average crystallite size of the C@ZFO NWs was estimated to be approximately 6.8 nm, which can be calculated from Scherrer equation. According to the XRD patterns, the calcination route of glucose-coated ZnFe2(C2O4)3 NWs successfully transformed ZnFe2(C2O4)3 into pure spinel ZnFe2O4 phase, which is further confirmed by Raman, TEM, XPS measurements. This result demonstrates that the crystal structure of ZnFe2O4 was not changed after the carbon decoration on the ZFO NWs through calcination in Ar atmosphere.36 The C@ZFO NWs did not show any XRD peaks from the carbon layers, indicating that the carbon layer could be formed on the ZFO NWs with low crystallinity or low content.42 Moreover, no notable peak shifts or intensity variations induced by secondary phases or impurities were detected, indicating high purity of the final products. Fig. 2b presents the Raman spectra of the C@ZFO NWs, which shows two strong peaks located at 1367.5 (D-band) and 1591.4 cm−1 (G-band). These peaks confirm definitely the presence of carbon layers on the ZFO NWs. Furthermore, TGA analysis reveals that the carbon content in the C@ZFO NWs is about 2.4 wt% (see Fig. S3a in the ESI†). Because the carbon layer makes it difficult to distinguish the spectrum of the spinel ZFO in C@ZFO NWs owing to the attenuation of the Raman signal, the Raman spectra of ZFO NWs were further recorded at a low wave number range.47,48 Inset of Fig. 2b shows the Raman spectrum of non-carbon-coated ZFO NWs in the 900 to 100 cm−1 wave number range. The mode at above 600 cm−1 corresponds to oxygen motion in tetrahedral AO4 groups, therefore, the mode at 633.8 cm−1 could be of A1g symmetry.49,50 The other low-frequency phonon modes like at 440.9 and 358.2 are due to vibrations of metal ions in octahedral BO6 groups, which are attributed to the symmetric and anti-symmetric bending of oxygen in M–O bond.51,52 These peak positions are in good agreement with those of previously reported ZnFe2O4 materials.49–52
m (227) with lattice parameters of a = 8.441 Å.45 The average crystallite size of the C@ZFO NWs was estimated to be approximately 6.8 nm, which can be calculated from Scherrer equation. According to the XRD patterns, the calcination route of glucose-coated ZnFe2(C2O4)3 NWs successfully transformed ZnFe2(C2O4)3 into pure spinel ZnFe2O4 phase, which is further confirmed by Raman, TEM, XPS measurements. This result demonstrates that the crystal structure of ZnFe2O4 was not changed after the carbon decoration on the ZFO NWs through calcination in Ar atmosphere.36 The C@ZFO NWs did not show any XRD peaks from the carbon layers, indicating that the carbon layer could be formed on the ZFO NWs with low crystallinity or low content.42 Moreover, no notable peak shifts or intensity variations induced by secondary phases or impurities were detected, indicating high purity of the final products. Fig. 2b presents the Raman spectra of the C@ZFO NWs, which shows two strong peaks located at 1367.5 (D-band) and 1591.4 cm−1 (G-band). These peaks confirm definitely the presence of carbon layers on the ZFO NWs. Furthermore, TGA analysis reveals that the carbon content in the C@ZFO NWs is about 2.4 wt% (see Fig. S3a in the ESI†). Because the carbon layer makes it difficult to distinguish the spectrum of the spinel ZFO in C@ZFO NWs owing to the attenuation of the Raman signal, the Raman spectra of ZFO NWs were further recorded at a low wave number range.47,48 Inset of Fig. 2b shows the Raman spectrum of non-carbon-coated ZFO NWs in the 900 to 100 cm−1 wave number range. The mode at above 600 cm−1 corresponds to oxygen motion in tetrahedral AO4 groups, therefore, the mode at 633.8 cm−1 could be of A1g symmetry.49,50 The other low-frequency phonon modes like at 440.9 and 358.2 are due to vibrations of metal ions in octahedral BO6 groups, which are attributed to the symmetric and anti-symmetric bending of oxygen in M–O bond.51,52 These peak positions are in good agreement with those of previously reported ZnFe2O4 materials.49–52
 | ||
| Fig. 2 (a) XRD patterns of the glucose-coated ZnFe2(C2O4)3 NWs and C@ZFO NWs. (b) Raman spectra of the C@ZFO NWs. Inset in panel (b) shows the Raman spectra of ZFO NWs in the 900 to 100 cm−1 wave number range. | ||
The detailed structural properties of the C@ZFO NWs were further examined by TEM analysis. As shown in Fig. 3a, the C@ZFO NWs are uniformly coated with amorphous carbon (approximately 10 nm in thickness), unlike non-carbon-coated ZFO NWs (see Fig. S4a in the ESI†). Furthermore, the image shows that the C@ZFO NWs consisted of many primary ZnFe2O4 nanoparticles with diameters of 5–20 nm which form the 1D NW morphology. From the presence of multiple diffraction rings in the selected area electron diffraction (SAED) pattern of the inset of Fig. 3a, the polycrystalline nature of the C@ZFO NWs was confirmed. These diffraction rings in the SAED correspond to the diffraction peaks of (220), (311), and (400) lattice planes of spinel ZnFe2O4 phases, which is in line with the d-spacing of XRD patterns. Lattice fringes with d spacings of 0.244, 0.254, and 0.49 nm were observed from HRTEM image (Fig. 3b), which is in accordance with the (222), (311), and (111) interplane spacings of the spinel ZnFe2O4 phase, respectively.
