Cross-coupling reactions catalyzed by an N-heterocyclic carbene–Pd(II) complex under aerobic and CuI-free conditions†
Hongfei Lu*a,
Lin Wanga,
Feifei Yanga,
Runze Wua and
Wei Shen*b
aSchool of Biological and Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang, China. E-mail: zjluhf1979@hotmail.com
bJiangsu Key Laboratory of Pesticide Science, College of Science, Nanjing Agricultural University, Nanjing, China. E-mail: swgdj@njau.edu.cn
First published on 20th June 2014
Abstract
A Pd-complex, (Cat. 3), has been successfully employed as a highly efficient and recyclable catalyst for the Sonogashira and Heck reactions of aryl bromides with various terminal acetylenes and olefins. The catalytic reactions proceed with excellent yields with a low catalyst loading (1.0 mol%) under aerobic and CuI-free conditions. A plausible mechanism has also been proposed for the reaction.
The palladium catalyzed cross-coupling reaction is one of the most powerful tools for the construction of C–C bonds.1 The exemplary cross-coupling methodologies encompass Suzuki reactions,1e,2 Heck reactions and Sonogashira reactions.3 In the Heck and Sonogashira reactions, despite numerous other metals proved to be effective for such reactions,5 conventional palladium complexes such as PdCl2(PPh3)2,3,4 often in combination with copper(I) salts, are still the most preferred ones. Although the ligands of those palladium complexes are air-sensitive phosphine-based structures of limited practicality, the catalysts are still being widely employed because the cross-coupling conditions are generally compatible with a broad library of functionalities. Most noticeably, a large number of modified protocols have also been developed to expand the scope of the applications.5a,6
Ever since the initial discovery of the cross-coupling reactions, tremendous endeavor has recently been focused on searching for alternatives, among which electrophiles and aryl tosylates have been proven to be a new class of practical surrogates to aryl halides.1c,7 Compared to arylhalides, aryl tosylates are usually less expensive and more readily available from phenols.8 Nonetheless, more efficient, moisture- and air-insensitive palladium catalysts6a,9c,e,10 are still being called for to affect the Sonogashira and Heck reaction with low catalyst loadings under copper-free6e,9 and less rigorously anhydrous and anaerobic conditions.9f,11
During the past twenty years, significant advances have been made to develop N-heterocyclic carbenes (NHCs) and their metal complexes to facilitate various organic transformations. Organ highlighted that bulky yet flexible NHC ligands were able to circumvent various limitations of cross-coupling reactions.9c,12 Encouraged by the excellent yields of [Pd–NHC] complex (IPr*–Pd(II)–Py, Catalyst 3) mediated Suzuki couplings of aryl bromides and arylsulfonates with arylboronic acids under aerobic conditions, we further proceeded to investigate its application in other cross-couplings.
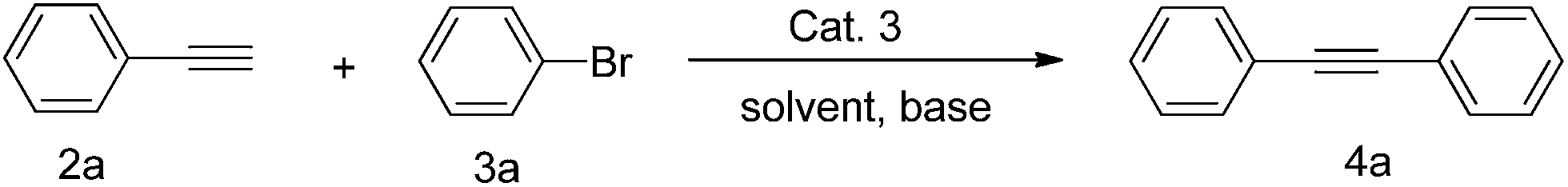
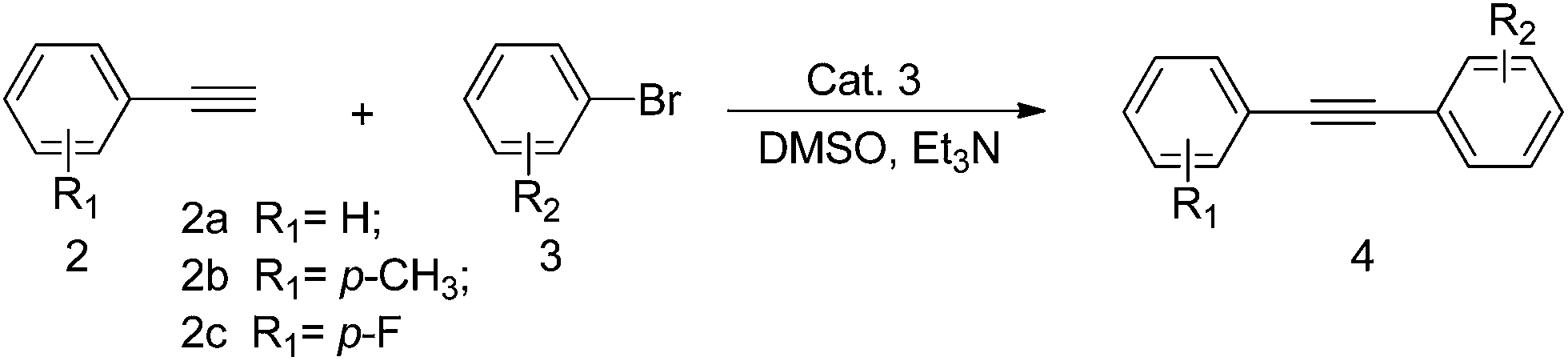
We report herein that Cat. 3 efficiently catalyzes Heck and Sonogashira reactions under CuI-free condition with low catalyst loading (1.0 mol%) in the absence of the protection of inert atmosphere (Schemes 1 and 2).
