Localized synthesis of gold nanoparticles in anisotropic alginate structures†
Elisa Mele*a,
George C. Anyfantisa,
Despina Fragoulia,
Roberta Ruffillib and
Athanassia Athanassioua
aSmart-Materials Nanophysics, Istituto Italiano di Tecnologia (IIT), Via Morego 30, 16163 Genoa, Italy. E-mail: elisa.mele@iit.it
bNanochemistry, Istituto Italiano di Tecnologia (IIT), Via Morego 30, 16163 Genoa, Italy
First published on 25th April 2014
Abstract
We present a method to create alginate nanocomposite objects having two regions with distinct chemical composition by spatially confining the synthesis of gold nanoparticles (Au NPs) in the polysaccharide matrix. Controlling the floating of sodium alginate drops on calcium chloride solution containing specific concentrations of gold ions, we demonstrate the formation of bicolour structures with a mushroom-like shape. Their immersed portion (the cap) was exclusively involved in the uptake and reduction of gold precursor, acquiring the typical purple colour of Au NPs; whereas, the emerged portion (the stem) did not participate in the in situ synthesis and retained the white colour of calcium alginate. Moreover, we noted that the localized growth of Au NPs was strongly related to the initial amount of gold precursor. In fact for high concentrations, the gold ions were just encapsulated inside the gel matrix and no reduction was observed in the wet structures neither in the cap nor in the stem. The reported procedure is simple and capable of directing the synthesis of Au NPs into selected areas of the alginate matrix, making possible the fabrication of a novel class of alginate structures with an anisotropic character and potential applications in the area of drug delivery and biosensors.
Introduction
Inspired by natural systems, such as bones, horn, nacre in pearls and shells, a promising class of composites based on the incorporation or in situ synthesis of inorganic nanomaterials within biopolymers has been proposed.1,2 These bionanocomposites are typically composed of a combination of fillers, including clays, hydroxyapatite, silica and metal nanoparticles, with naturally occurring materials, like polysaccharides, aliphatic polyesters, polypeptides and proteins, and polynucleic acids. The use of biopolymers instead of synthetic ones offers remarkable advantages in terms of biocompatibility, biodegradability and environmentally friendliness. This makes biohydrids attractive in applications that include innovative biomedical devices, tissue engineering, surgical implants, drug and pesticides delivery, food processing and packaging, water cleaning, automotive and construction products.Among the various natural polymers of interest for the production of bionanocomposites, alginic acid is widely studied for its metal uptake capability, deriving from the role that it plays in nature.3 In fact, this polysaccharide is abundant in brown algae, where it works as biosorbent for removing metal ions from seawater.4 Typically, alginate bionanocomposites are processed in form of films or spherical beads by crosslinking the polysaccharide with multivalent cations, mostly divalent such as Ca2+ or Ba2+. The ion-chelation mechanism involves the α-L-guluronic (G) acid residues that constitute the polysaccharide chain together with the 1,4 linked-β-D-mannuronic (M) ones. In particular, M and G blocks are arranged in a non regular blockwise pattern with homopolymeric regions of M and G interspersed with regions of alternating MG units.5 Divalent cations are coordinated with the oxygen atoms of two consecutive G sequences that belong to different polymer chains. In this way, cavities are created for accommodating the ions in a configuration known as “egg-box”.6–8 The final hydrogel structure, which results from intra-cluster association of alginate dimers, possesses metal sorption capacity due to the existing carboxylic groups and it can be used for metal ion incorporation.