 | ||
| Fig. 3 (a) TEM and (b) HRTEM image of the C@ZFO NWs. The inset in panel (a) shows a corresponding SAED pattern. (c) Dark field TEM image with corresponding elemental mappings of C, Zn, Fe, and O for the C@ZFO NWs. | ||
The image shows many tiny holes throughout the NWs, originating from the inter-nanoparticle voids, which could enhance the surface-to-volume ratio and thus ensure a high electrode/electrolyte contact area for efficient Li+ transport.34,35 The 1D C@ZFO NWs show porous structure with a specific surface area of 120.1 m2 g−1 (see Fig. S5 in the ESI†) from the type IV BET isotherms. The average pore size of 7.5 nm was obtained using the Barrett–Joyner–Halenda (BJH) method. The elemental distribution in the C@ZFO NWs was examined by elemental mappings of the electron energy loss spectra. As depicted in Fig. 3c, the elements of C, Zn, Fe, and O are distributed uniformly across the C@ZFO NWs. Well-dispersed C element demonstrates the carbon layers were uniformly decorated onto the ZFO NWs. This carbon layer could improve the electrical connection in the electrode network system, which could improve the rate capability at a relatively high current density.37
To further examine the oxidation state of C, Zn, and Fe elements in the C@ZFO NWs, XPS analysis was performed. The XPS survey spectra of the C@ZFO NWs are depicted in Fig. S3b (ESI†). As shown in Fig. 4a, the C 1s peaks can be resolved into three peak components: main peak at 284.5 eV is assigned to non-oxygenated carbon (C–C), whereas minor peaks at 285.9 and 288.4 eV can be attributed to oxidized carbons such as C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.42,53 In Fig. 4b, the peaks of 1044.4 and 1021.3 eV can be attributed to Zn 2p1/2 and Zn 2p3/2, respectively. The binding energy separation between two peaks is 23.1 eV, which is in line with previous reports.29,54,55 Fig. 4c shows Fe 2p spectrum at the binding energies of 724.0 and 710.4 eV, which can be ascribed to Fe 2p1/2 and Fe 2p3/2, respectively. The binding energy difference between the Fe 2p1/2 and Fe 2p3/2 peaks is 13.6 eV, which is consistent with that of ZnFe2O4 materials.28,29,55 As shown in Fig. 4d, the O 1s peak is deconvoluted into two peaks: one peak at 529.6 eV corresponds to the lattice oxygen in ZnFe2O4, while the other peak at 530.9 eV is ascribed to oxygen in carbonate species on the C@ZFO NWs,37,56 which is in a good agreement with results of Fig. 4a. Consequently, the XPS spectra further demonstrate that the C@ZFO NWs are composed of ZnFe2O4 and carbon in this study. In addition, EDX spectra shown in Fig. S6 (ESI†) also indicate the presence of C, Zn, Fe, and O elements in the sample.
O, respectively.42,53 In Fig. 4b, the peaks of 1044.4 and 1021.3 eV can be attributed to Zn 2p1/2 and Zn 2p3/2, respectively. The binding energy separation between two peaks is 23.1 eV, which is in line with previous reports.29,54,55 Fig. 4c shows Fe 2p spectrum at the binding energies of 724.0 and 710.4 eV, which can be ascribed to Fe 2p1/2 and Fe 2p3/2, respectively. The binding energy difference between the Fe 2p1/2 and Fe 2p3/2 peaks is 13.6 eV, which is consistent with that of ZnFe2O4 materials.28,29,55 As shown in Fig. 4d, the O 1s peak is deconvoluted into two peaks: one peak at 529.6 eV corresponds to the lattice oxygen in ZnFe2O4, while the other peak at 530.9 eV is ascribed to oxygen in carbonate species on the C@ZFO NWs,37,56 which is in a good agreement with results of Fig. 4a. Consequently, the XPS spectra further demonstrate that the C@ZFO NWs are composed of ZnFe2O4 and carbon in this study. In addition, EDX spectra shown in Fig. S6 (ESI†) also indicate the presence of C, Zn, Fe, and O elements in the sample.
