Modification of graphene oxide by a facile coprecipitation method and click chemistry for use as a drug carrier†
Guoqiang Xu,
Pengwu Xu,
Dongjian Shi and
Mingqing Chen*
Key Laboratory of Food Colloids and Biotechnology, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, 1800 Lihu Road, Wuxi, 214122, China. E-mail: mqchen@jiangnan.edu.cn; Fax: +86-510-85917763; Tel: +86-510-85917019
First published on 19th June 2014
Abstract
A ternary composite based on graphene oxide (GO), Fe3O4 nanoparticles and poly[2-(dimethylamino) ethyl methacrylate] (PDMAEMA) was synthesized via anchoring Fe3O4 nanoparticles onto a GO nanosheet by a facile coprecipitation method and subsequently grafting PDMAEMA to the surface by click chemistry. It was shown that the composite had a defined structure and morphology. Thermogravimetric analysis (TGA) results showed that the weight ratio of attached Fe3O4 nanoparticles was above 10.86 wt% and the surface grafting density reached above 2 chains per 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 carbon atoms of GO sheet. The composite exhibited good water dispersive stability depending on surface grafting density and grafted chain length. Loading and release behaviours of levofloxacin (LOFX) demonstrated that the composites had high loading capacity and pH triggered controlled release properties.
000 carbon atoms of GO sheet. The composite exhibited good water dispersive stability depending on surface grafting density and grafted chain length. Loading and release behaviours of levofloxacin (LOFX) demonstrated that the composites had high loading capacity and pH triggered controlled release properties.
Introduction
Graphene oxide (GO) has been widely studied in such fields as paper,1 photovoltaic devices,2 catalysts,3 and biotechnology.4 Due to its ultrahigh specific surface area and huge aromatic domains, aromatic molecules can be absorbed on its surface via van der Waals, π–π stacking interaction and other noncovalent interactions.5–8 Common covalent modification methods suffer from several drawbacks, such as low efficiency, tedious processes, lack of proper control of polymer molecular weight and architecture, requiring rigorous reacting conditions.9 Therefore, it is necessary to adopt approaches to solve these adverse factors.Click chemistry, especially Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition between azides and alkynes (CuAAC), has advantages in high reactivity and efficiency,9,10 mild reacting condition,11 chemoselectivity and regioselectivity.12,13 Therefore, it has aroused comprehensive applications in synthesis14 and modification,11 architecture design,15,16 biochemistry17–20 and medical engineering.21–23 Recently, click chemistry has been used in surface modification of GO and graphene. For instance, Wu et al.9 firstly combined click chemistry and atom transfer radical polymerization (ATRP) to graft polystyrene (PS) onto graphene surface, which improved grafting efficiency and significantly enhanced the dispersity of graphene sheets in organic solvents in contrast to in situ polymerization.24 Yang et al.25,26 also confirmed that a combination of click chemistry and reversible addition fragmentation chain transfer (RAFT) polymerization could be a powerful tool in functionalization of GO. Given these explorations, we believe that the combination of click chemistry and control radical polymerization methods will offer versatile, high efficient and facile approaches to modify GO with well-defined polymeric components.
Poly[2-(dimethylamino) ethyl methacrylate] (PDMAEMA) is a biocompatible and stimuli-responsive polymer for the potential application in biomedical fields.27–31 It is water soluble at room temperature, but undergoes a coil to globule transition above the low critical solution temperature (LCST) which fluctuates from 32 °C to 53 °C due to the impact of molecular weight, pH value and salt concentration of solution.32,33 Thus, functionalization of GO by PDMAEMA is promising to prepare materials with unique properties for various applications.
Magnetic nanoparticles, including Fe3O4, γ-Fe2O3, can be magnetically driven to the desired positions inside the body, which helps to improve the therapeutic efficiency and protect the normal organs or tissues from drug toxicity. Hence, a few of researches about targeted delivery were reported based on Fe3O4.34–37 With the interaction between GO sheets and Fe3O4 nanoparticles preventing themselves from agglomerating, Fe3O4/GO composites could be used as sensor, switch, electrode, energy storage and drug carrier.38–45 However, the Fe3O4/GO composites were generally prepared above 180 °C in the previous researches. Thus, it is meaningful that Fe3O4 nanoparticles are directly anchored onto GO platform by a facile method. In addition, water solubility and dispersive stability of the Fe3O4/GO composite still need to be enhanced for further applications. Consequently, modification of the Fe3O4/GO composite with a water soluble polymer like PDMAEMA seems to be feasible.
Herein, for the first time, we reported the preparation of a ternary composite Fe3O4/PDMAEMA-GO via a facile chemical coprecipitation method and a combination of click chemistry and ATRP. Firstly, alkynyl groups were introduced onto the GO surface by esterification between hydroxyl of propargyl alcohol and carboxyl of GO. Secondly, the in situ decoration of the alkynyl pendant GO sheets (alkynyl-GO) with Fe3O4 nanoparticles was performed based on a facile chemical coprecipitation method. Finally, PDMAEMA that synthesized by ATRP was grafted onto the as-prepared Fe3O4/alkynyl-GO composite via click chemistry after polymeric substitution of halide end groups by azide groups, as schematically illustrated in Fig. 1. A series of composites were prepared and the structures and morphologies were determined by Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, thermogravimetric analysis (TGA) and transmission electron microscopy (TEM). After the procedures, the influence of the surface grafting density and the grafted chain length on the aqueous dispersive stability of the composites was discussed. As the domains of GO unoccupied by PDMAEMA chains or Fe3O4 nanoparticles can absorb aromatic drugs, the application of Fe3O4/PDMAEMA-GO in loading and releasing behaviours of levofloxacin (LOFX), a broad-spectrum antibacterial drug, was investigated. Effects of factors on the loading capacity and release rate were also discussed. It is believed that, with a higher aqueous dispersive stability than Fe3O4/GO composites and with magnetic control compared to nonmagnetic GO–polymer composites, the ternary composite will have a greater potential in targeted delivery, controlled release and other biomedical applications.