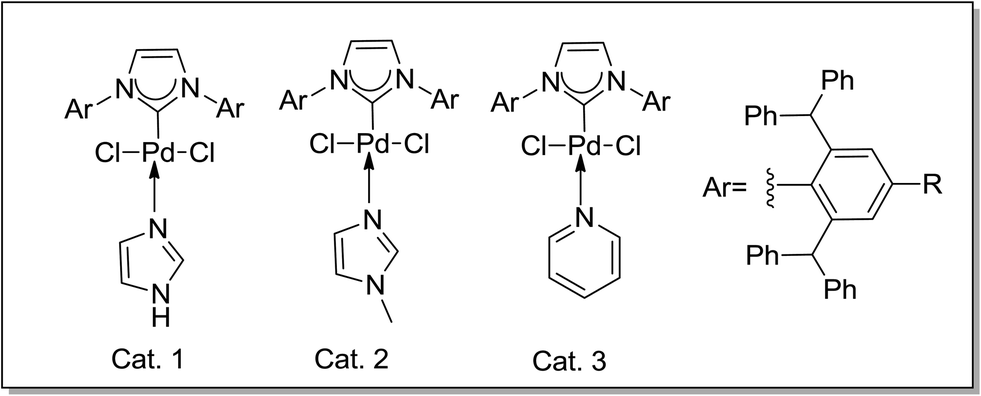 | ||
| Scheme 1 Catalysts. | ||
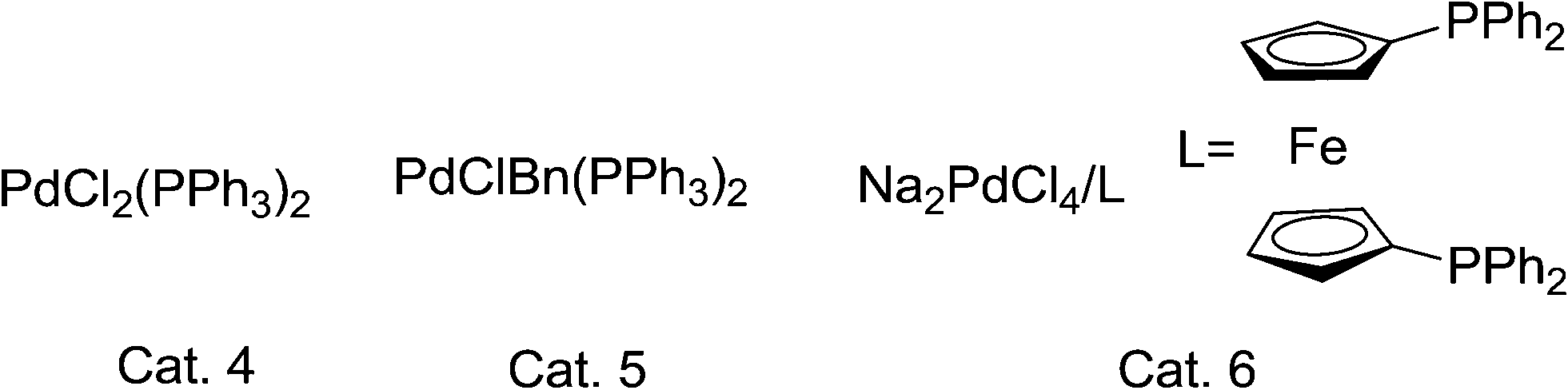 | ||
| Scheme 2 Pd(II) catalysts. | ||
In our previous study of cross-coupling reactions (Scheme 3), it was found that the catalyst Cat. 3 is highly efficient for the Suzuki cross-coupling reaction, and the yield of the idea products are measured up to 93%.13 Considering the good catalytic performance of Cat. 3, we were encouraged to investigate the catalytic activity of the present catalyst for the Sonogashira and Heck reactions.
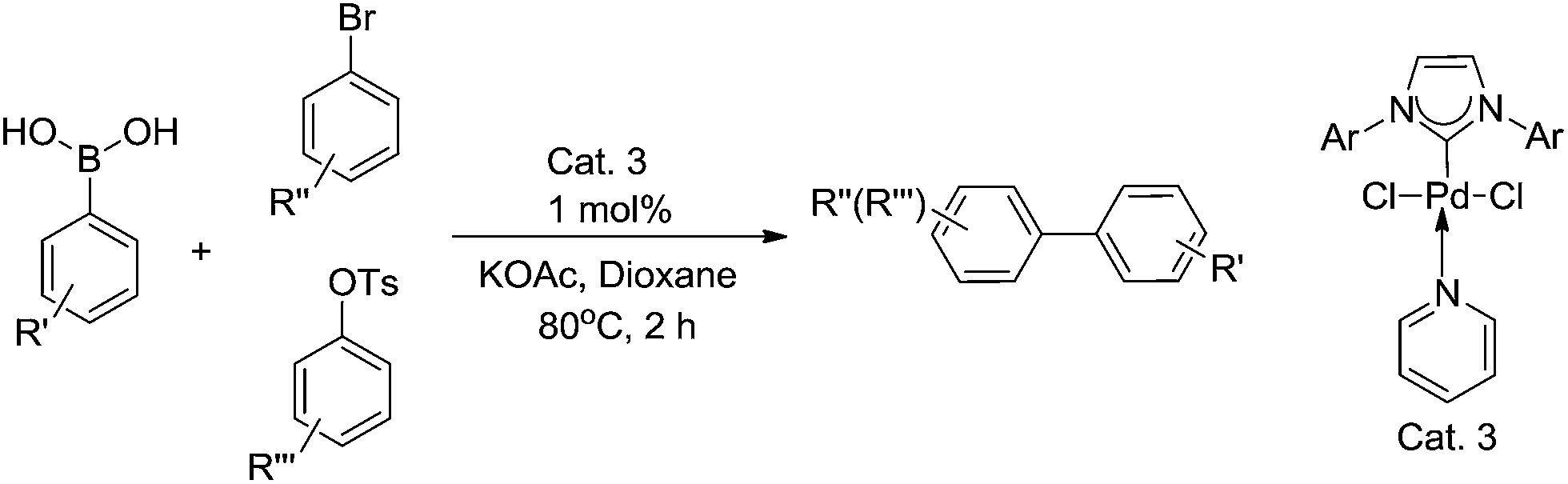 | ||
| Scheme 3 Suzuki cross-coupling reactions. | ||
In the presence of substrate, Pd(II) complex catalytic system is efficient in the reactions (Fig. 1). Great attention should be paid to Cat. 3, which performed much better than that of the others, and the Pd(II) catalysts (Cat. 4, Cat. 5 and Cat. 6) without co-catalyst Cu(I) performed not well than Cat. 3. We proposed that this phenomenon may attribute to the electron-withdrawing group, which could improve the catalyze activity of the complex system (Fig. 1, 4d). Therefore, in this article, we chose Cat. 3 as the catalyst model to the following research.