 | ||
| Fig. 4 XPS spectra of (a) C 1s, (b) Zn 2p, (c) Fe 2p, and (d) O 1s regions of the C@ZFO NWs. | ||
The electrochemical properties of as-prepared C@ZFO NWs were investigated by CVs, galvanostatic charge/discharge, and GITT measurements. Fig. 5 shows the CV curves for the C@ZFO NWs electrode measured at a scan rate of 0.05 mV s−1 over the potential range of 3.0–0.01 V (versus Li/Li+) whose CV profiles are similar to previously reported ZnFe2O4-based materials.28–30 The following equations represent sequential Li+ storage reactions with the ZnFe2O4.28–30,32–34
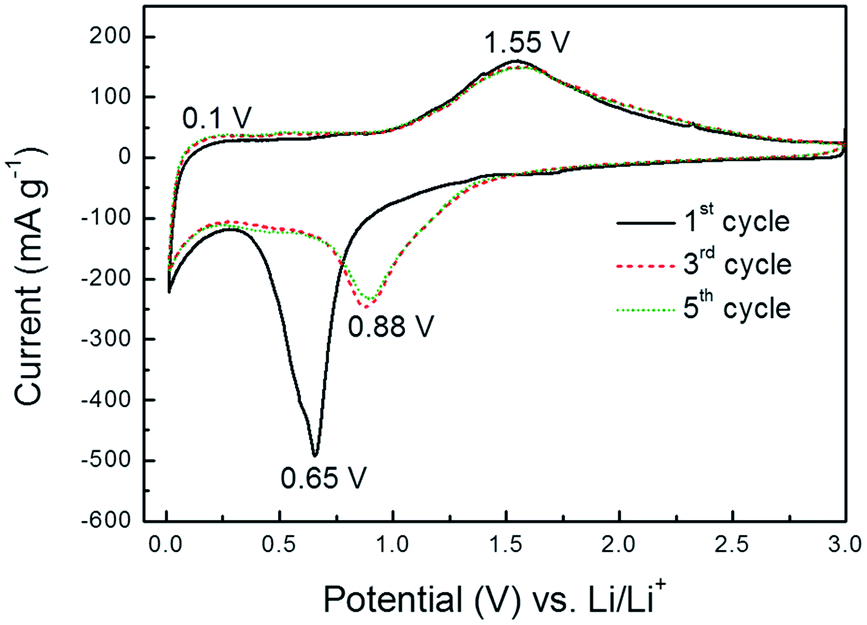 | ||
| Fig. 5 CVs of C@ZFO NWs electrode between 3.0 and 0.01 V at a scan rate of 0.05 mV s−1. | ||
The first discharge:
| ZnFe2O4 + 9Li+ + 9e− → ZnLi + 2Fe + 4Li2O | (1) |
Reversible reactions:
| ZnO + 2Li+ + 2e− ⇄ Zn + Li2O | (2) |
| Fe2O3 + 6Li+ + 6e− ⇄ 2Fe + 3Li2O | (3) |
| Zn + Li+ + e− ⇄ ZnLi | (4) |
As shown in CV profiles, there is a significant difference between the first and subsequent loops, suggesting that a different Li+ storage reaction takes place in the first scan compared with the following scans. In the first cathodic sweep, the CVs show a broad peak at approximately 0.65 V, which could be attributed to the reduction of Zn2+ and Fe3+ to Zn0 and Fe0, respectively, and an irreversible electrolyte decomposition to form solid electrolyte interphase (SEI) layers. Moreover, peaks near 0 V are ascribed to ZnLi alloy formation like eqn (1)28–30 and a Li–C intercalation reaction.37 In the first anodic sweep, the wide oxidation peaks near 0.1 V correspond to the Li+ de-alloying from the ZnLi alloy and de-intercalation from Li–C compound formed in the reductive sweep, according to eqn (4).28–30,37 The peaks at approximately 1.55 V can be attributed to the oxidation of metallic Fe0 and Zn0 to Fe3+ and Zn2+ along with the decomposition of the Li2O, as indicated in eqn (2) and (3).32–34 In subsequent cycles, the cathodic peak shifts from 0.65 to 0.88 V, which could be indicative of a structural rearrangement in the subsequent cycles.57 Following anodic peaks are very close to the first one, suggesting identical electrochemical reactions are involved for the anodic scans. More importantly, there is no notable variation of the oxidation/reduction peaks after the first cycle, indicating high reversibility and structural stability of the electrode reactions.