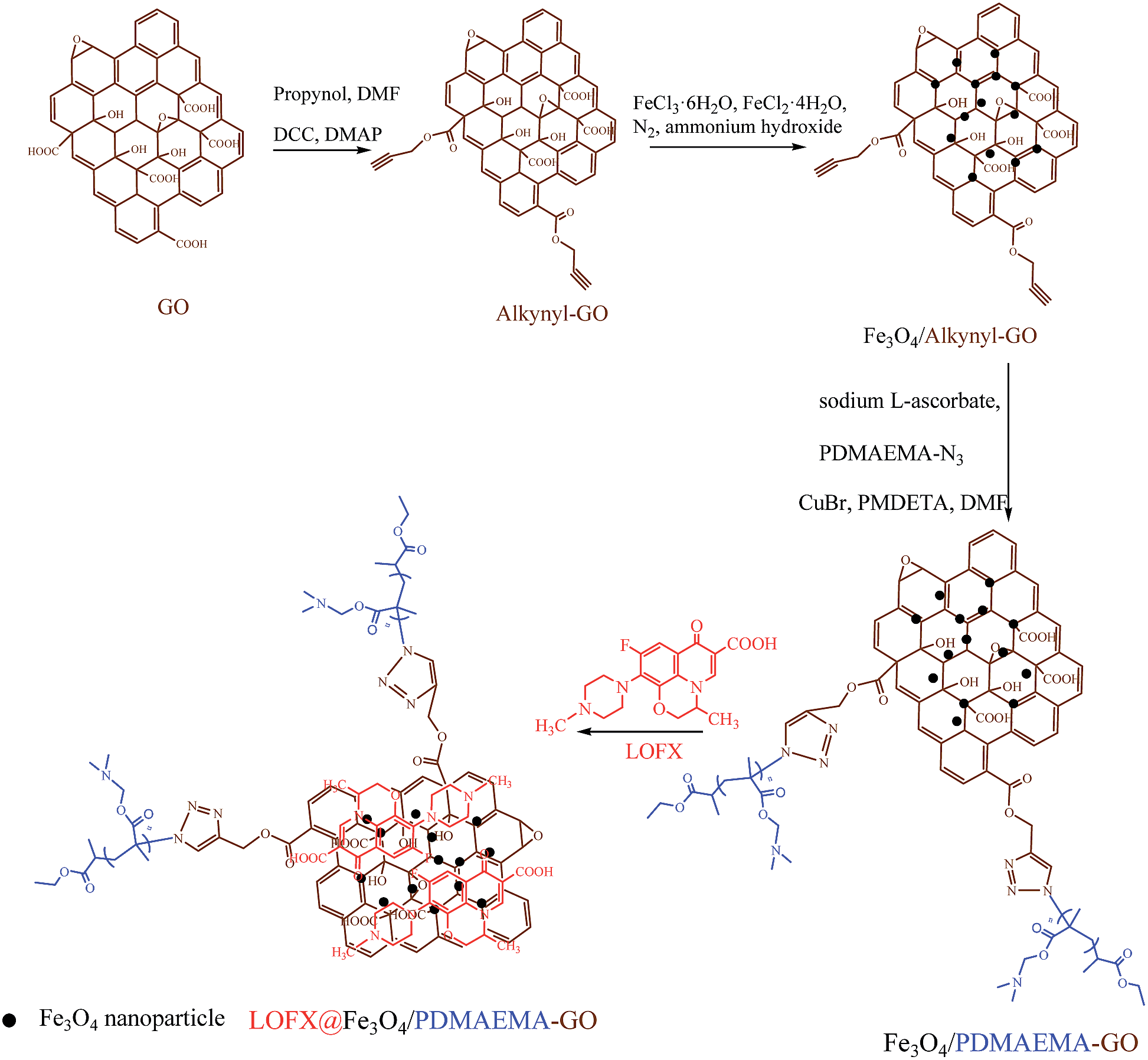 | ||
| Fig. 1 Schematic illustration for the synthesis of Fe3O4/PDMAEMA-GO and loading of drugs. | ||
Experimental
Materials
N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA), CuCl and CuBr were obtained from Tokyo Chemical Industry Co., Ltd. NaN3 was purchased from Xiya Reagent Company (Chengdu, China). LOFX (99%) was purchased from Maya Reagent Company (Jiaxing, China). Dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), sodium L-ascorbate were analytical grade and purchased from Aladdin Chemical Reagent Company (Shanghai, China). Dimethyl formamide (DMF), propargyl alcohol, ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ammonium hydroxide (28 wt%) were all analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). N2 (99.9%) was purchased from China Resources Co., Ltd. (Wuxi, China).Synthesis procedures
GO was prepared through a modified Hummers' method.46 Azido-terminated PDMAEMA (PDMAEMA-N3) homopolymers with different molecular weights were synthesized by ATRP and substitution reaction47–51 (Fig. S1 in (ESI)†). The structure was confirmed by FTIR spectra, and the results showed that GO and PDMAEMA-N3 were synthesized successfully (Fig. S2A and S3A in ESI†). Molecular weights of PDMAEMA-N3 with different molar ratios were determined by GPC measurements (Fig. S2B and Table S1 in ESI†).The brown solid GO (1 g) was suspended in 100 mL dry DMF by ultrasonication. Propargyl alcohol (2.68 g, 47 mmol), DCC (2 g, 10.44 mmol) and DMAP (0.3 g, 2.46 mmol) were then added into the GO suspension. The mixture was reacted at room temperature for 24 h with magnetic stirring. Alkynyl-GO was obtained after filtered, washed by DMF and deionized water and then dried. Alkynyl-GO (0.5 g) was dispersed in 100 mL deionized water in a 250 mL three-necked round bottom flask by ultrasonication. The flask was then purged with N2 for 60 min. A mix solution of FeCl3·6H2O (300 mg) and FeCl2·4H2O (4 g) in deionized water (25 mL) was added into the flask after pre-purged with N2. The mixture was stirred for 1 h under N2 atmosphere. Then, 2 mL of ammonium hydroxide was rapidly added into the solution. The suspension was kept at 65 °C for 6 h. The product was collected with a magnet and thoroughly washed by deionized water. Fe3O4/alkynyl-GO was obtained after being dried in vacuum at 65 °C overnight. Finally, Fe3O4/alkynyl-GO (0.1 g, containing ∼5.34 mg and ∼0.097 mmol alkynyl group) and PDMAEMA-N3 (Mn,(GPC) = 5710) (0.51 g, 0.089 mmol) was dissolved in 25 mL DMF and degassed. Under N2 atmosphere, CuBr (64 mg, 0.445 mmol), PMDETA (77 mg, 0.445 mmol), and sodium L-ascorbate (88.1 mg, 0.445 mmol) were added into the flask. The mixture was stirred for 48 h at room temperature, and the product named as S1 was collected by additional magnet, washed with DMF and deionized water, and then dried. Other products (S2–S5) with various feed weights were then prepared by the same method (Table S3 in ESI†).
