
Structure and stability of two dimensional phosphorene with ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O or NH functionalization†
O or NH functionalization†
Jun Dai and
Xiao Cheng Zeng*
Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA. E-mail: xzeng1@unl.edu
First published on 24th September 2014
Abstract
We investigate the stability and electronic properties of oxy-(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or imine-(NH) functionalized monolayer phosphorene with either single-side or double-side functionalization based on density-functional theory calculations. Our thermodynamic analysis shows that oxy-functionalized phosphorene can be formed under the conditions ranging from ultrahigh vacuum to high concentrations of molecular O2, while the imide-functionalized phosphorene can be formed at relatively high concentrations of molecular N2H2. In addition, our Born–Oppenheimer molecular dynamics (BOMD) simulation shows that under ambient conditions both O2 and N2H2 can etch phosphorene away.
O) or imine-(NH) functionalized monolayer phosphorene with either single-side or double-side functionalization based on density-functional theory calculations. Our thermodynamic analysis shows that oxy-functionalized phosphorene can be formed under the conditions ranging from ultrahigh vacuum to high concentrations of molecular O2, while the imide-functionalized phosphorene can be formed at relatively high concentrations of molecular N2H2. In addition, our Born–Oppenheimer molecular dynamics (BOMD) simulation shows that under ambient conditions both O2 and N2H2 can etch phosphorene away.
Recently, a new two-dimensional (2D) semiconductor, phosphorene, has been successfully isolated through mechanical cleavage of crystalline black phosphorus.1–4 It has been demonstrated that phosphorene-based field-effect transistors (FETs) can exhibit a high on/off ratio (∼105) and relatively high carrier mobility (up to 1000 cm2 V−1 s−1),1,2,4 suggesting potential applications of phosphorene in nano-electronic devices. Black phosphorus, the bulk counterpart of phosphorene, is the most stable form of phosphorus and was discovered by Bridgman in 1914.5 Like graphite, black phosphorus is also a layered material with weak interlayer van der Waals (vdW) interaction. In each layer, the phosphorus atom is bonded with three adjacent phosphorus atoms, forming a puckered honeycomb structure.6,7 The three bonds take up all three valance electrons of phosphorus atom. Thus, a monolayer phosphorene is a 2D semiconductor with a direct bandgap of about 0.3 eV.8–12 More interestingly, the direct bandgap feature of few-layer phosphorene is dependent on the thickness. Previous theoretical calculations show that the bandgap can be tuned from 1.5 eV for monolayer to 0.6 eV for 5-layer phosphorene.13 Furthermore, either in-plane or out-of-plane strain can significantly change the bandgap of monolayer phosphorene. Indeed, a ∼5% in-plane strain can convert the monolayer phosphorene from a direct-gap to an indirect-gap semiconductor,2 while a vertical compression can induce the semiconductor-to-metal transition.14 The in-plane anisotropic optical properties of phosphorene15,16 and its potential applications in solar-cell systems17 have also been reported.
Although black phosphorus is the most stable allotrope of phosphorus, it is still reactive under ambient condition.5,18–20 Especially, it has been reported that phosphorene flakes can be etched away at ambient condition.20 We also note that for graphene, chemical functionalization can be an effective way to tune its electronic properties.21–29 Therefore, it is useful to study stability and properties of chemical functionalized phosphorene sheets. Note also that carbon and phosphorus have different valence electron configuration, namely, 2s22p2 for carbon and 3s23p3 for phosphorus. In graphene, three valence electrons in carbon form sp2 orbitals and the remaining valence electrons, one in each carbon atom, form delocalized π orbital. On the other hand, in phosphorene, phosphorus forms sp3 bonding with a lone pair of valence electrons in each phosphorus atom. Therefore, the chemical species used for chemical functionalization of graphene may not be suitable for functionalization of phosphorene.
Here, we consider divalent electron donors such as O, S, NH and CH2 for possible phosphorene functionalization in view of successful synthesis of organophosphonates whose simplest forms include OPH3, SPH3, HNPH3 and H2CPH3.30,31 The structures and thermodynamic stabilities of these divalent ligands functionalized phosphorene are carefully examined. Effect of the functionalization on the electronic structure of phosphorene is also discussed. We show that oxy-(O) functionalized phosphorene can be automatically formed in the presence of O2 with either high or low O2 concentration, while imide-(NH) functionalized phosphorene can be automatically form in relatively high concentration of N2H2. Our BOMD simulations show that both oxy- and imide-functionalization can etch phosphorene away at the ambient condition in the presence of either O2 or N2H2.