Fig. 6a shows the voltage profiles of the C@ZFO NWs electrode, which were obtained at a current density of 100 mA g−1 in the potential window of 0.01 to 3.0 V (versus Li/Li+). During the first discharge, the voltage shows a pronounced plateau at approximately 0.8 V arising from the formation of metallic Zn0 and Fe0 in the Li2O matrix and of SEI layers, which is consistent with the earlier CV profiles. Note that the C@ZFO NWs electrode shows a higher initial coulombic efficiency (82.1%) than that of non-carbon-coated ZFO NWs (76.1%) and ZFO NPs (55.6%) (Table S1 in ESI†). Because the outer carbon layer could reduce the side reactions with the electrolyte, the reversibility of the charge/discharge process is further enhanced for the C@ZFO NWs electrode with a high coulombic efficiency.42 Thereafter, the coulombic efficiency of the C@ZFO NWs electrode increases to 94.1% in the second cycle and then is stabilized until the end of the measurement (see Fig. S7a in the ESI†), indicating a high reversibility of the electrode reactions, which is also consistent with CV profiles. Fig. 6b shows the capacity-cycle number curves for the C@ZFO NWs electrode up to 100 cycles at a rate of 100 mA g−1 in a potential range from 0.01 to 3.0 V at room temperature. The initial discharge capacity of the C@ZFO NWs was 1285.1 mA h g−1, and a high discharge capacity of 1292.1 mA h g−1 was maintained even after 100 cycles. Such a high discharge capacity could be ascribed to increased electrical conductivity in the presence of the outer carbon layer, by improving the kinetic properties of the materials. Furthermore, we observed that the C@ZFO NWs maintained their 1D configuration even after 100 charge/discharge cycles (see Fig. S7b in the ESI†), showing high structural durability of C@ZFO NWs. For comparison, we also tested the non-carbon-coated ZFO NWs and ZFO NPs under the identical test conditions. The C@ZFO NWs and ZFO NWs showed higher stability than ZFO NPs, indicating that the 1D NWs structure can alleviate the strain energy during repeated charge/discharge processes, compared with NPs structure. Since theoretical specific capacity of carbon (ca. 372 mA h g−1) is smaller than that of ZnFe2O4 (ca. 1072 mA h g−1), the presence of carbon on the ZFO NWs could reduce the Li+ storage capacity in terms of total mass of C@ZFO NWs compared with non-carbon-coated ZFO NW. Interestingly, however, the discharge capacity of C@ZFO NWs is higher than that of ZFO NWs, which could be explained that a greater quantity of ZnFe2O4 particles could participate in Li+ storage reactions effectively in the