Different groups have profitably exploited this property of alginate to direct the synthesis of nanoparticles, such as gold,9,10 silver,11–13 nickel and cobalt,14 in the gel matrix. E. Torres and coworkers have reported on the growth of gold and silver nanoparticles on dried calcium alginate beads by metal biosorption from aqueous solutions of chloroauric acid (HAuCl4) and silver nitrate (AgNO3).12 Moreover, S. Saha et al. have demonstrated the preparation of Ca-alginate beads embedded with mono or bimetallic Au and Ag clusters by incubating them in HAuCl4 and AgNO3 aqueous solutions and subsequent photochemical reduction of the metal.11,13 V. Jaouen and collaborators have used Au ions as cationic cross-linkers instead of Ca2+, and they have impregnated sodium alginate films in solutions of HAuCl4 followed by Au reduction through glucose.9 R. Brayner et al. have reported on a similar approach by replacing Au ions with Ni2+ and Co2+ in order to control the growth of Co and Ni nanoparticles and CoNi nanoalloys in the polysaccharide matrix. To this aim, they extruded alginate beads in solutions of NiCl2 and CoCl2, followed by thermal reduction at 350 °C.14
Recently, we have presented the fabrication of small objects having a “mushroom”-like shape by exploiting the floating of alginate drops onto the surface of calcium chloride solutions.15 Herein, we demonstrate that this method can be advantageously used to localize the synthesis of colloidal gold in selected regions of these alginate complex structures. By adding specific amounts of gold precursor in the CaCl2 bath, we observed that during the floating state the growth of gold nanoparticles (Au NPs) was confined inside the cap of the mushroom-like structure, without involving the stem. On the contrary, during the drying of the alginate nanocomposites, not-reacted gold ions, entrapped inside the core of the structures, were preferentially released from the stem, which was consequently decorated with gold. Thus, in the dried structures Au NPs were located in the whole inner volume of both the cap and the stem, and selectively on the surface of the latter. In this way, we were able to realize a novel class of alginate bionanocomposites, characterized by anisotropy both in shape and chemistry.
Experimental
Materials
Solutions of sodium alginate at 3 wt% were prepared by mixing alginic acid sodium salt from brown algae (Sigma-Aldrich) and Milli-Q double distilled water (resistivity >18.2 MΩ cm). Solutions containing Ca2+ and Au3+ ions were obtained by adding 0.1 or 1 mg mL−1 of HAuCl4 (Sigma-Aldrich) to a water bath with 0.2 g mL−1 of calcium chloride (CaCl2, Sigma-Aldrich). All chemicals were used without further purification.Characterization of the bionanocomposite structures
Videos of the formation of the asymmetric architectures were recorded by a high speed camera IDT MotionXtra NX4S2, working at a maximum frame rate of 1000 Hz. Photographs of the alginate structures were taken by a Canon EOS 5D (macrolens 100 mm).Absorption spectra (ultraviolet-visible light) were acquired by a Varian spectrophotometer (Cary 300 Scan). The size of the Au NPs was characterized by Jeol Jem 1011 Transmission Electron Microscope (TEM) operating at an accelerating voltage of 100 kV and equipped with a digital camera Gatan Orius 1000. The samples for TEM were prepared by dissolving the alginate structures in a supersaturated aqueous solution of NaCl. A dilute solution of the collected NPs was then dropped on carbon-coated copper grids and the imaging was carried out after the complete evaporation of water.