For density-functional theory (DFT) calculations, the generalized gradient approximation (GGA) for the exchange-correlation potential is adopted. The plane-wave cutoff energy for wave function is set to 500 eV. The ion-electron interaction is treated with the projected augmented wave (PAW)32,33 method as implemented in the Vienna ab initio simulation package (VASP 5.3).34,35 For geometry optimization, a 8 × 10 × 1 Monkhorst–Pack k-mesh is adopted for functionalized phosphorene systems. A vacuum spacing of ∼20 Å is used so that the interaction between adjacent layers can be neglected. During the geometric optimization, both lattice constants and atomic positions are relaxed until the residual force on atoms are less than 0.01 eV Å−1 and the total energy change is less than 1.0 × 10−5 eV. In addition, a combination of optB88-vdW36,37 for geometry optimization and HSE06 (ref. 38) for band structure calculation (based on the optB88-vdW optimized structure) is used, which has been proven very reliable for few-layer phosphorene systems.2 Our benchmark calculations for bulk black phosphorus also confirm reliability of the selected two DFT methods (see ESI Fig. S1†).
Four divalent adsorbates, namely, O, S, NH and CH2 are initially considered for functionalization of the phosphorene monolayer. However, our calculations show that S atoms cannot effectively bond to P atoms while CH2 can disrupt the integrity of phosphorene (see ESI Fig. S2†). Hereafter, we only focus on O and NH functionalized phosphorene. Both single-side and double-side functionalization are taken into account. For simplicity, we use P–O-half and P–O notations to denote single-side and double-side O functionalized phosphorene, and P–NH-half and P–NH for the single-side and double side NH functionalized phosphorene. The optimized structures are shown in Fig. 1 and the structural parameters of P–O-half, P–O, P–NH-half, and P–NH are summarized in Table 1. One can see that the functionalization with four different patterns on phosphorene share some common features. Firstly, the functionalization with O and NH results in an in-plane structural expansion, where the in-plane lattice constant a expands over 4.9–12.5% while b expands over 11.7–21.0% with different functionalization patterns. As a result, the in-plane P–P bond length also expands. The P–P bond length that is out of the xy plane expands over 1.3–5.6%. These expansions stem from the weakening of bond strength since these adsorbates are electron acceptors which can fetch electrons from P atoms (see ESI Table S1†). Another intriguing feature is that in P–O-half, the difference in bond length for two types of P–P bonds is almost negligible.
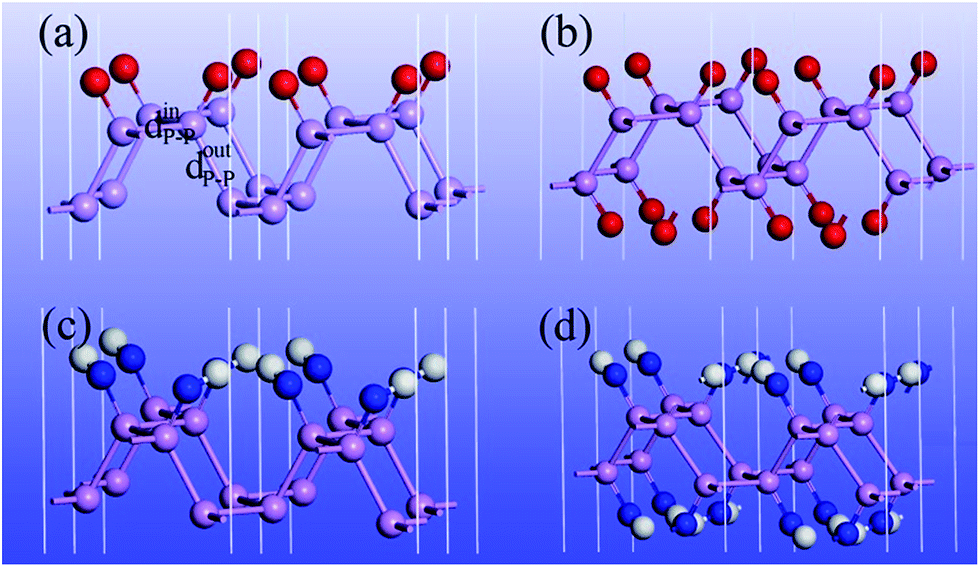 | ||
| Fig. 1 Optimized structure of (a) P–O-half, (b) P–O, (c) P–NH-half and (d) P–NH. Red, white, blue and pink spheres denote oxygen, hydrogen, nitrogen and phosphorus atoms, respectively. dinP–P and doutP–P denotes the P–P bond length in and out of the xy plane. | ||