C@ZFO NWs electrode system, because the carbon layer could enhance the integration abilities of the each ZnFe2O4 particle or charge transfer through the 1D NWs.36,37,42 Note that the discharge capacity of C@ZFO NWs and ZFO NWs appears to be increased gradually. It has been reported that transition-metal oxides, such as CoO, Co3O4, Fe3O4, and Mn3O4, often show a gradual capacity increase during charge/discharge cycles. It is known that this phenomenon is attributed to the reversible formation/dissolution of a polymeric gel-like film or reversible interfacial Li+ storage reactions.57
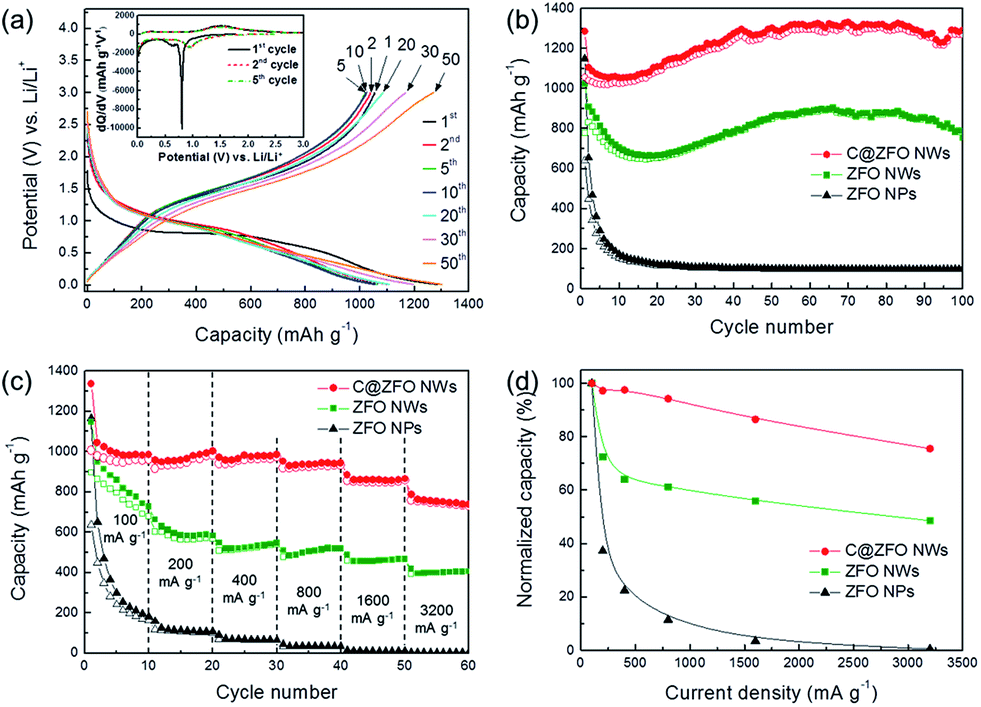 | ||
| Fig. 6 (a) Voltage profiles of the C@ZFO NWs electrode at a current density of 100 mA g−1 between 0.01 and 3.0 V. Inset shows the plot of the differential capacity versus the voltage. (b) Capacity-cycle number curves of C@ZFO NWs, ZFO NWs and ZFO NPs in the potential window of 0.01–3.0 V. (c) Cycling performance of C@ZFO NWs, ZFO NWs and ZFO NPs at various current rates (100, 200, 400, 800, 1600, and 3200 mA g−1) between 0.01 and 3.0 V. (d) Normalized capacity at each step by the average capacity values at a current density of 100 mA g−1 in the first step. | ||
Besides the high specific capacity and long-term cyclability, the good rate capability is also of great importance for high-power applications. We further investigated the rate performance of the C@ZFO NWs electrode at various current densities from 100 up to 3200 mA g−1, as shown in Fig. 6c. The non-carbon-coated ZFO NWs and ZFO NPs electrode were also tested under identical conditions. For the current densities of 100, 200, 400, 800, 1600, and 3200 mA g−1, the C@ZFO NWs delivered average discharge capacities of 997.1, 969.5, 972.4, 939.5, 862.3, and 753.1 mA h g−1, respectively. On the other hand, the ZFO NWs delivered significantly decreased discharge capacities of 829.8, 601.2, 531.6, 508.1, 464.0, and 403.1 mA h g−1, and the ZFO NPs delivered very small discharge capacities of 317.9, 118.4, 71.3, 36.4, 11.2, and 1.5 mA h g−1, respectively. As shown in Fig. 6d, the normalized capacity of the C@ZFO NWs electrode (75.5%) is much higher than that of the ZFO NWs electrode (48.6%) and ZFO NPs electrode (0.5%) at a high current density of 3200 mA g−1 (or 14.7 min per half-cycle). Consequently, as the charge/discharge rate increased, the capacity fade of the C@ZFO NWs was considerably decreased as compared with that of the ZFO NWs and ZFO NPs, indicating good Li+ storage reversibility and structural stability of C@ZFO NWs electrode system even at relatively high current densities.