The surface morphology of dried calcium alginate “mushrooms” was investigated by Scanning Electron Microscopy (SEM), using a variable pressure Jeol JSM-6490LA microscope equipped with a W thermionic electron source. The SEM imaging was performed by both secondary electron (SE) and backscattered electron (BE) signal. The latter was used in conjunction with low vacuum, in order to reveal the compositional differences between the gold NPs and the polymeric matrix. Energy-dispersive spectroscopy (EDS) measurements were carried out by a Jeol JED 2300 Si(Li) detector in the SEM instrument. Before the imaging, the structures were dried in air at room temperature overnight.
Results and discussion
To spatially control the synthesis of Au NPs in the alginate structures, we used a method based on the floating of aqueous drops on liquid surfaces.15 Briefly, drops of sodium alginate solution (3 wt%) with a volume of about 8 μL were generated at the tip of a needle (Fig. 1a) and released into a reservoir containing the CaCl2 solution with 0.1 mg mL−1 of HAuCl4 (Fig. 1b). When the falling height (the distance between the needle tip and the target surface) was smaller than 8 mm, we observed the floating of the drop due to the balance between the gravitational force and the surface tension. As presented in Fig. 1c, 0.1 s after the collision, the drop acquired a hemispherical shape with only one portion immersed in the CaCl2/HAuCl4 bath. The gelation of this immersed part starts occurring immediately after the impact, mainly due to the calcium ions.16 In fact, the binding affinity of alginate for Ca2+ is stronger than for Au3+ and, moreover, in the aqueous bath calcium is three orders of magnitude more concentrated than gold. After the polymerization of the alginate layer in direct contact with the CaCl2/HAuCl4 solution, the reaction front propagated in time and determined the vertical elongation of the emerged part (Fig. 1d). During the formation of the final Ca-alginate architectures in the floating state (8–10 hours), we observed a progressive colour change of the immersed part from white to pink, eventually becoming red-purple; the emerged one instead maintained the white colour typical of alginate. In Fig. 1e and f, photographs of the realized bicolour mushroom-like structures are shown, still in the wet state. It is clearly visible the sharp separation between the immersed (red) and emerged (white) regions, corresponding to the immersion level of the structures in the CaCl2/HAuCl4 bath. The red colour is associated to the growth of colloidal Au NPs into the alginate matrix as consequence of the reduction of gold ions to the elemental state. During the formation of the structures, Au3+ ions diffuse through the cross-linked hydrogel. As known, the carboxylic groups of the polysaccharide participate in the sorption of [AuCl4]− complex.12 The metal uptake is favoured by the high number of protonated carboxylic acid moieties that are present in alginate cross-linked with CaCl2 and act to lower the electrostatic repulsion between COO− and anionic gold species. Then, the reduction of Au3+ to Au0 takes place by the hydroxylic groups that have also the role to stabilize the particles into the calcium alginate matrix.13 Since only the immersed portion of the alginate structure was in direct contact with the CaCl2/HAuCl4 solution, the sorption of gold and the consequent nucleation and growth of nanoparticles was selectively confined in this region. On the contrary, it is also possible to create anisotropic structures without spatial confinement of the Au NPs synthesis (red mushroom in Fig. 1f) by immersing already formed pure alginate objects (white mushroom in Fig. 1f) in an aqueous solution of HAuCl4 (0.1 mg mL−1). In this way, the Au NPs are uniformly present both in the cap and in the stem. | ||
| Fig. 1 (a–d) Time sequence of the formation of one asymmetric alginate structure (scale bar: 1.5 mm). Photographs of (e) one single bicolour mushroom and (f) three different bionanocomposite structures in the wet state with uniform dispersion of gold nanoparticles both in the cap and in the stem (left), of pure alginate without gold (centre), and with gold nanoparticles only in the cap (right). Scale bar: 1 mm. | ||