O, N) bond length (dP–x), in-plane lattice constants (latt. const.), and adsorption energies for functionalized phosphorene monolayer. P–O-half, P–O, P–NH-half, P–NH and P denote monolayer phosphorene with single-side O functionalization, double-side O functionalization, single-side NH functionalization, double-side –NH functionalization and pristine monolayer phosphorene, respectively
| dinP–P (Å) | doutP–P (Å) | dP–x (Å) | latt. const. (Å) | Ead (eV) | |
|---|---|---|---|---|---|
| P–O-half | 2.283 | 2.283 | 1.478 | a = 5.091, b = 3.469 | 4.132 |
| P–O | 2.338 | 2.380 | 1.483 | a = 5.514, b = 3.690 | 4.184 |
| P–NH-half | 2.288 | 2.304 | 1.555 | a = 5.136, b = 3.492 | 2.634 |
| P–NH | 2.386 | 2.367 | 1.562 | a = 5.466, b = 3.719 | 2.754 |
| P | 2.220 | 2.253 | N/A | a = 4.556, b = 3.305 | N/A |
Adsorption energies for the functional groups are computed at the HSE06 level, which is defined as: Ead = − (Etot − EP − nEx)/n, where Etot is the total energy of the functionalized phosphorene with nX (X = O or NH) as ligands, EP is the energy of pristine phosphorene monolayer, and Ex is the energy of an isolated X. According to this definition, a larger value of Ead means stronger adsorption. As shown in Table 1, the average adsorption energy in the double-side functionalization is slightly greater than that in the single-side functionalization. Besides, the adsorption of O is significantly stronger than that of NH, because O is a stronger electron acceptor than NH. Thus, the P–O bonding is stronger than P–N.
Similar to the definitions used in the discussion of the thermodynamic stability of functionalized graphene nanoribbons39,40 and graphene oxides,41 we define the zero temperature formation energies of the functionalized phosphorene as:
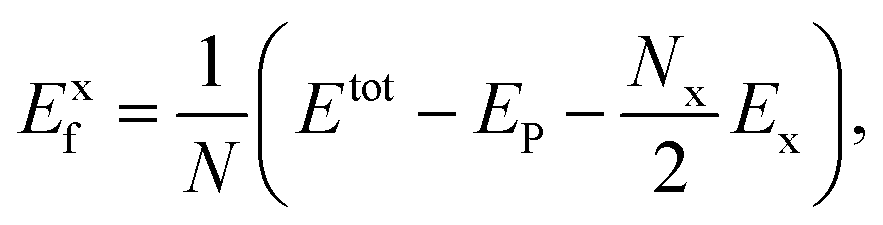 or NH in a supercell, and N is the number of phosphorus atom in a supercell. The stability of different structures can be determined by Exf under different experimental conditions. For example, in the presence of molecular O2 gas, the relative stability can be obtained by comparing
or NH in a supercell, and N is the number of phosphorus atom in a supercell. The stability of different structures can be determined by Exf under different experimental conditions. For example, in the presence of molecular O2 gas, the relative stability can be obtained by comparingwhere ρO = NO/N, at the absolute temperature T, and for a partial O2 pressure P, the chemical potential of O2 can be obtained as:
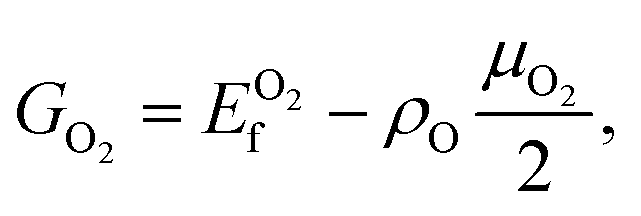
where H°(T) and S°(T) are the enthalpy and entropy at the pressure P° = 1 bar (taken from the JANAF thermochemical tables).42 For a given value of μ, the structure with a lower value of G is more stable.