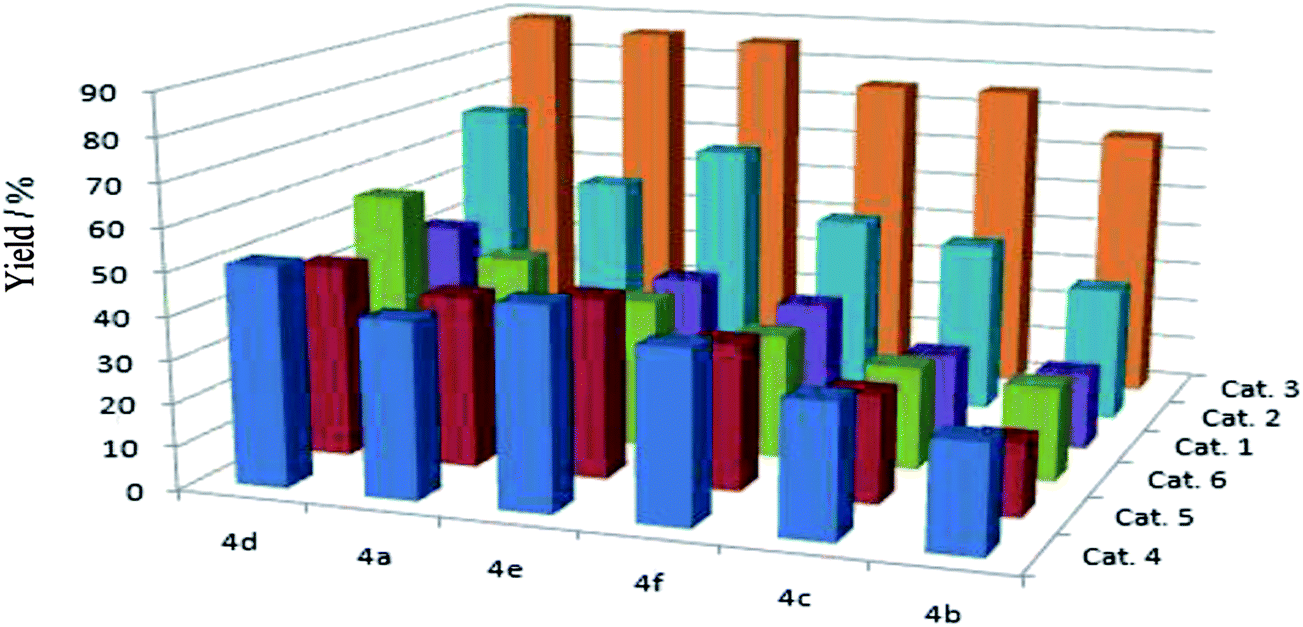 | ||
| Fig. 1 Parallel multisubstrate for the reactions of phenylacetylene with six aryl bromides catalyzed by Pd(II) catalysts. | ||
To verify the solvent and base effects on Sonogashira coupling reactions, we investigated the Sonogashira coupling reaction with the present catalyst using the coupling of bromobenzene with phenylacetylene as a model reaction to evaluate the reaction conditions (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Base | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 2a (1.2 mmol), 3a (0.8 mmol), base (2 equiv.), solvent (2 mL), Cat. 3 (1.0 mol%), aerobic condition.b Isolated yields. | |||||
| 1 | DMSO | Et3N | 25 | 1 | Trace |
| 2 | DMSO | Et3N | 40 | 1 | 21 |
| 3 | DMSO | Et3N | 60 | 1 | 63 |
| 4 | DMSO | Et3N | 80 | 1 | 86 |
| 5 | DMSO | Et3N | 90 | 1 | 89 |
| 6 | Dioxane | Et3N | 80 | 3 | 78 |
| 7 | DMF | Et3N | 80 | 2.5 | 76 |
| 8 | Toluene | Et3N | 80 | 4 | 20 |
| 9 | DMSO | K2CO3 | 80 | 1 | 42 |
| 10 | DMSO | Piperidine | 80 | 1 | 81 |
| 11 | DMSO | Pyridine | 80 | 1 | 82 |
| 12 | DMSO | No base | 80 | 1 | No reaction |
We initiated our investigation by screening for reaction conditions under which the Sonogashira reaction would proceed in the presence of oxygen, and in the absence of copper and phosphine. We observed that the yields of the desired coupling products could be isolated after 1.5 h at 25 °C to 80 °C in DMSO using Cat. 3 (catalyst loading 1.0 mol%), and TEA as base under aerobic conditions (Table 1, entries 1–4). Nevertheless, when the temperature was set up to 90 °C (Table 1, entry 5), the increasing trend of the isolated yield was dramatically decreased (only 3%). Measurable yields were also observed with the different solvents including Dioxane, DMF and Toluene (Table 1, entries 6, 7 and 8), however, Toluene as the solvent, gave a much lower amount of conversions than that of other solvents (Table 1, entry 8).
The base often plays an important role in the Sonogashira cross-coupling reactions, different kinds of bases were then screened (entries 10–12). As shown in the Table 1, organic bases are effective in the present catalytic system, especially TEA. However, inorganic bases are less effective and gave moderate yield of the target product. No conversion was observed in the absence of base.