Drug load
5 mL of LOFX solutions with different initial concentrations (from 4 to 36 μg mL−1) were sonicated with 0.5 mL Fe3O4/PDMAEMA-GO (0.1 mg mL−1) suspension for overnight at room temperature. The complex (LOFX@Fe3O4/PDMAEMA-GO) was collected by a magnet, washed with deionized water and then dried for further use. The amount of unloading LOFX in the remained solution was determined by a UV-vis spectrophotometer with a standard LOFX concentration curve. The amount of LOFX loaded on the Fe3O4/PDMAEMA-GO was determined based on eqn (1).| Φ = (MLOFX − MLOFX′)/Mcarrier | (1) |
In vitro drug release
To examine drug release behaviours, LOFX@Fe3O4/PDMAEMA-GO composites were dialyzed in beakers against 20 mL phosphate buffer solution (PBS) with pH at 5.0, 7.2 and 9.18, respectively. All the beakers were placed in water baths at 37 °C for a period of 3 days. At each predetermined time, 3 mL medium was drawn out and an equal volume of fresh PBS was replenished to the beaker. LOFX release profiles were obtained by measuring the amount of released LOFX at each time point via a standard LOFX concentration curve generated by a UV-vis spectrophotometer. All the drug release experiments were repeated at least three times.Characterization
Fourier transform infrared (FTIR) spectra were recorded in KBr pellets on an ABB Boman FALA 2000-104 FTIR spectrophotometer. Gel permeation chromatography (GPC) was performed by a Waters 1515 system equipped with a refractive index and a photodiode array detector, with tetrahydrofuran (THF) used as eluent (elution rate, 1.0 mL min−1) and PS standards used for calibration. Thermogravimetric analysis (TGA) was performed on a Mettler-Toledo TGA-1100SF thermogravimetric analyzer from 25 to 800 °C at a heating rate of 10 °C min−1 and N2 flow rate of 50 mL min−1. Raman spectra were recorded by a Renishaw Invia Raman spectrometer using a 532 nm argon ion laser. Scanning electron microscopic (SEM) images were acquired by a Hitachi S-4800 field emission scanning electron microscopy. Transmission electron microscopic (TEM) images were obtained using a JEOL JEM-2100 transmission electron microscopy at an accelerating voltage of 200 kV. One drop of sample solution (at a concentration of 0.01 mg mL−1) was placed on a copper-mesh coated with carbon and then air-dried before measurement. Fluorescence emission spectra were acquired on a Shimadzu RF5301PC fluorescence spectrophotometer with a excitation wavelength of 300 nm. Zeta potential and size of the composites were determined by a Brookhaven ZetaPALS zeta potential and nanosize analyzer. Ultraviolet-visible (UV-vis) absorbance spectra were recorded on a Rayleigh UV-1100 spectrophotometer. A standard LOFX concentration curve was generated by the UV-vis spectrophotometer from a series of LOFX solutions with different concentrations at a wavelength of 290 nm. The standard curve was used to determine the amount of LOFX loaded on the Fe3O4/PDMAEMA-GO and the release rate of the loaded LOFX.Results and discussion