To understand the reasons for the enhanced rate capability and cycling durability of the C@ZFO NWs with respect to the ZFO NWs electrode, total internal resistance was obtained from the GITT measurements after 10 cycles, as shown in Fig. 7a. The total internal resistance was calculated by the potential difference between closed-circuit voltage and quasi-open-circuit voltage divided by the input current.58–60 The total internal resistance involves interface resistance, electronic resistance, and Li+ transfer resistance, etc.61,62 As shown in Fig. 7b, the total internal resistance decreases during the lithiation, while it increases during the de-lithiation period. During the lithiation period, consecutive resistance decrease is related to volume expansion of C@ZFO NWs by Li+ storage reactions, which leads to an extension of the contact area between C@ZFO NWs and carbon additives. On the other hand, the increase of the internal resistance over the de-lithiation could result from loss of contact between C@ZFO NWs and carbon additives by contraction of the C@ZFO NWs. More importantly, the internal resistance of the C@ZFO NWs electrode is much smaller than that of the ZFO NWs electrode at both the lithiation and de-lithiation periods except the full lithiation regions, as shown in Fig. 7b, because a larger amount of Li2O phase with low conductivity was formed in the C@ZFO NWs electrode compared with ZFO NWs electrode in the low potential regions. This result indicates the loss of conductive network is less serious in the C@ZFO NWs electrode compared to the ZFO NWs electrode. Accordingly, the C@ZFO NWs are able to show enhanced rate performance compared with ZFO NWs at rather high current densities. The enhanced kinetic properties of C@ZFO NWs electrode, as compared to the ZFO NWs electrode, were further elucidated by electrochemical impedance spectroscopy measurements in Fig. S8d (ESI†). Another issue of 3d transition-metal oxide-based electrodes is a high voltage hysteresis during charge/discharge cycles as compared with the intercalation electrodes, which could decrease a round-trip efficiency.63,64 As shown in Fig. S8 (ESI†), the voltage hysteresis decreased from 0.81 V of ZFO NPs and 0.79 V of ZFO NWs to 0.77 V of C@ZFO NWs, which is based on the second charge/discharge profiles at a current density of 100 mA g−1. This reduction of voltage hysteresis is a further indication of the enhanced Li+ transport kinetics in the C@ZFO NWs network electrode system.
 | ||
| Fig. 7 (a) GITT curves of C@ZFO NWs and ZFO NWs electrodes by charging/discharging for 30 min and relaxing for 1 h after 10th cycle. (b) Total internal resistance of C@ZFO NWs and ZFO NWs electrodes obtained from GITT measurements. | ||
The results show that C@ZFO NWs electrode has improved properties like larger lithium storage capacity, higher cyclability, higher coulombic efficiency, and better rate performance, compared to non-carbon-decorated ZFO NWs and ZFO NPs electrode. As depicted in Fig. 1a(iii), this excellent performance of C@ZFO NWs electrode could be attributed to its unique structural features as follows: (i) carbon coated layers could improve the conductivity of the electrode system, which enhances electron transfer and decreases ohmic resistance. (ii) Large surface-to-volume ratio of 1D C@ZFO NWs in electrode system could enable electrolyte permeability to increase for faster Li+ transport. (iii) Porosity characteristics from the polycrystalline 1D NWs also could contribute to the improved electrochemical performance of the electrode, because they facilitate Li+ diffusion during repeated charge/discharge processes.
Conclusions
In conclusion, we have reported on carbon-coated ZnFe2O4 nanowires via a calcination of glucose-coated ZnFe2(C2O4)3 nanowires, which were prepared in glucose containing microemulsion solutions. When the carbon-coated ZnFe2O4 nanowires were used as LIBs anode, they showed a high initial discharge capacity (1285.1 mA h g−1), high initial coulombic efficiency (82.1%), excellent cyclability, and high rate capability compared with non-carbon-coated ZnFe2O4 nanowires and ZnFe2O4 nanoparticles. The advanced lithium storage properties of carbon-coated ZnFe2O4 nanowires are related to their unique structural features such as carbon layers that improve the electron transport and interspacing formation in the nanowire network structure that facilitates Li+ transfer rate. Therefore, the C@ZFO NWs electrode, with high long-term durability and high energy density, could be a promising potential anode system for next-generation LIBs.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2013029776 (Mid-career Researcher Program)) and by the Global Frontier R&D Program (0420-20120126) on Center for Multiscale Energy System through NRF. We also appreciate the financial support by the Core Technology Development Program from the Research Institute for Solar and Sustainable Energies (RISE/GIST).Notes and references
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- M. R. Palacín, Chem. Soc. Rev., 2009, 38, 2565 RSC.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488 CrossRef CAS PubMed.
- S. H. Nam, Y. S. Kim, H.-S. Shim, J. G. Kim and W. B. Kim, Nanoscale Res. Lett., 2011, 6, 292 CrossRef PubMed.
- G. Zhang, H. B. Wu, H. E. Hoster and X. W. Lou, Energy Environ. Sci., 2014, 7, 302 CAS.
- B. Wang, J. S. Chen and X. W. Lou, J. Mater. Chem., 2012, 22, 9466 RSC.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- Z. Xiao, Y. Xia, Z. Ren, Z. Liu, G. Xu, C. Chao, X. Li, G. Shen and G. Han, J. Mater. Chem., 2012, 22, 20566 RSC.
- Z. Wang, D. Luan, S. Madhavi, C. M. Li and X. W. Lou, Chem. Commun., 2011, 47, 8061 RSC.