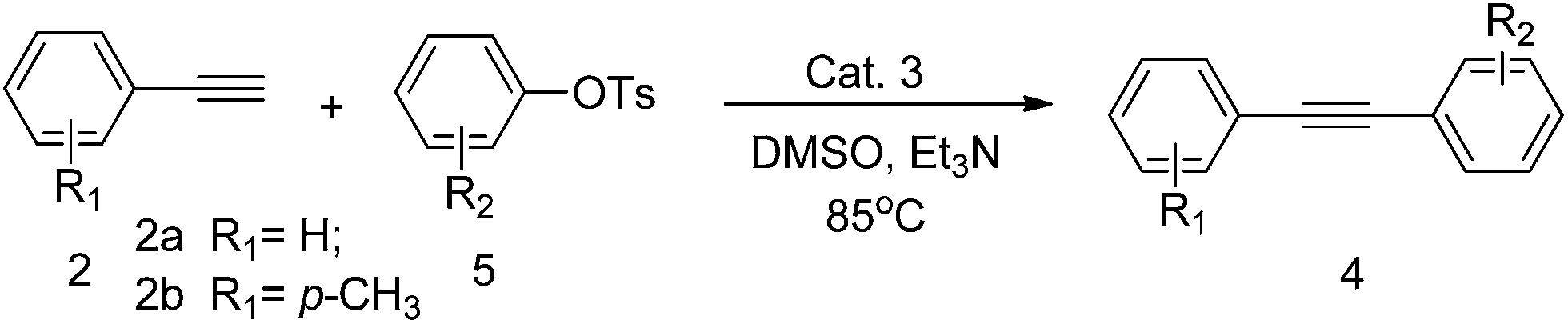
With the optimized reaction conditions in hand, a variety of aryl bromides, aryl tosylates and terminal alkynes have been examined for their generality. The results are summarized in Tables 2 and 3.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Terminal acetylene | Substrate | Products | Time (h) | Temperature (°C) | Yieldb (%) |
| a Conditions: 2 (1.2 mmol), 3 (0.8 mmol), TEA (2 equiv.), DMSO (2 mL), Cat. 3 (1.0 mol%), aerobic condition.b Isolated yield after flash chromatography.c Inert atmosphere. | ||||||
| 1 | 2a | 3a (Ph-Br) | 4a | 1 | 80 | 86 |
| 2c | 2a | 3b (p-OHPh-Br) | 4b | 6 | 85 | 65 |
| 3 | 2a | 3c (o-Me-m-MePh-Br) | 4c | 3 | 85 | 75 |
| 4 | 2a | 3d (p-CNPh-Br) | 4d | 0.7 | 80 | 89 |
| 5 | 2a | 3e (2-Br-thiophene) | 4e | 1.5 | 80 | 85 |
| 6 | 2a | 3f (1-Br-naphthalene) | 4f | 3 | 80 | 75 |
| 7c | 2b | 3b | 4g | 6 | 85 | 60 |
| 8 | 2b | 3c | 4h | 5 | 85 | 63 |
| 9 | 2b | 3d | 4i | 1.5 | 80 | 83 |
| 10 | 2b | 3e | 4j | 2 | 80 | 76 |
| 11 | 2b | 3f | 4k | 4 | 85 | 65 |
| 12 | 2c | 3a | 4l | 1.5 | 80 | 88 |
| 13 | 2c | 3g (p-CH3Ph-Br) | 4m | 2 | 80 | 74 |
| 14 | 2a | 3h (2-NH2-5-Br-pyrazin) | 4s | 4.5 | 80 | 72 |
| 15 | 2b | 3h | 4t | 5 | 80 | 67 |
|
|||||
|---|---|---|---|---|---|
| Entrya | Terminal acetylene | Substrate | Product | Time (h) | Yieldb (%) |
| a Conditions: 2 (1.2 mmol), 5 (0.8 mmol), TEA (2 equiv.), DMSO (2 mL), Cat. 3 (1.0 mol%), aerobic condition.b Isolated yield after flash chromatography. | |||||
| 1 | 2a | 5a (Ph-OTs) | 4a | 5 | 82 |
| 2 | 2a | 5b (1-OTs-naphthalene) | 4f | 7 | 73 |
| 3 | 2a | 5c (p-ClPh-OTs) | 4n | 5 | 83 |
| 4 | 2a | 5d (p-NO2Ph-OTs) | 4o | 4 | 86 |
| 5 | 2b | 5a | 4p | 6 | 72 |
| 6 | 2b | 5e (p-CH3Ph-OTs) | 4q | 7 | 61 |
| 7 | 2b | 5b | 4k | 9 | 60 |
| 8 | 2b | 5c | 4r | 5 | 76 |
| 9 | 2b | 5d | 4u | 5 | 77 |
As demonstrated in Table 2, all the reactions proceeded smoothly under the optimum reaction conditions, and the desired products were formed in good yields (63–89%), showing its wide substrate tolerance. Reaction of aryl bromides with electron-withdrawing substituents such as cyano (Table 2, entries 4 and 9) with phenylacetylene and 4-ethynyltoluene gave excellent yields of expected products (83–89%), while aryl bromides with electron-donating groups such as hydroxy and methyl (Table 2, entries 2, 7 and 13) gave the corresponding coupling products in a slightly lower yield. Even sterically hindered substrates, such as 1-bromo-2,3-dimethylbenzene, were also successfully coupled in good yields (Table 2, entries 3 and 8), although a slight higher temperature and a longer reaction time were required. To our satisfaction, heterocycle, such as 2-bromothiophene and 5-bromopyrazin-2-amine (Table 2, entries 5, 10, 14 and 15) also gave good yields (67–85%).
Since only few examples of Sonogashira cross-coupling reactions utilizing aryl tosylates have been reported,8 we next wanted to investigate the coupling of various aryl tosylates with terminal alkynes. As shown in Table 3, high catalytic activity was observed in the coupling of aryl tosylates possessing electron-donating groups such as fused ring (Table 3, entries 2 and 7), and 1-bromo-4-nitrobenzene (Table 3, entries 4 and 9) has electron-deficient aromatic rings also gave the corresponding coupling product under the reaction conditions identified above.
Generally speaking, the synthesis of skew symmetric tolanes can be realized by employing two different measures (Scheme 4). From a synthetic point of view it is important to ask which of the two leads to better yields when R or R′ are sterically demanding or undemanding or when R or R′ possess electron-releasing or -donating character.