The molecular weight of PDMAEMA-N3 polymers largely increased from 5710 to 17130 with increment of monomer amounts from 1:50 to 1:200 (Fig. S2B and Table S1 in ESI†), meaning the polymers with various chain lengths. A series of Fe3O4/PDMAEMA-GO composites were synthesized via the chemical deposition method and “grafting to” method by click chemistry, as shown in Fig. 1 and Table S3 in ESI.† As shown in Table S3,† S1–S3 were prepared with different feed amounts of PDMAEMA-N3 by the same component. S3–S5 were prepared by the PDMAEMA-N3 with different molecular weights but with the same molar ratio. Thus, products S1–S3 had the same grafted chain length but different surface grafting densities, and products S3–S5 should have the same surface grafting density but different grafted chain lengths. FTIR spectrum of representative Fe3O4/PDMAEMA-GO (S1) showed the characteristic features (Fig. 2a), 582 cm−1 (Fe–O stretching), 1090 cm−1 (C–O bending), 1434 cm−1 (–CH2– stretching), 1700 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 2918 cm−1 (C–H stretching), which existed in FTIR spectra of its intermediates (Fig S2A and S3A, ESI†). Driven by external magnetic field indicated potential of the composite (S1) in targeted delivery (Fig. 2b).
O stretching), 2918 cm−1 (C–H stretching), which existed in FTIR spectra of its intermediates (Fig S2A and S3A, ESI†). Driven by external magnetic field indicated potential of the composite (S1) in targeted delivery (Fig. 2b).
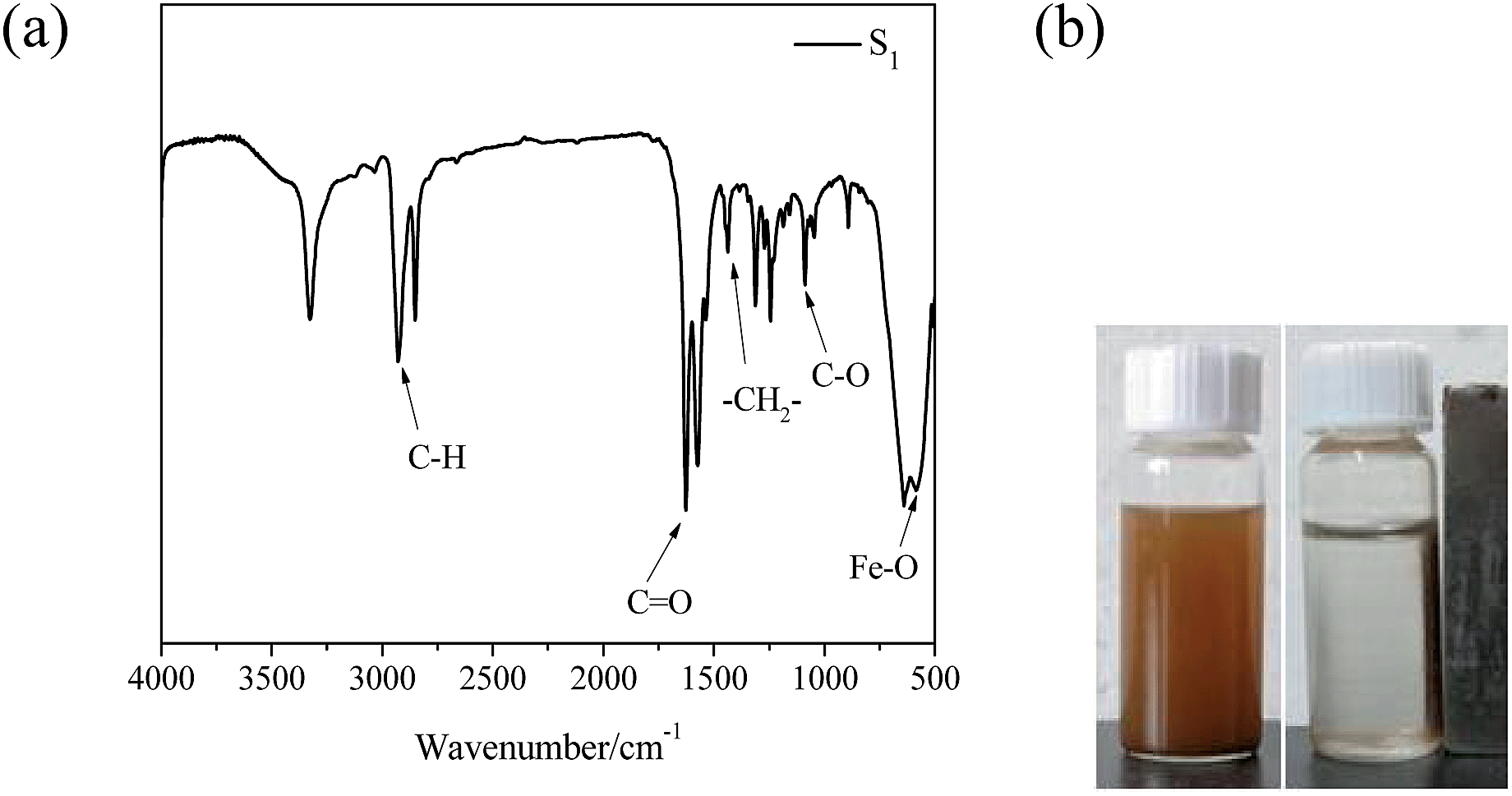 | ||
| Fig. 2 (a) FTIR spectrum of Fe3O4/PDMAEMA-GO (S1) and (b) photographs of Fe3O4/PDMAEMA-GO (S1) with and without external magnetic field. | ||
Weight ratio of each constituent of the ternary composites was determined by TGA (Fig. 3, S2C, S4, Table 1and S2, ESI†). The grafting density could be calculated by eqn (2) and (3) (ref. 52), which was then proved to be one of factors affecting dispersive stability of composites in PBS.
|
Dp = 10000MCWp/MpWC
| (2) |
| Dc = 108MCWp/MpAbWC | (3) |
000 carbon atoms), Dc is the grafting density (grafted chains per μm2 sheet), MC is the relative molar mass of carbon (MC = 12 g mol−1), Mp represents Mn,(GPC) of the grafted polymer, WC and Wp are the weight fractions of the GO backbone and the grafted polymer. Ab represents the area of a benzene ring (5.24 Å2).
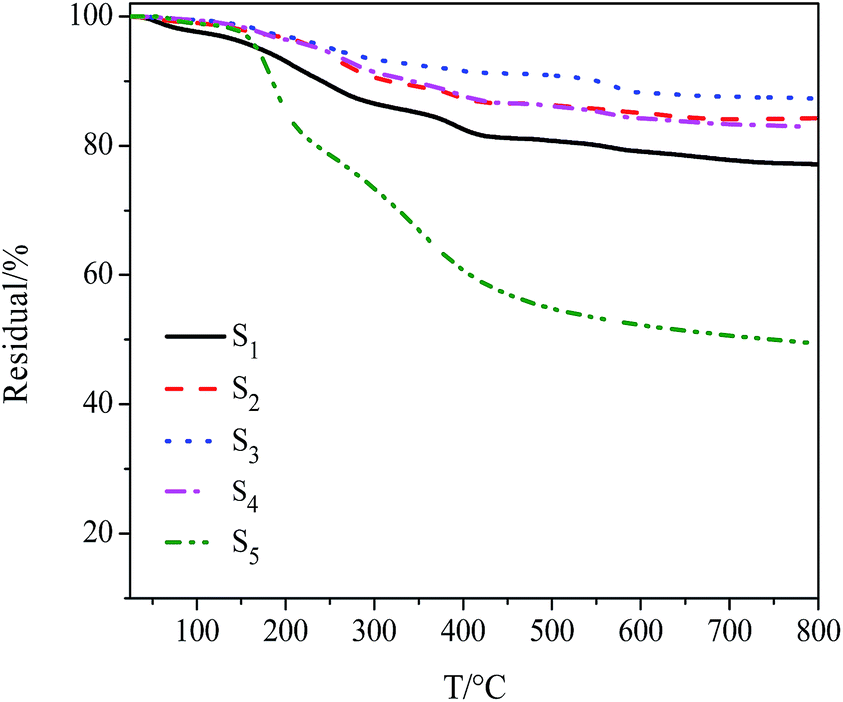 | ||
| Fig. 3 TGA curves of Fe3O4/PDMAEMA-GO composites at a heating rate of 10 °C min−1 in nitrogen. | ||
Structures of products were further confirmed by Raman spectroscopy (Fig. S6 and Table S4, ESI†). The morphological analyses of the ternary composites were investigated by TEM (Fig. 4). As shown in Fig. 4, lots of Fe3O4 nanoparticles with a diameter of ∼20 nm were deposited on the surface of the GO sheets randomly without aggregation. Even though a long time of reaction process in preparation of Fe3O4/PDMAEMA-GO, the Fe3O4 nanoparticles were still firmly anchored on the surface of GO, implying the strong interaction between the two substances.41–43 By contrast, the bare Fe3O4 nanoparticles aggregated to clusters (Fig. S5, ESI†).