In order to analyze the size and the shape of the synthesised nanoparticles, we treated the bionanocomposite alginate mushrooms with a supersaturated aqueous solution of NaCl. In this way, we dissolved the calcium alginate matrix and the synthesised Au NPs were characterized. TEM analysis revealed that the nanoparticles were spherical with an average diameter of about 14 nm (Fig. 2a). As shown in Fig. 2b, almost 90% of the population has a size in the range of 7–21 nm. UV-vis absorption analysis of the colloidal solution also confirmed the presence of Au NPs. An intense peak at 526 nm in the absorption spectrum was observed (inset of Fig. 2b), due to the surface plasmon resonance of the Au NPs. The position of the absorption peak is related to the size of the particles, as confirmed by other studies, where it has been demonstrated that colloidal gold nanodots and particles with size of 10–20 nm exhibit the maximum of plasmon absorption at 520–530 nm.10,17,18 The produced gold particles are durably encapsulated within the calcium alginate matrix even after months of storage in pure water.
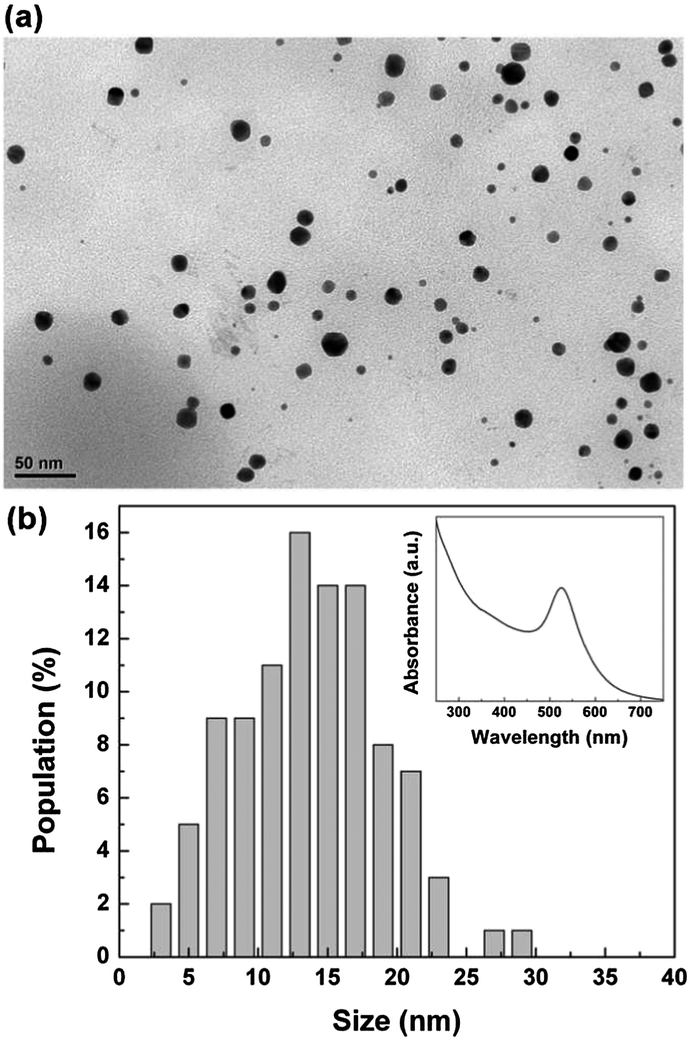 | ||
| Fig. 2 (a) TEM image of Au NPs in situ synthesised in the cap of the alginate structures, and (b) corresponding size distribution analysis. Inset: UV-vis spectrum of the water-based Au NPs dispersion. | ||
After the realized structures were dried over night, their morphology and chemical composition were analysed by SEM and EDS. From Fig. 3 we can note that the surface of the immersed portion was characterized by hierarchical micro- and nano-protrusions (Fig. 3a), whereas the emerged part was smooth and did not present evident features (Fig. 3b). This difference in the roughness was mainly due to the long exposure to chloride ions that were especially aggressive towards the hydrogel.12 This determined the degradation of the external superficial layers and the consequent increment of the roughness. On the contrary, the gelation of the stem mainly occurred when it was in air, due to the diffusion of Ca2+ ions and the propagation of the reaction front from the inner to the outer region of the alginate structure.15
 | ||
| Fig. 3 Secondary electron SEM images showing the morphology of the dried alginate structures for an area of (a) the cap and (b) the stem. The sample was coated with 10 nm thick carbon film, in order to prevent charging effects. | ||
The dried bionanocomposite structures were investigated by SEM working with back-scattered electrons. In the dried state, the mushroom cap still exhibited the purple colour that characterized its wet form; whereas the stem from originally white became slightly pink. As shown in Fig. 4a, bright microspots, corresponding to Au NPs or aggregates, uniformly covered the emerged portion of the Ca-alginate architecture; instead these features were not present on the immersed one. Despite the red-purple colour of the mushroom cap was an indication of the existence of colloidal gold (as shown in Fig. 1e), this was hardly detected on its surface, but it was predominant on the surface of the stem. High magnification SEM images better prove the presence of gold particles on the stem (left inset in Fig. 4b) and not on the cap (right inset in Fig. 4a). This was also confirmed by EDS microanalysis (Fig. 4b). As expected, C, O, Au and Ca were the main elements on the surfaces of the realized alginate bionanocomposites. From the elemental microanalysis, we can observe that for the stem the peaks of gold (Austem, black line) were considerably more intense than for the cap (Aucap, purple line). In particular, the ratio Aucap/Austem is 1/7 for the peak at 2.1 keV that corresponds to the Mα line of gold. Moreover, from SEM analysis of the cross-section of the realized structures the presence of Au NPs is clearly visible on the surface on the structures but it is difficultly detectable in the inner part (Fig. S1†).