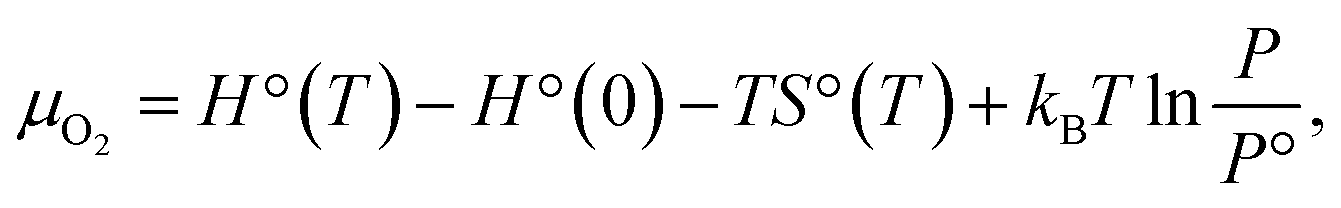
In Fig. 2, G vs. μ is plotted for the functionalized phosphorenes. First, we can see that P–O is more stable than P–O-half for all negative values of the O2 chemical potential, suggesting that the P–O is the more stable specie thermodynamically under experimental conditions ranging from ultrahigh vacuum to atmospheric concentration of molecular O2. For P–NH and P–NH-half, we can see that P–NH-half is more stable than P–NH at relatively low values of N2H2 chemical potential, while at relatively high values of N2H2 chemical potential, P–NH is more stable than P–NH-half. Thus, under the experimental condition of ultralow concentration of N2H2, P–NH-half is more stable, while under the condition of relatively high N2H2 concentration, P–NH is more stable than P–NH-half. Second, Fig. 2 suggests that phosphorene is unstable in the presence of O2 even in the ultrahigh vacuum concentration of molecular O2 (G is negative), and oxy-functionalized phosphorene can be automatically formed. In the presence of N2H2, phosphorene is unstable under condition of relatively high concentration of N2H2, and the imine-functionalized phosphorene can be automatically formed as well. To address the substrate induced lattice change in real systems, we computed the formation energies of P–O and P–NH with a uniaxial strain of 5% or −5% along a or b axis, or a biaxial strain of 5% or −5% along both a and b. The results are summarized in ESI Table S2.† One can see that a net effect of the strain on the G–μ curves as shown in Fig. 2 is a shift along the y axis in a range of +0.001 to +0.067 eV, therefore, will not change the conclusion regarding the thermal stability of P–O and P–NH.
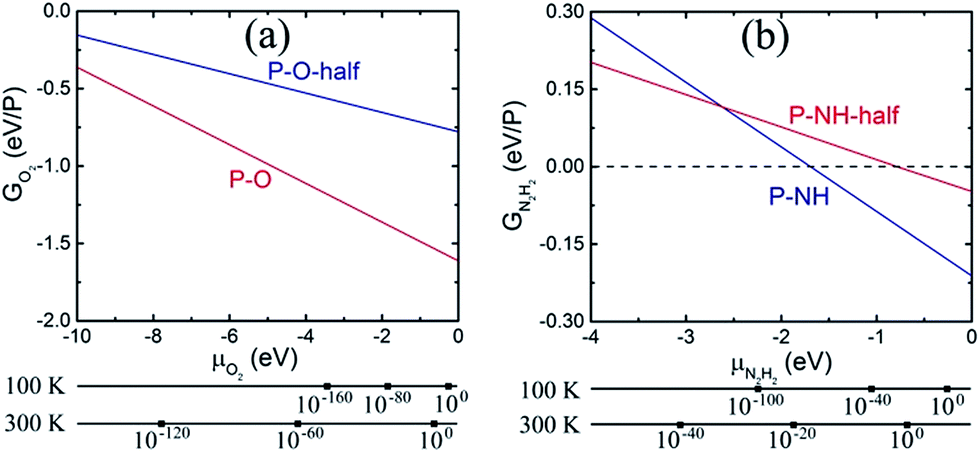 | ||
| Fig. 2 Formation energies versus chemical potential for (a) P–O-half and P–O, (b) P–NH-half and P–NH. The alternative axes show the pressure, in bar, of molecular O2 and N2H2 gases corresponding to the chemical potential at T = 100 K and 300 K. | ||
We have also studied thermal stability of the functionalized phosphorene by means of the BOMD simulations with the constant-pressure and constant-temperature (NPT) ensemble. Here, the pressure is set to 1 atm, while the temperature is controlled at either 70 K or 300 K. The time step is 2 fs, and the total simulation time is 8 ps. As shown in Fig. 3, at the low temperature (70 K), structures of P–O-half, P–O and P–NH-half are still robust and intact, indicating their stability near liquid nitrogen temperature. But for P–NH, the structure is partially destroyed, indicating at relatively high concentration of N2H2, phosphorene can be etched away. Near the room temperature (300 K), none of the four functionalized phosphorene sheets can retain their structure integrity, especially for the P–NH one which would decompose into several clusters with a huge volume expansion. The BOMD simulations indicate that these functionalized phosphorene sheets can be possibly observed at low temperature in the presence of either O2 or N2H2, while at high temperature, phosphorene can be etched away.