- S. H. Nam, K. S. Kim, H.-S. Shim, S. H. Lee, G. Y. Jung and W. B. Kim, Nano Lett., 2011, 11, 3656 CrossRef CAS PubMed.
- B. Wang, J. S. Chen, H. B. Wu, Z. Wang and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 17146 CrossRef CAS PubMed.
- B.-S. Lee, S.-B. Son, K.-M. Park, G. Lee, K. H. Oh, S.-H. Lee and W.-R. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 6702 CAS.
- Y. Ma, G. Ji and J. Y. Lee, J. Mater. Chem., 2011, 21, 13009 RSC.
- L. Ji, Z. Tan, T. R. Kuykendall, S. Aloni, S. Xun, E. Lin, V. Battaglia and Y. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 7170 RSC.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252 CAS.
- W. Wang, M. Tian, Y. Wei, S.-H. Lee, Y.-C. Lee and R. Yang, Nano Energy, 2013, 2, 943 CrossRef CAS PubMed.
- H. Xia, D. Zhu, Y. Fu and X. Wang, Electrochim. Acta, 2012, 83, 166 CrossRef CAS PubMed.
- S. Liu, J. Xie, C. Fang, G. Cao, T. Zhu and X. Zhao, J. Mater. Chem., 2012, 22, 19738 RSC.
- Z. Zhang, Y. Wang, M. Zhang, Q. Tan, X. Lv, Z. Zhong and F. Su, J. Mater. Chem. A, 2013, 1, 7444 CAS.
- Z. Xing, Z. Ju, J. Yang, H. Xu and Y. Qian, Electrochim. Acta, 2013, 102, 51 CrossRef CAS PubMed.
- Y. Fu, Q. Chen, M. He, Y. Wan, X. Sun, H. Xia and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 11700 CrossRef CAS.
- P. Lavela and J. L. Tirado, J. Power Sources, 2007, 172, 379 CrossRef CAS PubMed.
- Y. Fu, Y. Wan, H. Xia and X. Wang, J. Power Sources, 2012, 213, 338 CrossRef CAS PubMed.
- Y. Ding, Y. Yang and H. Shao, Electrochim. Acta, 2011, 56, 9433 CrossRef CAS PubMed.
- Y. Deng, Q. Zhang, S. Tang, L. Zhang, S. Deng, Z. Shi and G. Chen, Chem. Commun., 2011, 47, 6828 RSC.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513 CrossRef CAS.
- F. Mueller, D. Bresser, E. Paillard, M. Winter and S. Passerini, J. Power Sources, 2013, 236, 87 CrossRef CAS PubMed.
- W. Song, J. Xie, S. Liu, G. Cao, T. Zhu and X. Zhao, New J. Chem., 2012, 36, 2236 RSC.
- Z. Xing, Z. Ju, J. Yang, H. Xu and Y. Qian, Nano Res., 2012, 5, 477 CrossRef CAS.
- L. Yao, X. Hou, S. Hu, Q. Ru, X. Tang, L. Zhao and D. Sun, J. Solid State Electrochem., 2013, 17, 2055 CrossRef CAS PubMed.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999 RSC.
- X. Guo, X. Lu, X. Fang, Y. Mao, Z. Wang, L. Chen, X. Xu, H. Yang and Y. Liu, Electrochem. Commun., 2010, 12, 847 CrossRef CAS PubMed.
- Y. Sharma, N. Sharma, G. V. S. Rao and B. V. R. Chowdari, Electrochim. Acta, 2008, 53, 2380 CrossRef CAS PubMed.
- J. Sui, C. Zhang, D. Hong, J. Li, Q. Cheng, Z. Li and W. Cai, J. Mater. Chem., 2012, 22, 13674 RSC.
- J. G. Kim, S. H. Nam, S. H. Lee, S. M. Choi and W. B. Kim, ACS Appl. Mater. Interfaces, 2011, 3, 828 CAS.
- M.-H. Ryu, K.-N. Jung, K.-H. Shin, K.-S. Han and S. Yoon, J. Phys. Chem. C, 2013, 117, 8092 CAS.
- Z. Yang, G. Du, Q. Meng, Z. Guo, X. Yu, Z. Chen, T. Guo and R. Zeng, J. Mater. Chem., 2012, 22, 5848 RSC.
- G. Zhang, B. Y. Xia, C. Xiao, L. Yu, X. Wang, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 8643 CrossRef CAS PubMed.
- Z. Yang, G. Du, Q. Meng, Z. Guo, X. Yu, Z. Chen, T. Guo and R. Zeng, RSC Adv., 2011, 1, 1834 RSC.