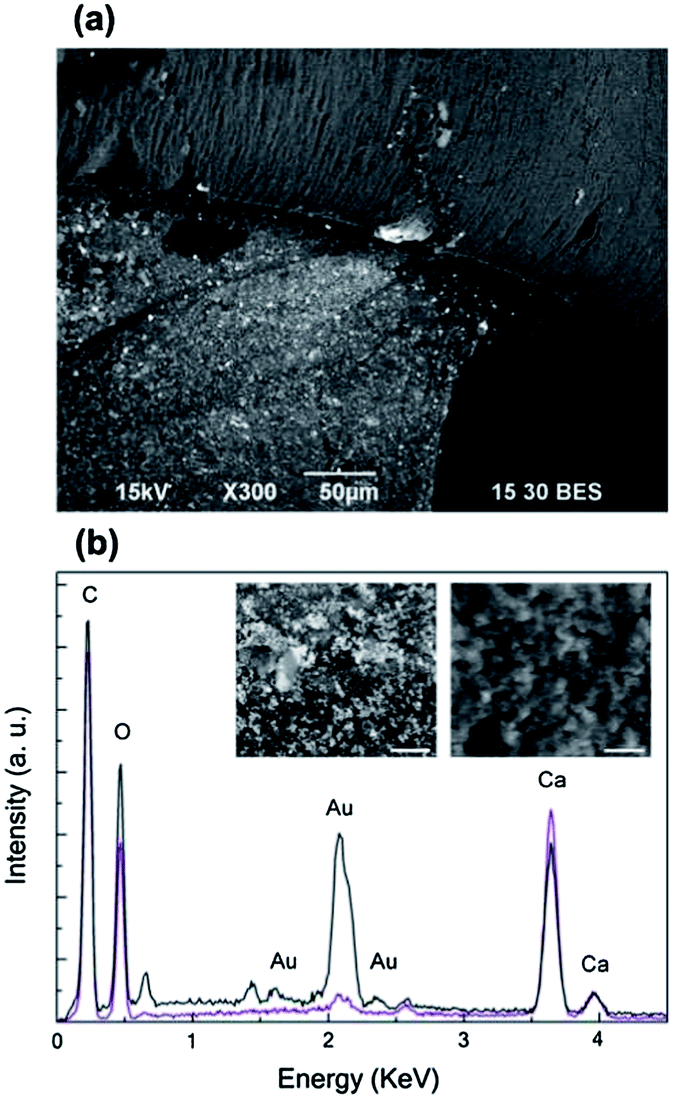 | ||
| Fig. 4 (a) Backscattered electron SEM image showing compositional contrast in a dried mushroom-like structure: the upper part corresponds to the cap and the lower one is the stem. Bright particles covering the stem are evident. (b) EDS spectra for the cap (purple line) and the stem (black line). In the insets: BE SEM images at high magnification of a sample of the stem (left) and the cap (right). Scale bar = 5 μm. | ||
We hypothesize that Au NPs are not exposed on the mushroom cap most likely because they were synthesised in the inner layers. Other studies have reported that Au ions tend to diffuse inside Ca-alginate beads and not to be concentrated on their surface.11 During the floating state the reaction between Ca2+ and sodium alginate occurs before the gold uptake, mainly because of the high affinity of alginate for Ca ions. Hence, a primary gelled Ca-alginate shell is formed immediately after the contact of the cap with the electrolyte solution.19 While the definition of the asymmetric structure proceeds, the gold ions diffuse inside the alginate matrix. We assume that a large part of them is involved in the nucleation and growth of colloidal nanoparticles inside the mushroom cap; whereas a certain amount is transported through the gel and trapped in the mushroom core.20 In this innermost zone, typically consisted of a dense gel, the gold anionic species are not complexed with alginate, probably because high concentrations of gold precursor are reached.12 During the dehydration of the structures, these species are transported outwards together with water. The preferential migration of the Au ions from the stem can be related to the different degree of cross-linking of the two parts of the alginate anisotropic structures. Since the emerged portion was not directly exposed to the calcium ions, it is expected to consist of a less compact gel network compared to the immersed portion. We believe that throughout the drying step the functional sites (mainly hydroxyl groups) located on the surface of the stem are responsible for the metal reduction, together with the environment parameters (such as humidity and light).21 This results in the decoration of this portion of the dried complex structures with Au NPs.
In order to investigate the effect of the amount of gold precursor on the localized synthesis of NPs, we realized mushroom-like structures using high concentration of HAuCl4 (1 mg mL−1) in the water bath. In this case, we did not observe the formation of bicolour objects but in the wet state both the cap and the stem exhibited a uniform yellow colour that is typical of the not-reacted gold precursor. On the contrary in the dried state the surface of the asymmetric particles were uniformly decorated with gold particles without detectable differences between the immersed and emerged region, as shown in Fig. 5. In fact, high magnification SEM images revealed the presence of gold on the surface of the cap (Fig. 5a) and the stem (Fig. 5b). Due to the high amount of gold ions present in the liquid bath during the formation of the anisotropic alginate particles, the precursor was likely encapsulated inside the whole alginate structure included its surface. Nevertheless, it did not undergo complexation and reduction in the wet state, a phenomenon already reported when high gold ion concentrations are used.12,22 Instead, when the particles were dried, Au NPs are created on the entire surface of the immersed and emerged region as a consequence of the reduction of the gold exposed to the ambient conditions. Hence, we can state that when the concentration of gold ions is extremely high, no control in the spatial localization of the gold synthesis can be achieved.