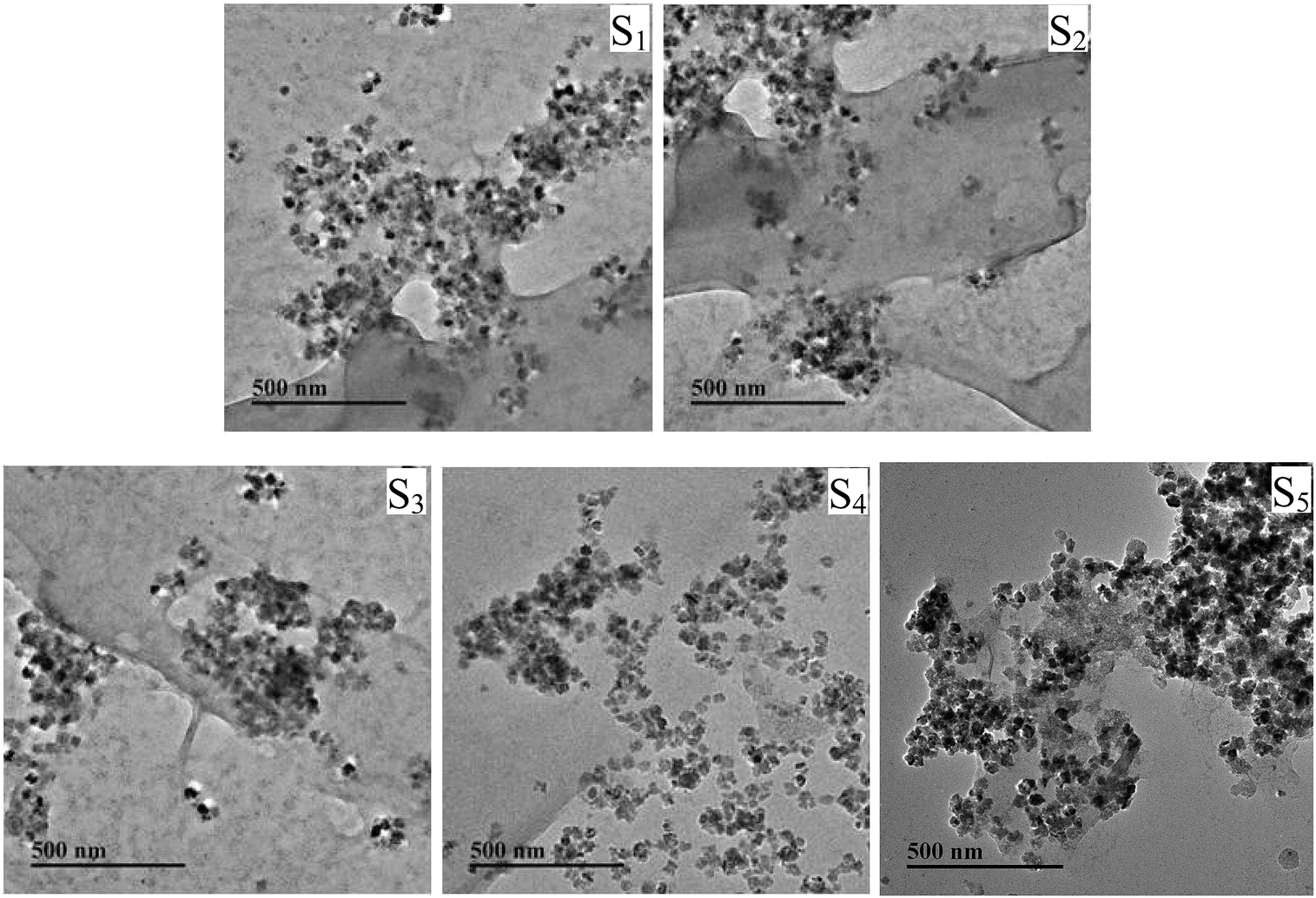 | ||
| Fig. 4 TEM images of Fe3O4/PDMAEMA-GO. | ||
The dispersive stability of the composites in PBS was tested according to the fact that when composites disperse well in PBS, the transmittance will stay low. As was shown in Fig. 5A, the stability depended on either the grafting density for the composites with the same grafted chain length (S1–S3) or the grafted chain length for the composites with the same grafting density (S3–S5). The photographs of samples (Table S5 and S6, ESI†) displayed that S1 and S5 had the best stability in each group. In addition, it was evident that the ternary composites had better dispersive stability than Fe3O4/GO (Fig. S7, ESI†). Since the interaction between the nanoparticles and GO avoided restacking of the exfoliated GO sheets, the grafted polymers helped to prevent the agglomeration of the ternary composite. Zeta potential values of samples matched with the stability test, as nitrogen atoms of PDMAEMA chains can capture the protons,30,33 thus S1 and S5 with more nitrogen atoms had higher positive zeta potential values (Table S7, ESI†). Accordingly, S1 and S5 displayed better stability than others. Due to the thermosensitivity of the grafted polymer,29 the ternary composites exhibited slight size change when environmental temperature varied (Fig. 5 B), as the polymer chains extend at low temperature and shrink at high temperature (Fig. 5C). The stretch of surface polymer chains promoted water solubility and dispersity of the ternary composites in PBS, which could explain the slight decline of transmittance in Fig. 5A during 22nd and 23rd day.
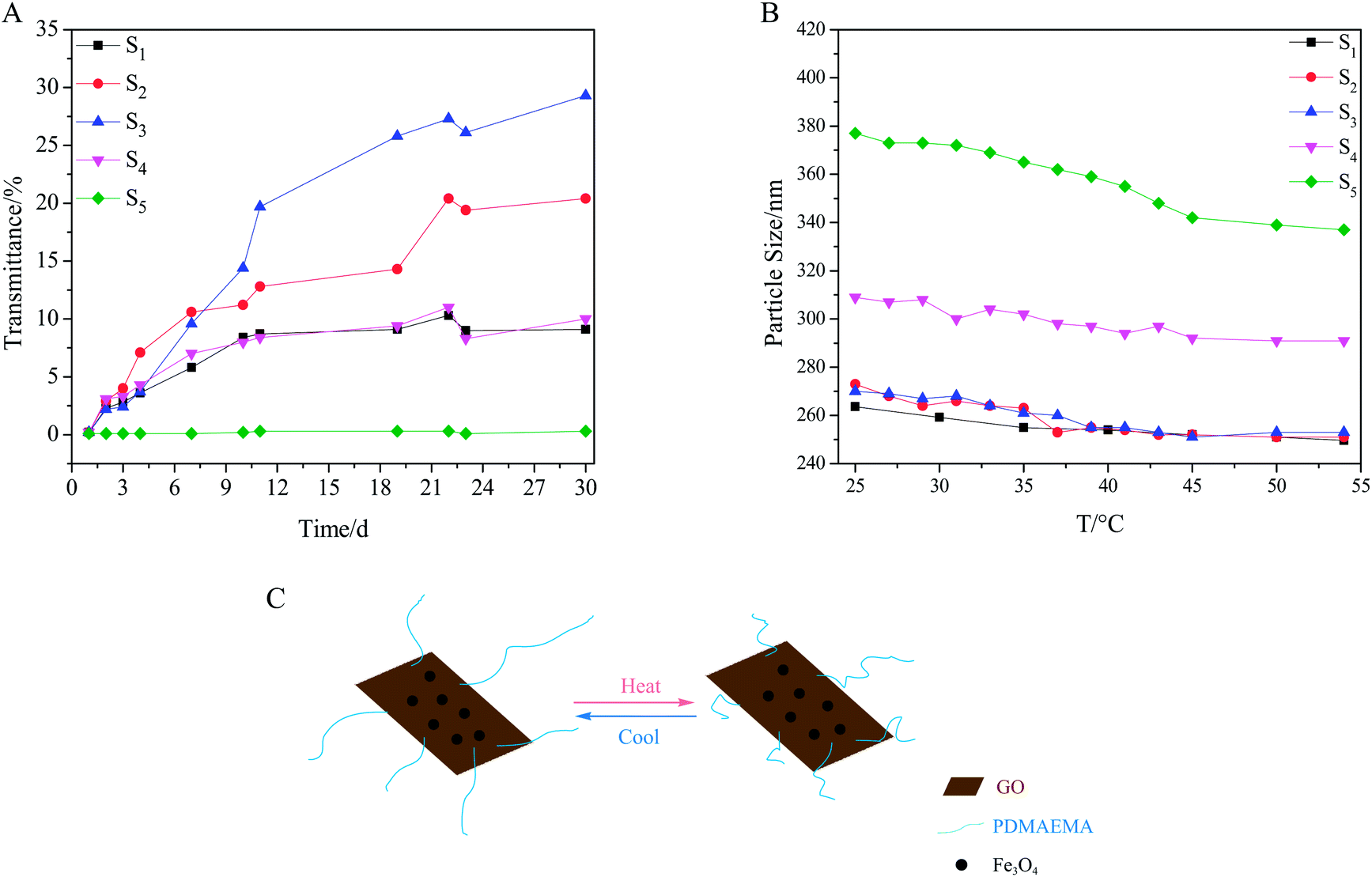 | ||
| Fig. 5 (A) Effects of grafting density and grafted chain length on stability of samples in PBS (pH = 7.2). (B) Effect of temperature on particle size. (C) Schematic plot of thermosensitivity of Fe3O4/PDMAEMA-GO composite. | ||
The composites were as carriers for loading and releasing the drug, such as LOFX. LOFX was loaded on the composite via strong π–π stacking interaction between LOFX and GO platform,53 which was confirmed by the fluorescence spectra (Fig. 6). At the same LOFX concentration, free LOFX exhibited a fluorescence emission peak at ∼490 nm, while LOFX@Fe3O4/PDMAEMA-GO (as S1 as example) exhibited drastic quenching of emission band at the same excitation wavelength. The