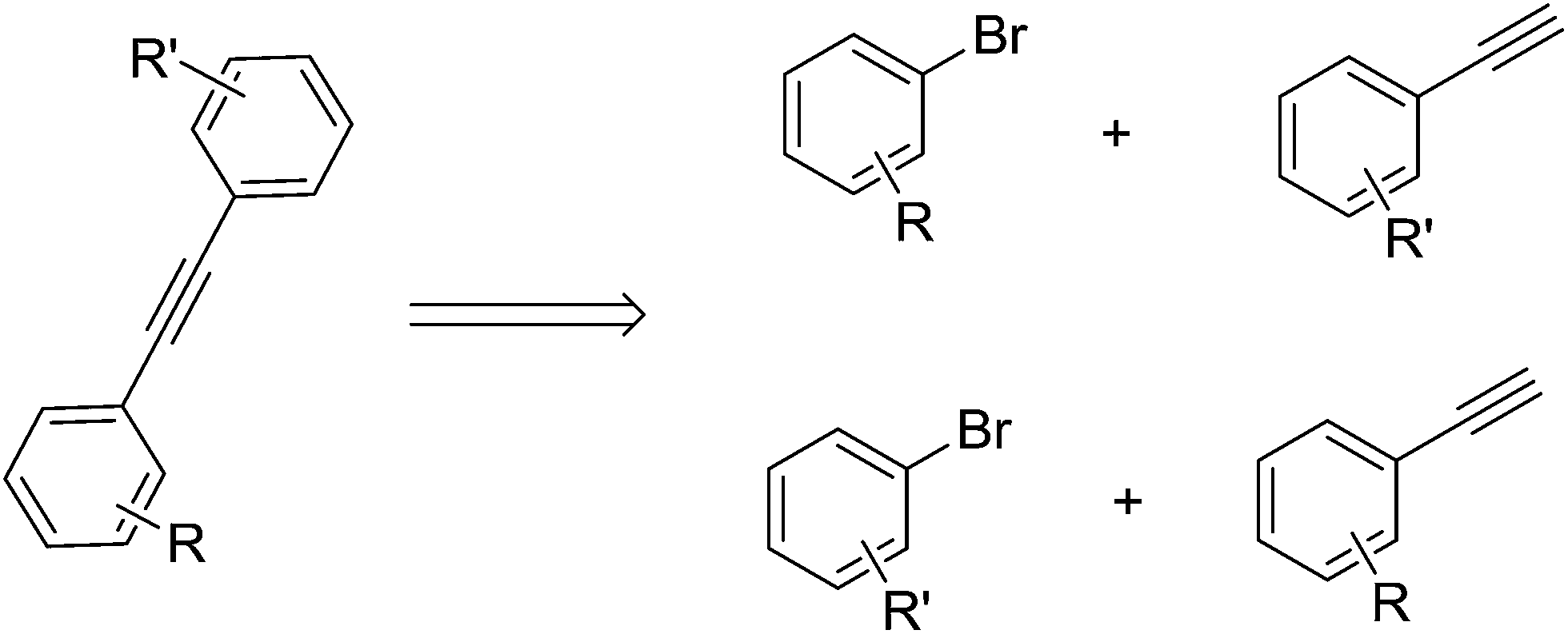 | ||
| Scheme 4 Potential Sonogashira routes to tolanes. | ||
In the first place, we decided to investigate which reaction (Scheme 5) would be more strongly influenced by electronic effects. As the above statement, electron withdrawing groups have an effect on increasing the reactivity of both substrates. The reaction C (Scheme 5, Fig 2) attains 71% yield via 30 min, at the same time, while the reaction D reaches 46%. However, when the reaction time was prolonged to 90 min, both reactions acquired almost 90% yields. When the reaction B achieved 81% yield after 90 min's reaction, reaction A only exhibited 56% yield. Nonetheless, they all reached to 80% yields after 180 min reaction. For this catalyst system, the reactivity of aryl bromide is higher than that of aryl tosylates. After 10 min's reaction, F only gave 9% yield, and, the reaction E has no conversion at all. For all this, they all could reach 80% yields after 150 min's reaction. Finally, the yield of reaction E is lightly lower than that of F.