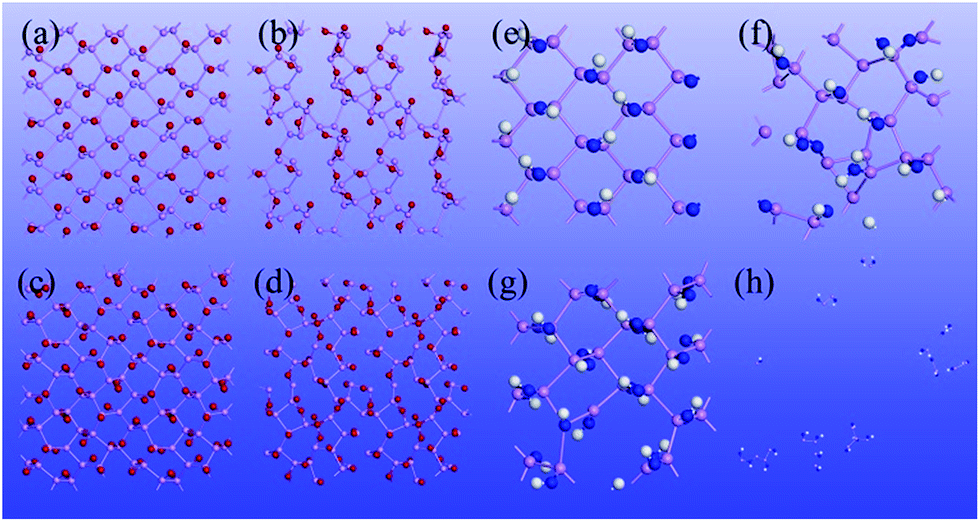 | ||
| Fig. 3 Snapshots of functionalized phosphorene monolayer at 8 ps of the Born–Oppenheimer molecular dynamics simulation in the NPT ensemble, (a) P–O-half at 70 K, (b) P–O-half at 300 K, (c) P–O at 70 K, (d) P–O at 300 K, (e) P–NH-half at 70 K, (f) P–NH-half at 300 K, (g) P–NH at 70 K and (h) P–NH at 300 K. | ||
Lastly, we examine effects of the chemical functionalization on the electronic structures of phosphorene monolayer. Our benchmark calculations show that unlike half-hydrogenated or half-fluorinated graphene systems43,44 for which their magnetic properties can be tuned by surface functionalization, here the four functionalized phosphorene sheets appear to be non-spin-polarized. This is because phosphorus atoms in phosphorene adopt the sp3 hybridization; the adsorbates act as electron acceptor and bond with P atoms with the electron lone pairs on P atoms. Hence, there are no unpaired electrons and spin polarization in these systems. The HSE06 band structures and atomic projected density of states (pDOS) are plotted in Fig. 4. Except for P–O, the direct bandgap feature is not retained in P–O-half, P–NH-half and P–NH. The bandgap of P–O-half (1.55 eV) is close to pristine monolayer phosphorene (∼1.5 eV).13 The bandgap in other three functionalized phosphorene sheets are slightly reduced to 1.03 eV, 1.44 eV and 1.24 eV for P–O, P–NH-half and P–NH, respectively. Moreover, in functionalized phosphorene sheets, the VBM is a hybrid state in which O or NH contributes the most part, while the CBM is nearly contributed by P atoms. It is worthy of mentioning that although previous theoretical calculations have shown that the ∼5% in-plane compression can convert monolayer phosphorene from being a direct-gap to an indirect-gap semiconductor,2 the studied phosphorene-based FET devices still have relatively good properties. Possible contamination of phosphorene by O2 or N2H2, especially O2, should also be carefully monitored since the contamination can harm performance of the devices.