quenching might be the result of a photoinduced electron-transfer from LOFX to GO, which was induced by strong π–π stacking interaction between GO platform and LOFX.53–56 Except for the π–π stacking interaction, there exist hydrogen bond and electrostatic interaction between GO plane and LOFX. Since there is a carboxyl and nitrogen atoms which could be positively charged interacting with the remaining oxygen-containing groups in GO plane, and the hydrogen bond and electrostatic interaction might contribute to the fluorescence quenching as well. The loading mount of LOFX on Fe3O4/PDMAEMA-GO was investigated in different initial LOFX concentrations with respect to the same concentration of Fe3O4/PDMAEMA-GO (0.1 mg mL−1). The loading capacity of LOFX was faintly affected by the grafting density and the grafted chain length (Fig. 7). The loading amount of LOFX could reach 1.75–1.95 mg mg−1, higher than many other drug carrier materials.6 Comparing the composites with different grafting densities (Fig. 7A), sample S3 with the lowest grafting density had the highest loading amount for LOFX. However, the effect of chain length on the loading amount was more notable at low concentration and fainter at high concentration than that of the grafting density (Fig. 7B). This phenomenon might result from the loading capacity of the samples (S3–S5) tending to saturation at high concentration.
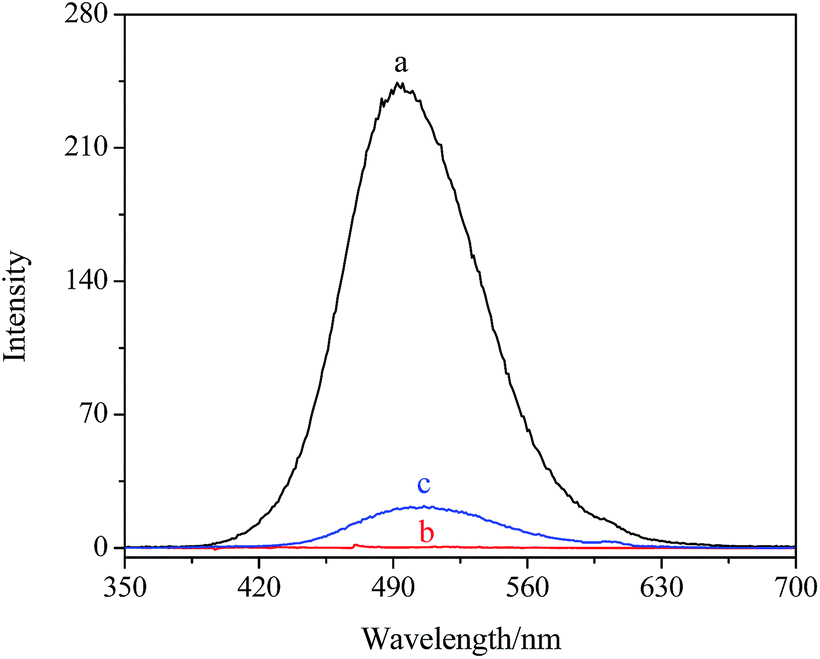 | ||
| Fig. 6 Fluorescence spectra of (a) LOFX, (b) Fe3O4/PDMAEMA-GO and (c) LOFX@Fe3O4/PDMAEMA-GO. | ||
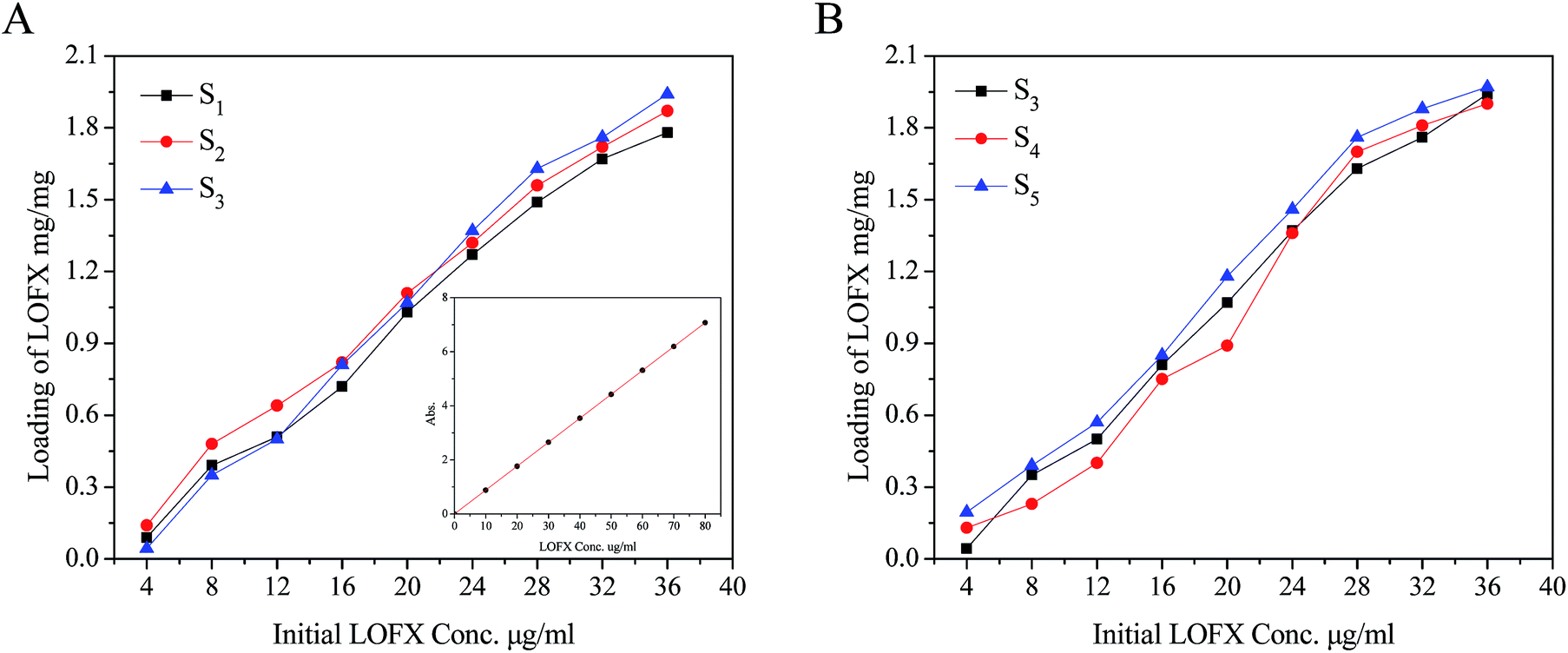 | ||
| Fig. 7 (A) Loading capacity of samples with different grafting densities. The inset figure is standard curve of LOFX. (B) Loading capacity of samples with different grafted chain lengths. | ||
The release behaviours of LOFX from Fe3O4/PDMAEMA-GO (S1) at 37 °C in different pH buffers were investigated (Fig. 8). In the initial period, LOFX was released fast from the carrier, and the release rates slowed down after 24 h. The property of controlled release under neutral was considerable. It was found that ∼88% of the total bound LOFX were released from the carrier under basic condition, which was much higher than the other two conditions. It might attribute to the dissociation of hydrogen bond between LOFX and GO plane under basic condition, and higher solubility of LOFX under basic condition.57 Hence, the amount of released LOFX was much greater. Protonation of nitrogen atoms of LOFX under acidic condition might lead to faster release rate and greater release amount than neutral condition.