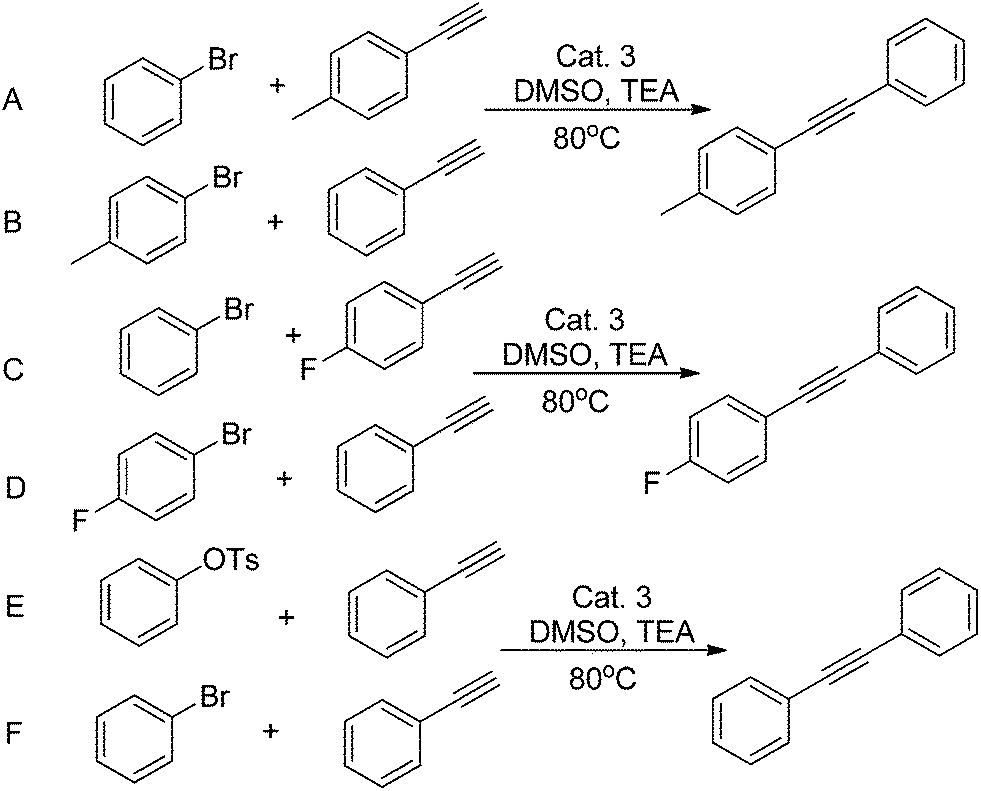 | ||
| Scheme 5 Electronic modifications of substrates and aryl tosylates or aryl bromides for Sonogashira coupling. | ||
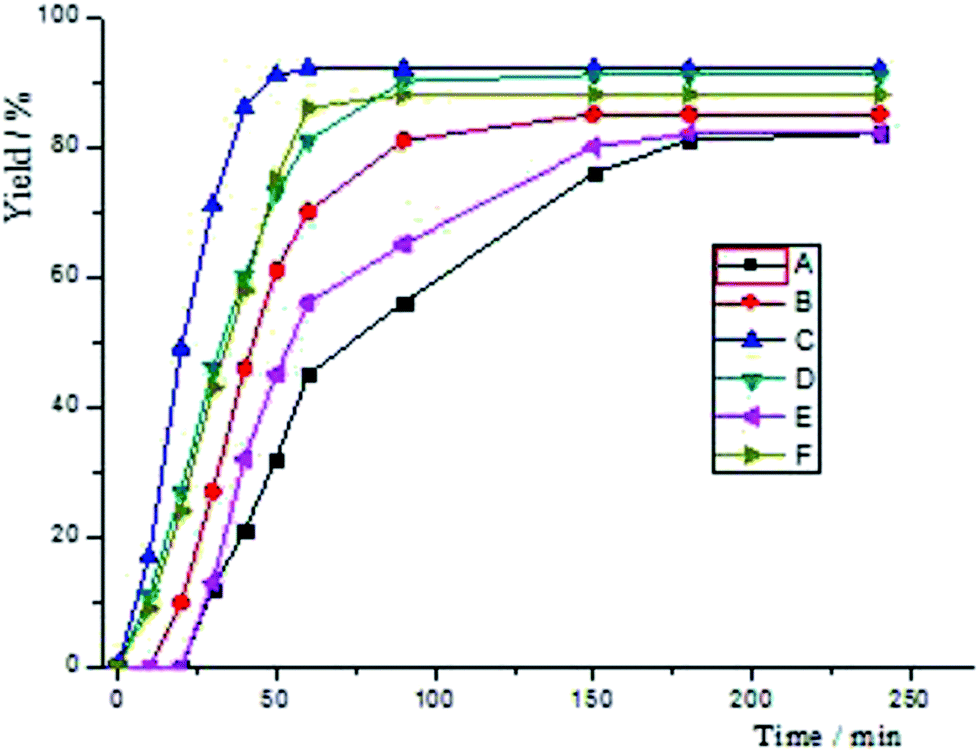 | ||
| Fig. 2 Time yield curves for the reactions of Scheme 5. | ||
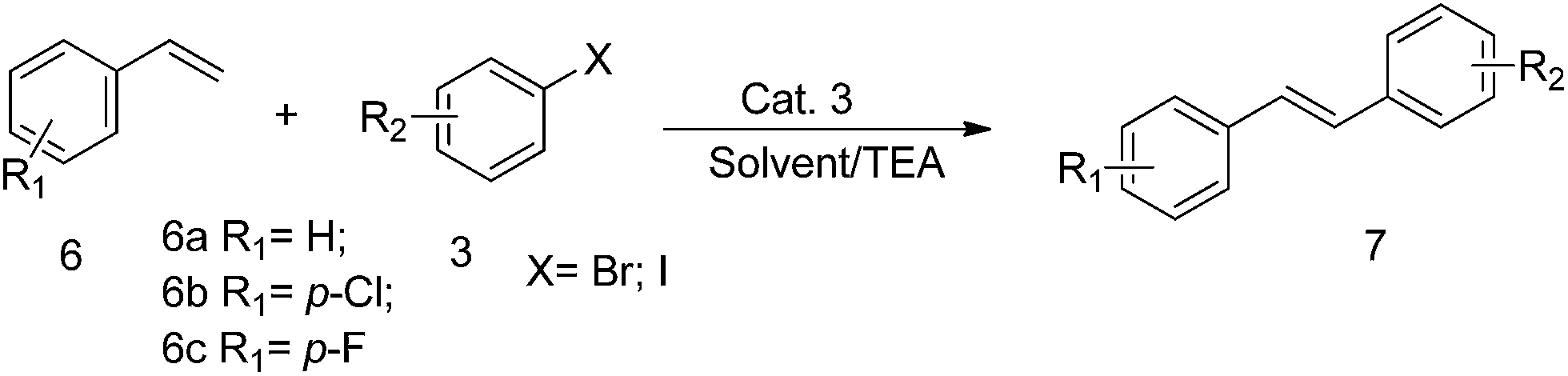
Encouraged by the achievements in the Suzuki and Sonogashira reactions, we further tested the catalytic activity of Cat. 3 in the Heck reactions using various aryl bromides and terminal olefins as substrates. The results of the Heck reactions were summarized in the Table 4. Taking bromobenzene and styrene as substrates, Cat. 3 at a loading of 1.0 mol% afforded 75% yield (Table 4, entry 1). For bromobenzene with electron-withdrawing groups, including –F, –Cl, the reaction proceeded smoothly and 77–85% yields of the corresponding coupling product were acquired. However, for bromobenzene with an electron-rich group, such as –OCH3, –OH, –CH3, the reaction proceeded much slower, and only 57–73% yields were obtained.