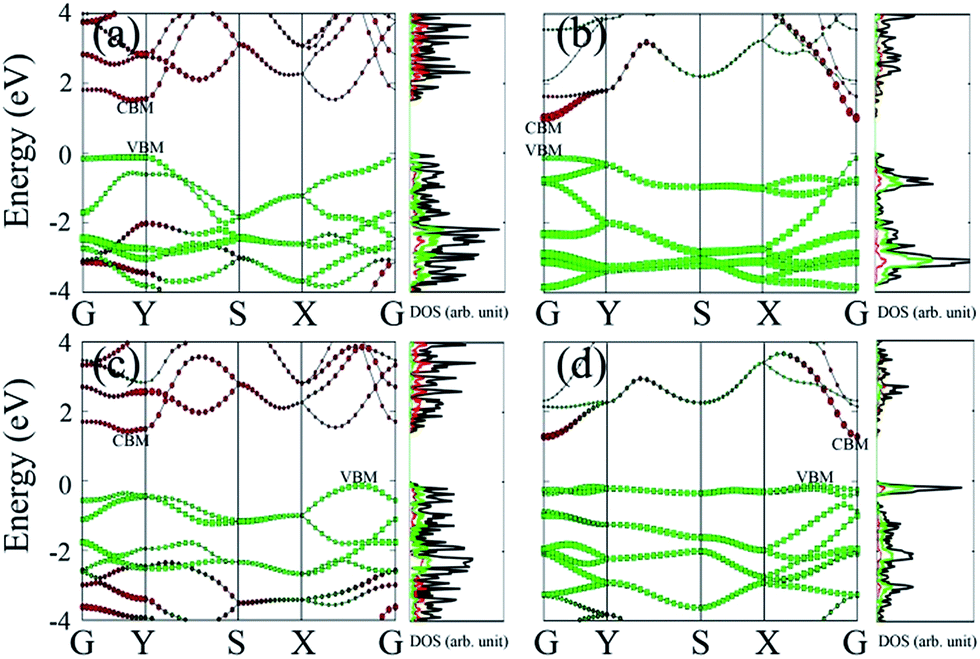 | ||
| Fig. 4 HSE06 band structure and atomic projected partial density of states (pDOS) of (a) P–O-half, (b) P–O, (c) P–NH-half and (d) P–NH. The Fermi level is set to 0. In band structures, the size of red spheres and green rectangles (lines) denote the contribution from P and O (or NH), and valence band maximum and conduction band minimum and labelled as VBM and CBM, while in pDOS, black, red and green lines denote the total DOS, pDOS of P and O (or NH), respectively. | ||
Conclusions
We investigate properties of oxy-(O) and imine-(NH) functionalized monolayer phosphorene with either single-side or double-side functionalization. Our thermodynamic analysis shows that in the presence of molecular O2 with either high or low concentration, oxy-functionalized phosphorene will be automatically formed while imide-functionalized phosphorene will be formed at relatively high concentration of N2H2. Moreover, our BOMD simulation suggests that at ambient conditions the phosphorene will be etched away.
Notes and references
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- E. S. Reich, Nature, 2014, 506, 19 CrossRef PubMed.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 Search PubMed.
- P. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS.
- J. C. Jamieson, Science, 1963, 139, 1291–1292 CAS.
- A. Brown and S. Rundqvist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS.
- R. W. Keyes, Phys. Rev., 1953, 92, 580 CrossRef CAS.
- H. Asahina and A. Morita, J. Phys. C: Solid State Phys., 1984, 17, 1839 CrossRef CAS.