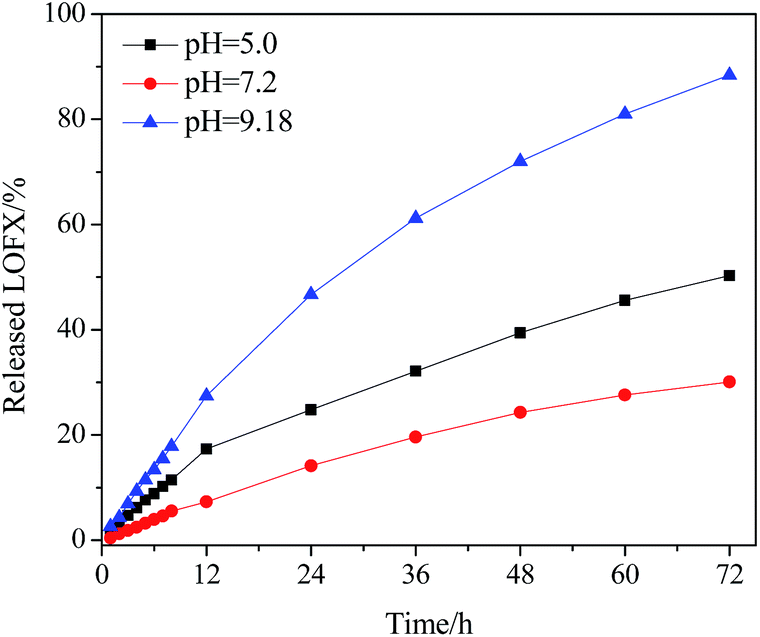 | ||
| Fig. 8 Release profiles of LOFX@Fe3O4/PDMAEMA-GO. | ||
Conclusions
In summary, the GO based ternary composites have been successfully prepared with well-defined grafted polymers and considerable quantity of firmly anchored Fe3O4 nanoparticles via a facile coprecipitation method and click chemistry. This combination prompts the feasibility of synthesis of polybasic GO or graphene based composites, leading to further development of multifunctional materials based on GO and facilitating GO based materials closer to real applications. The as-prepared composite has magnetic control property and good aqueous dispersive stability depending on grafting density and grafted chain length. As a drug carrier, the composite has high loading capacity and pH triggered controlled release properties for LOFX in different buffer solutions. With these advantages, the composite has the potential applications in targeted delivery, controlled release and other applications.Acknowledgements
The authors are grateful for financial aid from the National Natural Science Foundation of China (Grant no. 21341009 and no. 51173072) and the Fundamental Research Funds for the Central Universities (JUSRP51408B).Notes and references
- S. Park, K. S. Lee, B. Gulay, W. W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- I. P. Murray, S. J. Lou, L. J. Cote, S. Loser, C. J. Kadleck, T. Xu, J. M. Szarko, B. S. Rolczynski, J. E. Johns, J. X. Huang, L. P. Yu, L. X. Chen, T. J. Marks and M. C. Hersam, J. Phys. Chem. Lett., 2011, 2, 3006–3012 CrossRef CAS.
- S. Wang, C. T. Nai, X. F. Jiang, Y. H. Pan, C. H. Tan, M. Nesladek, Q. H. Xu and K. P. Loh, J. Phys. Chem. Lett., 2012, 3, 2332–2336 CrossRef CAS.
- J. Huang, C. Zong, H. Shen, M. Liu, B. Chen, B. Ren and Z. J. Zhang, Small, 2012, 8, 2577–2584 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- D. Depan, J. Shah and R. D. K. Misra, Mater. Sci. Eng., C, 2011, 31, 1305–1312 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- S. T. Sun, Y. W. Cao, J. C. Feng and P. Y. Wu, J. Mater. Chem., 2010, 20, 5605–5607 RSC.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876 CrossRef CAS PubMed.