|
|||||
|---|---|---|---|---|---|
| Entrya | Terminal olefin | Substrate | Products | Time (h) | Yieldb (%) |
| a Conditions: 6 (1.2 mmol), 3 (0.8 mmol), TEA (2 equiv.), dioxane (2 mL), Cat. 3 (1.0 mol%), aerobic condition.b Isolated yield after flash chromatography.c DMF (2 mL), inert atmosphere. | |||||
| 1 | 6a | 3a | 7a | 8 | 75 |
| 2c | 6a | 3b | 7b | 16 | 28 |
| 3k (p-OHPh-I) | 12 | 62 | |||
| 3 | 6a | 3h (p-OCH3Ph-Br) | 7c | 24 | 32 |
| 3l (p-OCH3Ph-I) | 15 | 60 | |||
| 4 | 6a | 3g | 7d | 10 | 67 |
| 5 | 6a | 3i (p-ClPh-Br) | 7e | 8 | 79 |
| 6 | 6a | 3j (m-FPh-Br) | 7f | 10 | 77 |
| 7 | 6b | 3h | 7g | 13 | 57 |
| 8 | 6b | 3i | 7h | 7 | 80 |
| 9 | 6b | 3j | 7i | 9 | 80 |
| 10 | 6c | 3h | 7j | 20 | 34 |
| 3l | 14 | 62 | |||
| 11 | 6c | 3g | 7k | 12 | 73 |
| 12 | 6c | 3a | 7l | 8 | 78 |
| 13 | 6c | 3j | 7m | 9 | 81 |
| 14 | 6c | 3i | 7n | 7 | 85 |
We proposed a plausible mechanism for this Cat. 3 mediated Sonogashira and Heck coupling reactions (Scheme 6). The Pd(0) species could be generated readily from the reaction of Cat. 3 with substrates, ligands, and TEA,2a,4,9e,14 and, followed by the oxidative addition of Pd(0) with R1X to form intermediate (1). On account of alkalinity of TEA is feeblish, the acetylene is not able to deprotonation, therefore, acetylene and Pd complex form intermediate (2), which was further involved in the typical catalytic cycle of Sonogashira coupling reaction. The mechanism is also suitable for Heck cross-coupling reactions.
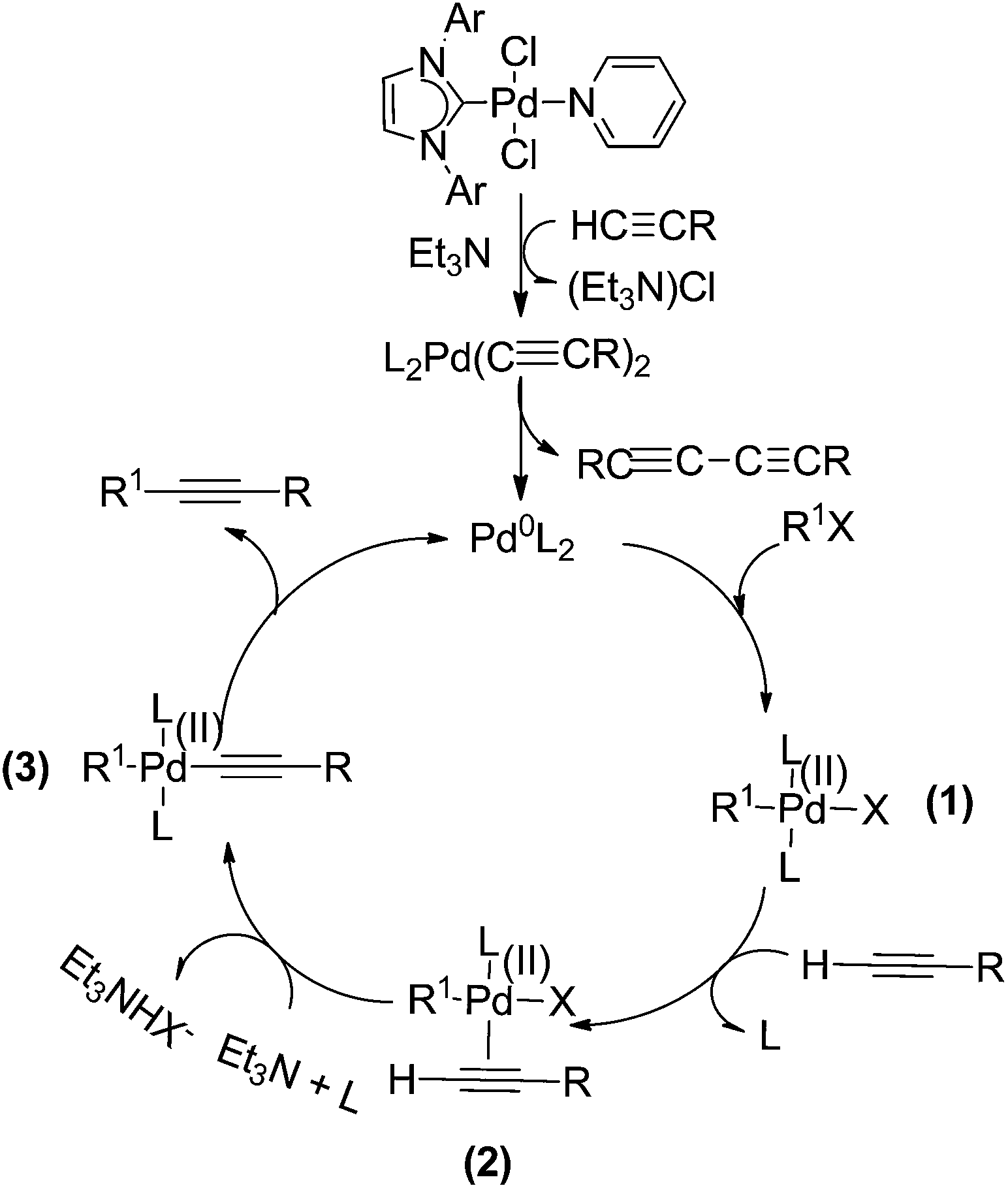 | ||
| Scheme 6 Proposed mechanism for Sonogashira reactions catalyzed by Cat. 3. | ||
In summary, the NHC–Pd complex featured with air-insensitivity, was proved to be a highly efficient catalyst for the Suzuki, Sonogashira and Heck coupling reactions. In a simple one-pot procedure free of co-catalyst, this complex is available for a wide range of substrates under mild conditions with good to excellent product yields and a low catalyst loading.
Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China (no. 21302098).References
- (a) J. D. Senra, L. F. B. Malta, M. E. H. M. da Costa, R. C. Michel, L. C. S. Aguiar, A. B. C. Simas and O. A. C. Antunes, Adv. Synth. Catal., 2009, 351, 2411–2422 CrossRef CAS; (b) R. Bernini, S. Cacchi, G. Fabrizi, G. Forte, F. Petrucci, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, Org. Biomol. Chem., 2009, 7, 2270–2273 RSC; (c) J. F. Cívicos, D. A. Alonso and C. Njera, Adv. Synth. Catal., 2013, 355, 203–208 CrossRef; (d) L. Zhou, F. Ye, Y. Zhang and J. B. Wang, J. Am. Chem. Soc., 2010, 132, 13590–13591 CrossRef CAS PubMed; (e) K. H. Chung, C. M. So, S. M. Wong, C. H. Luk, Z. Y. Zhou, C. P. Lau and F. Y. Kwong, Chem. Commun., 2012, 48, 1967–1969 RSC; (f) Z. Y. Wang, G. Q. Chen and L. X. Shao, J. Org. Chem., 2012, 77, 6608–6614 CrossRef CAS PubMed.
- (a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS; (b) W. J. Tang, S. Keshipeddy, Y. D. Zhang, X. D. Wei, J. Savoie, N. D. Patel, N. K. Yee and C. H. Senanayake, Org. Lett., 2011, 13, 1366–1369 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- Y. Liang, Y. X. Xie and J. H. Li, J. Org. Chem., 2006, 71, 379–381 CrossRef CAS PubMed.
- (a) H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954–6956 CrossRef CAS PubMed; (b) T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed; (c) Z. Gonda, G. L. Tolnai and Z. Novák, Chem.–Eur. J., 2010, 16, 11822–11826 CrossRef CAS PubMed; (d) N. E. Leadbeater, Nat. Chem., 2010, 2, 1007 CrossRef CAS PubMed.
- (a) H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (b) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (c) T. Schulz, C. Torborg, S. Enthaler, B. Schaeffner, A. Dumrath, A. Spannenberg, H. Neumann, A. Boerner and M. Beller, Chem.–Eur. J., 2009, 15, 4528–4533 CrossRef CAS PubMed; (d) A. D. Finke, E. C. Elleby, M. J. Boyd, H. Weissman and J. S. Moore, J. Org. Chem., 2009, 74, 8897–8900 CrossRef CAS PubMed; (e) J. L. Bolligera and C. M. Frech, Adv. Synth. Catal., 2009, 351, 891–902 CrossRef.
- (a) D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993–5996 CrossRef CAS PubMed; (b) O. R'kyek, N. Halland, A. Lindenschmidt, J. Alonso, P. Lindemann, M. Urmann and M. Nazaré, Chem.–Eur. J., 2010, 16, 9986–9989 CrossRef PubMed; (c) Y. Nishihara, D. Ogawa, S. Noyori and M. Iwasaki, Chem. Lett., 2012, 41, 1503 CrossRef CAS.
- D. A. Alonso, C. Nájera, I. Pastor and M. Yus, Chem.–Eur. J., 2010, 16, 5274–5284 CrossRef CAS PubMed.
- (a) Z. Gu, Z. Li, Z. Liu, Y. Wang, C. Liu and J. Xiang, Catal. Commun., 2008, 9, 2154–2157 CrossRef CAS PubMed; (b) M. H. Sie, Y. H. Hsieh, Y. H. Tsai, J. R. Wu, S. J. Chen, P. V. Kumar, J. H. Lii and H. M. Lee, Organometallics, 2010, 29, 6473–6481 CrossRef CAS; (c) X. Cui, Z. Li, C. Z. Tao, Y. Xu, J. Li, L. Liu and Q. X. Guo, Org. Lett., 2006, 8, 2467–2470 CrossRef CAS PubMed; (d) E. Wuttke, B. Nägele, B. Weibert and F. Kessler, Organometallics, 2011, 30, 6270–6282 CrossRef CAS; (e) A. Kamal, V. Srinivasulu, B. N. Seshadri, N. Narkandeya, A. Alarifi and N. Shankaraiah, Green Chem., 2012, 14, 2513–2522 RSC; (f) D. Méry, K. Heuzé and D. Astruc, Chem. Commun., 2003, 15, 1934–1935 RSC; (g) M. Bakherad, A. Keivanloo and S. Samangooei, Tetrahedron Lett., 2012, 53, 5773–5776 CrossRef CAS PubMed.
- (a) Y. Zhang, T. F. Jamison, S. Patel and N. Mainolfi, Org. Lett., 2011, 13, 280–283 CrossRef CAS PubMed; (b) J. P. Huang, W. Wang and H. Li, ACS Catal., 2013, 3, 1526–1536 CrossRef CAS.
- (a) H. J. Xu, Y. Q. Zhao and X. F. Zhou, J. Org. Chem., 2011, 76, 8036–8041 CrossRef CAS PubMed; (b) A. Ohtaka, T. Teratani, R. Fujii, K. Ikeshita, T. Kawashima, K. Tatsumi, O. Shimomura and R. Nomura, J. Org. Chem., 2011, 76, 4052–4060 CrossRef CAS PubMed.
- C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- H. F. Lu, F. F. Yang, R. Z. Wu, J. T. Zhou, Z. B. Luo and Y. J. Hu, submitted for publication.
- R. R. Tykwinski, Angew. Chem., Int. Ed., 2003, 42, 1566–1568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02480j |
| This journal is © The Royal Society of Chemistry 2014 |