 | ||
| Fig. 5 (a) BE SEM image of a dried mushroom-like structure obtained with high concentration of gold precursor; relative high magnification pictures of a sample of (b) the cap and (c) the stem. | ||
Conclusions
In conclusion, we demonstrated the spatial localization of the synthesis of Au NPs in “mushroom” shaped alginate structures, by exploiting the floating of Na-alginate drops on the surface of CaCl2 solution containing specific concentrations of HAuCl4. We observed that for 0.1 mg mL−1 of HAuCl4, metal nanoparticles with an average diameter of 14 nm were synthesised exclusively inside the cap of the anisotropic structures, corresponding to the portion immersed in CaCl2/HAuCl4 solution. This conferred to the wet alginate “mushrooms” a double colouring: red (colloidal gold) and white (Ca-alginate) for the cap and the stem, respectively. SEM investigations on the dried anisotropic structures revealed that the Au NPs were synthesised in the inner layers of the cap and they were not exposed on the surface; on the contrary, during the dehydration process, Au ions selectively migrated outwards the stem, decorating its surface with gold particles. The here proposed drop-floating method allowed us to spatially control the nucleation and growth of Au NPs in the Ca-alginate matrix, depending on the hydration degree of the polysaccharide structure or the initial amount of gold precursor. In this way, we created bionanocomposite objects characterized by a mushroom-like shape and a double chemical composition, with potential application as sensing elements. Moreover, anisotropic structures with a microscale size can be produced by reducing the initial volume of the alginate droplets through microinjection systems or microfluidic platforms.Notes and references
- M. Darder, P. Aranda and E. Ruiz-Hitzky, Adv. Mater., 2007, 19, 1309 CrossRef CAS.
- F. Spano, A. Massaro, L. Blasi, M. Malerba, R. Cingolani and A. Athanassiou, Langmuir, 2012, 28, 3911 CrossRef CAS.
- N. Nestle and R. Kimmich, Colloids Surf., A, 1996, 115, 141 CrossRef CAS.
- T. A. Davis, B. Volesky and A. Mucci, Water Res., 2003, 37, 4311 CrossRef CAS.
- I. Donati, Y. A. Morch, B. L. Strand, G. Skjak-Break and S. Paoletti, J. Phys. Chem. B, 2009, 113, 12916 CrossRef CAS.
- Y. Fang, S. Al-Assaf, G. O. Phillips, K. Nishinari, T. Funami, P. A. Williams and L. Li, J. Phys. Chem. B, 2007, 111, 2456 CrossRef CAS.
- L. Li, Y. Fang, R. Vreeker and I. Appelqvist, Biomacromolecules, 2007, 8, 464 CrossRef CAS.
- I. Braccini and S. Perez, Biomacromolecules, 2001, 2, 1089 CrossRef CAS.
- V. Jaouen, R. Brayner, D. Lantiat, N. Steunou and T. Coradin, Nanotechnology, 2010, 21, 185605 CrossRef.
- A. Pal, K. Esumi and T. Pal, J. Colloid Interface Sci., 2005, 288, 396 CrossRef CAS.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885 CrossRef CAS.
- E. Torres, Y. N. Mata, M. L. Blazquez, J. A. Munoz, F. Gonzalez and A. Ballester, Langmuir, 2005, 21, 7951 CrossRef CAS.
- S. Saha, A. Pal, S. Pande, S. Sarkar, S. Panigrahi and T. Pal, J. Phys. Chem. C, 2009, 113, 7553 CAS.
- R. Brayner, M.-J. Vaulay, F. Fievet and T. Coradin, Chem. Mater., 2007, 19, 1190 CrossRef CAS.
- E. Mele, D. Fragouli, R. Ruffilli, G. L. De Gregorio, R. Cingolani and A. Athanassiou, Soft Matter, 2013, 9, 6338 RSC.
- E. Secchi, T. Roversi, S. Buzzaccaro, L. Piazza and R. Piazza, Soft Matter, 2013, 9, 3931 RSC.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410 CrossRef CAS.
- N. T. Anh, D. V. Phu, N. N. Duy, B. D. Du and N. Q. Hien, Radiat. Phys. Chem., 2010, 79, 405 CrossRef.
- R. M. Hassan, M. Th. Makhlouf and S. A. El-Shatoury, Colloid Polym. Sci., 1992, 270, 1237 CAS.
- T. Braschler, A. Valero, L. Colella, K. Pataky, J. Brugger and P. Renaud, Anal. Chem., 2011, 83, 2234 CrossRef CAS.
- S. Eustis, H.-Y. Hsu and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 4811 CrossRef CAS.
- P. Calcagnile, D. Fragouli, E. Mele, R. Ruffilli and A. Athanassiou, RSC Advances, 2014, 4, 19177 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02183e |
| This journal is © The Royal Society of Chemistry 2014 |