- Z. M. Guo, A. Lei, X. M. Liang and Q. Xu, Chem. Commun., 2006, 4512–4514 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- Z. Yuan, G. C. Kuang, R. J. Clark and L. Zhu, Org. Lett., 2012, 14, 2590–2593 CrossRef CAS PubMed.
- D. D. Di'az, S. Punna, P. Holzer, A. K. Mcpherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS PubMed.
- H. Su, Y. W. Li, K. Yue, Z. Wang, P. T. Lu, X. Y. Feng, X. H. Dong, S. Zhang, S. Z. D. Cheng and W. B. Zhang, Polym. Chem., 2014, 5, 3697–3706 RSC.
- P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS PubMed.
- N. S. Hatzakis, H. Engelkamp, K. Velonia, J. Hofkens, P. C. M. Christianen, A. Svendsen, S. A. Patkar, J. Vind, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2006, 2012–2014 RSC.
- B. L. Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef.
- Y. Xu, Y. Suzuki and M. Komiyama, Angew. Chem., Int. Ed., 2009, 48, 3281–3284 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- A. Adibekian, B. RMartin, C. Wang, K. L. Hsu, D. A. Bachovchin, S. Niessen, H. Hoover and B. F. Cravatt, Nat. Chem. Biol., 2011, 7, 469–478 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- Y. W. Cao, Z. L. Lai, J. C. Feng and P. Y. Wu, J. Mater. Chem., 2011, 21, 9271–9278 RSC.
- M. Fang, K. G. Wang, H. B. Lu, Y. L. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- Y. F. Yang, X. H. Song, L. Yuan, M. Li, J. C. Liu, R. Q. Ji and H. Y. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 329–337 CrossRef CAS.
- H. Y. Hong, Y. Y. Mai, Y. F. Zhou, D. Y. Yan and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 668–681 CrossRef CAS.
- K. Q. Chen, D. H. Liang, J. Tian, L. Q. Shi and H. Y. Zhao, J. Phys. Chem. B, 2008, 112, 12612–12617 CrossRef CAS PubMed.
- L. L. Zhou, W. Z. Yuan, J. Y. Yuan and X. Y. Hong, Mater. Lett., 2008, 62, 1372–1375 CrossRef CAS PubMed.
- V. Darcos, S. E. Habnouni, B. Nottelet, A. E. Ghzaoui and J. Coudane, Polym. Chem., 2010, 1, 280–282 RSC.
- X. L. Jiang, M. C. Lok and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2077–2084 CrossRef CAS PubMed.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- M. Vamvakaki, N. C. Billingham and S. P. Armes, Macromolecules, 1999, 32, 2088–2090 CrossRef CAS.
- Y. F. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- M. Y. Lou, D. P. Wang, W. H. Huang, D. Chen and B. Liu, J. Magn. Magn. Mater., 2006, 305, 83–90 CrossRef CAS PubMed.
- Z. L. Lei, Y. L. Li and X. Y. Wei, J. Solid State Chem., 2008, 181, 480–486 CrossRef CAS PubMed.
- Q. Xu, X. J. Bian, L. L. Li, X. Y. Hu, M. Sun, D. Chen and Y. Wang, Electrochem. Commun., 2008, 10, 995–999 CrossRef CAS PubMed.
- X. X. Liu, H. Zhu and X. R. Yang, Talanta, 2011, 87, 243–248 CrossRef CAS PubMed.
- D. B. Lu, Y. Zhang, L. T. Wang, S. X. Lin, C. M. Wang and X. F. Chen, Talanta, 2012, 88, 181–186 CrossRef CAS PubMed.
- J. J. Liang, Y. F. Xu, D. Sui, L. Zhang, Y. Huang, Y. F. Ma, F. F. Li and Y. S. Chen, J. Phys. Chem. C, 2010, 114, 17465–17471 CAS.
- C. Y. Hou, Q. H. Zhang, H. Z. Wang and Y. G. Li, J. Mater. Chem., 2011, 21, 10512–10517 RSC.
- P. C. Lian, S. Z. Liang, X. F. Zhu, W. S. Yang and H. H. Wang, Electrochim. Acta, 2011, 58, 81–88 CrossRef CAS PubMed.
- J. Su, M. H. Cao, L. Ren and C. W. Hu, J. Phys. Chem. C, 2011, 115, 14469–14477 CAS.
- J. J. Liang, Y. Huang, J. Y. Oh, M. Kozlov, D. Sui, S. L. Fang, R. H. Baughman, Y. F. Ma and Y. S. Chen, Adv. Funct. Mater., 2011, 21, 3778–3784 CrossRef CAS.
- X. Y. Li, X. L. Huang, D. P. Liu, X. Wang, S. Y. Song, L. Zhou and H. J. Zhang, J. Phys. Chem. C, 2011, 115, 21567–21573 CAS.
- H. A. Becerril, J. Mao, Z. F. Liu, R. M. Stoltenberg, Z. Bao and Y. S. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- M. Hua, T. Kaneko, X. Y. Liu, M. Q. Chen and M. Akashi, Polym. J., 2005, 37, 59–64 CrossRef CAS.
- W. Yang, T. Kaneko, X. Y. Liu, M. Q. Chen and M. Akashi, Chem. Lett., 2006, 35, 222–223 CrossRef CAS.
- N. Hu, J. H. Li, D. J. Shi, X. Y. Liu and M. Q. Chen, Polym. Sci., Ser. B, 2013, 55, 69–76 CrossRef CAS.
- N. Hu, G. Q. Xu, J. H. Li, D. J. Shi and M. Q. Chen, Polym. Mater. Sci. Eng., 2014, 30, 36–39 CAS.
- J. A. Johnson, D. R. Lewis, D. D. Di'az, M. G. Finn, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 6564–6565 CrossRef CAS PubMed.
- L. Y. Kan, Z. Xu and C. Gao, Macromolecules, 2011, 44, 444–452 CrossRef CAS.
- J. Gao, F. Bao, L. L. Feng, K. Y. Shen, Q. D. Zhu, D. F. Wang, T. Chen, R. Ma and C. J. Yan, RSC Adv., 2011, 1, 1737–1744 RSC.
- X. Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. Huang and Y. S. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CAS.
- K. P. Liu, J. J. Zhang, F. F. Cheng, T. T. Zheng, C. M. Wang and J. J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- H. Shen, M. Liu, H. X. He, L. M. Zhang, J. Huang, Y. Chong, J. W. Dai and Z. J. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 6317–6323 CAS.
- A. Frick, H. Moller and E. Wirbitzki, Eur. J. Pharm. Biopharm., 1998, 46, 305–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02219j |
| This journal is © The Royal Society of Chemistry 2014 |