- Z. Zhang, A. J. Rondinone, J.-X. Ma, J. Shen and S. Dai, Adv. Mater., 2005, 17, 1415 CrossRef CAS.
- N. Du, Y. Xu, H. Zhang, J. Yu, C. Zhai and D. Yang, Inorg. Chem., 2011, 50, 3320 CrossRef CAS PubMed.
- J. G. Kim, S. H. Lee, S. H. Nam, S. M. Choi and W. B. Kim, RSC Adv., 2012, 2, 7829 RSC.
- Z. Jia, D. Ren, Y. Liang and R. Zhu, Mater. Lett., 2011, 65, 3116 CrossRef CAS PubMed.
- M. Wang, Z. Ai and L. Zhang, J. Phys. Chem. C, 2008, 112, 13163 CAS.
- H. Zhu, X. Gu, D. Zuo, Z. Wang, N. Wang and K. Yao, Nanotechnology, 2008, 19, 405503 CrossRef PubMed.
- S. N. Rishikeshi, S. S. Joshi, M. K. Temgire and J. R. Bellare, Dalton Trans., 2013, 42, 5430 RSC.
- E. Markevich, R. Sharabi, O. Haik, V. Borgel, G. Salitra, D. Aurbach, G. Semrau, M. A. Schmidt, N. Schall and C. Stinner, J. Power Sources, 2011, 196, 6433 CrossRef CAS PubMed.
- Z. Jian, L. Zhao, H. Pan, Y.-S. Hu, H. Li, W. Chen and L. Chen, Electrochem. Commun., 2012, 14, 86 CrossRef CAS PubMed.
- Z. Wang, D. Schiferl, Y. Zhao and H. S. C. O'Neill, J. Phys. Chem. Solids, 2003, 64, 2517 CrossRef CAS PubMed.
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210 CrossRef CAS.
- M. Maletin, E. G. Moshopoulou, A. G. Kontos, E. Devlin, A. Delimitis, V. T. Zaspalis, L. Nalbandian and V. V. Srdic, J. Eur. Ceram. Soc., 2007, 27, 4391 CrossRef CAS PubMed.
- H. Xia, Y. Qian, Y. Fu and X. Wang, Solid State Sci., 2013, 17, 67 CrossRef CAS PubMed.
- J.-H. Zhou, Z.-J. Sui, J. Zhu, P. Li, D. Chen, Y.-C. Dai and W.-K. Yuan, Carbon, 2007, 45, 785 CrossRef CAS PubMed.
- R. Dom, R. Subasri, N. Y. Hebalkar, A. S. Chary and P. H. Borse, RSC Adv., 2012, 2, 12782 RSC.
- J. F. Moulder, W. F. Strickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation: Physical Electronics Division, Eden Prairie, MN, 1992 Search PubMed.
- Y. Kim, Y. Noh, E. J. Lim, S. Lee, S. M. Choi and W. B. Kim, J. Mater. Chem. A, 2014, 2, 6976 CAS.
- J. G. Kim, S. H. Lee, Y. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 11321 CAS.
- H. Zhang and P. V. Braun, Nano Lett., 2012, 12, 2778 CrossRef CAS PubMed.
- O. B. Chae, S. Park, J. H. Ku, J. H. Ryu and S. M. Oh, Electrochim. Acta, 2010, 55, 2894 CrossRef CAS PubMed.
- K. Zhong, X. Xia, B. Zhang, H. Li, Z. Wang and L. Chen, J. Power Sources, 2010, 195, 3300 CrossRef CAS PubMed.
- Y. J. Mai, D. Zhang, Y. Q. Qiao, C. D. Gu, X. L. Wang and J. P. Tu, J. Power Sources, 2012, 216, 201 CrossRef CAS PubMed.
- Y. J. Mai, S. J. Shi, D. Zhang, Y. Lu, C. D. Gu and J. P. Tu, J. Power Sources, 2012, 204, 155 CrossRef CAS PubMed.
- Y. Lu, X. Wang, Y. Mai, J. Xiang, H. Zhang, L. Li, C. Gu, J. Tu and S. X. Mao, J. Phys. Chem. C, 2012, 116, 22217 CAS.
- C. Chae, J. H. Kim, J. M. Kim, Y.-K. Sun and J. K. Lee, J. Mater. Chem., 2012, 22, 17870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEM, XRD, TGA, XPS, TEM, BET, EDX and electrochemical data. See DOI: 10.1039/c4ra02095b |
| This journal is © The Royal Society of Chemistry 2014 |