- T. Takahashi, H. Tokailin, S. Suzuki, T. Sagawa and I. Shirotani, J. Phys. C: Solid State Phys., 1985, 18, 825 CrossRef CAS.
- D. Warschauer, J. Appl. Phys., 2004, 34, 1853–1860 CrossRef PubMed.
- H. Guo, N. Lu, J. Dai, X. Wu and X. C. Zeng, J. Phys. Chem. C, 2014, 118, 14051–14059 CAS.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 Search PubMed.
- A. Rodin, A. Carvalho and A. Neto, Phys. Rev. Lett., 2014, 112, 176801 CrossRef CAS.
- R. Fei and L. Yang, Nano Lett., 2014, 14, 2884 CrossRef CAS PubMed.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- J. Dai and X. C. Zeng, J. Phys. Chem. Lett., 2014, 5, 1289–1293 CrossRef CAS.
- T. Nishii, Y. Maruyama, T. Inabe and I. Shirotani, Synth. Met., 1987, 18, 559–564 CrossRef CAS.
- S. P. Koenig, R. A. Doganov, H. Schmidt, A. Neto and B. Oezyilmaz, Appl. Phys. Lett., 2014, 104, 103106 CrossRef PubMed.
- A. Castellanos-Gomez, L. Vicarelli, E. Prada, J. O. Island, K. Narasimha-Acharya, S. I. Blanter, D. J. Groenendijk, M. Buscema, G. A. Steele and J. Alvarez, 2D Materials, 2014, 1, 025001 CrossRef.
- S. Ryu, M. Y. Han, J. Maultzsch, T. F. Heinz, P. Kim, M. L. Steigerwald and L. E. Brus, Nano Lett., 2008, 8, 4597–4602 CrossRef CAS PubMed.
- D. Elias, R. Nair, T. Mohiuddin, S. Morozov, P. Blake, M. Halsall, A. Ferrari, D. Boukhvalov, M. Katsnelson and A. Geim, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- J.-A. Yan, L. Xian and M. Chou, Phys. Rev. Lett., 2009, 103, 086802 CrossRef.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson and P. E. Sheehan, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- M. A. Ribas, A. K. Singh, P. B. Sorokin and B. I. Yakobson, Nano Res., 2011, 4, 143–152 CrossRef CAS PubMed.
- F. Withers, T. H. Bointon, M. Dubois, S. Russo and M. F. Craciun, Nano Lett., 2011, 11, 3912–3916 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- M. Z. Hossain, J. E. Johns, K. H. Bevan, H. J. Karmel, Y. T. Liang, S. Yoshimoto, K. Mukai, T. Koitaya, J. Yoshinobu and M. Kawai, Nat. Chem., 2012, 4, 305–309 CrossRef CAS PubMed.
- J. Dai, Y. Zhao, X. Wu, X. C. Zeng and J. Yang, J. Phys. Chem. C, 2013, 117, 22156–22161 CAS.
- N. A. Hosea, H. A. Berman and P. Taylor, Biochemistry, 1995, 34, 11528–11536 CrossRef CAS.
- I. Alkorta, G. Sánchez-Sanz, J. Elguero and J. E. Del Bene, J. Phys. Chem. A, 2014, 118, 1527–1537 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef PubMed.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, Phys. Rev. Lett., 2008, 101, 096402 CrossRef.
- A. P. Seitsonen, A. M. Saitta, T. Wassmann, M. Lazzeri and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115425 CrossRef.
- L. Wang, Y. Sun, K. Lee, D. West, Z. Chen, J. Zhao and S. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 161406 CrossRef.
- M. W. Chase Jr, C. A. Davis, J. R. Downey Jr, D. J. Fruip, R. A. McDonald, and A. N. Syverud, JANAF Thermochemical Tables, Journal of Physical and Chemical Reference Data, 3rd ed, 1985 Search PubMed.
- J. Zhou, Q. Wang, Q. Sun, X. Chen, Y. Kawazoe and P. Jena, Nano Lett., 2009, 9, 3867–3870 CrossRef CAS PubMed.
- J. Zhou, M. M. Wu, X. Zhou and Q. Sun, Appl. Phys. Lett., 2009, 95, 103108 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02850c |
| This journal is © The Royal Society of Chemistry 2014 |