 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shear and thermal effects in boundary film formation during sliding
Octavio
Furlong
a,
Brendan
Miller
b,
Peter
Kotvis
c,
Heather
Adams
c and
Wilfred T.
Tysoe
*c
aINFAP/CONICET, Universidad Nacional de San Luis, Ejercito de los Andes 950, 5700 San Luis, Argentina. E-mail: ojfurlong@unsl.edu.ar; Tel: +54 266 4436151
bChevron Oronite Company LLC, 100 Chevron Way, Richmond, CA 94802, USA. E-mail: Brendan.Miller@chevron.com; Tel: +1-510-242-2179
cDepartment of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, 3210 N Cramer Street, Milwaukee, WI 53211, USA. E-mail: wtt@uwm.edu; Fax: +1 414 229 5036; Tel: +1 414 229 5222
First published on 21st May 2014
Abstract
A prerequisite for understanding mechano- and tribochemical reaction pathways is that the interface be in thermodynamic equilibrium and that the temperature be well defined. It is suggested that this occurs in two regimes: when the surfaces are only slightly perturbed during sliding, leading to negligible frictional heating, and when the surface temperatures are very high (∼1000 K), in the so-called extreme pressure regime. The tribochemistry occurring in each regime is discussed in terms of the elementary steps leading to tribofilm formation, namely (i) a reaction of the additive or gas-phase lubricant on the surface to form an adsorbed precursor, (ii) decomposition of the molecular precursor, (iii) a process that causes the formation of a tribofilm that (iv) regenerates a clean surface that allows this tribochemical cycle to continue to form a thicker film. These steps are thermally driven in the extreme-pressure regime, while under milder conditions, they are induced by interfacial shear. In intermediate situations, the processes are likely to be a combination of those occurring at the extrema.
1. Introduction
Chemical reactions are most commonly driven thermally, by using an external potential (electrochemistry1) or photons (photochemistry2); energy can also be supplied mechanically to induce chemical reactions in a field known as mechanochemistry.3–15 Mechanochemical reactions have been known for millennia, for example when mercury was made in the 4th century BC by grinding cinnabar with acetic acid in a copper vessel,16 but the way in which chemistry is induced by the application of an external mechanical force is still not well understood even though quite a number of mechanically induced chemical reactions have been discovered.3,4,7,10,13–15,17–21 Analogous chemistry occurs at sliding interfaces during tribochemical reactions, where gas-phase lubricants or liquid lubricant additives react with surfaces to form a lubricious or anti-wear film.There are a number of mechanical processes that occur at a sliding interface that might accelerate a chemical reaction. The most obvious is the creation of high localised temperatures due to Joule heating at the contacting asperities on the sliding surfaces.22 These have been well documented and are known as “flash temperatures”.23–26 It should be emphasised that “temperature” is used in this case to indicate an energy density and may well not be a temperature in the sense that contacting interfaces may not be at thermodynamic equilibrium. Indeed, molecular dynamics (MD) simulations of a sliding contact suggest that the energy distribution deviates quite rapidly from a Boltzmann distribution with increasing sliding speed and applied load.27 Since theoretical chemical models are fundamentally based on statistical thermodynamics, describing systems that are not in thermodynamic equilibrium becomes a particular theoretical challenge. This could be addressed by using kinetic Monte Carlo simulations,28 for example, if the true energy distribution were known, but invariably it is not.
Therefore, in order to understand mechano- and tribochemical reactions, it is crucial to ensure that the interface is at thermodynamic equilibrium. Some of the earliest and most elegant methods for measuring interfacial temperatures used a sliding contact of two metals that formed a thermocouple29 and revealed that the interfacial temperature rises with increasing sliding speed and applied load, but reaches a limiting value at the melting point of the lowest-melting-point material (in this case, indium).29 Since, in this case, the measured temperature corresponds to a true “thermodynamic” temperature (a melting temperature), the interface is in thermodynamic equilibrium. More recent atomic force microscopy (AFM) measurements have confirmed this conclusion.30 Here, the conditions are sufficiently mild that the interface remains at thermodynamic equilibrium.
Paradoxically, another set of conditions for which the interface is found to be in thermodynamic equilibrium is when the applied loads and sliding speeds become extremely high, generally referred to as the “extreme-pressure” (or EP) lubrication regime.31
These effects are illustrated in the following using two examples. In the first the thermal effects in EP lubrication are illustrated using a model chlorinated hydrocarbon, methylene chloride, by comparing the reactions occurring at high temperatures on iron with their tribological behaviour. In the second example, the tribological behaviour is explored using frictional and surface chemistry experiments for dimethyl disulfide (DMDS) on copper. In this case, the tribological conditions are selected to be sufficiently mild such that the temperature rise on high-thermal-conductivity copper is negligible, and is much less than 1 K. In this case the interfacial temperature is well defined, being close to ambient temperature, and will remain close to thermodynamic equilibrium.
It is anticipated that the insights obtained from these results are not only relevant to mildly and severely perturbed interfaces, but will also provide insights into the mechanisms of lubricating film formation and tribochemical reactions in general.
2. Experimental methods
Surface chemistry experiments on iron were carried out using methylene chloride molecular beams in an ultrahigh vacuum (UHV) chamber as described previously.32 These were supplemented by measurements of the film growth kinetics using a vacuum microbalance as outlined elsewhere.33 The surface chemistry of DMDS was also explored in ultrahigh vacuum using an array of surface sensitive techniques34 and friction measurements were made in an UHV-compatible tribometer as described elsewhere.35 The normal loads used in these experiments was ∼0.44 N, with sliding speeds from ∼1 to 4 × 10−3 m s−1, leading to maximum flash temperature rise of ∼0.34 K.36–393. Results and discussion
3.1 Extreme-pressure tribochemistry of methylene chloride on iron
The interfacial temperatures attained in a pin and V-block apparatus at extreme pressures can be calibrated by measuring the wear rate as a function of applied load, where it is found that interfacial temperatures can reach ∼1000 K, and that an asymptotically high rate of wear is found as the temperature approaches the melting point of the material in the contact.40 Here the correspondence between the measured temperature and a thermodynamically well-defined temperature (the melting point) indicates that the interface is at thermodynamic equilibrium. In order to monitor the high-temperature surface chemistry of methylene chloride with iron, an experiment was carried out in which a beam of methylene chloride impinged on a clean iron surface in UHV while monitoring the 49 (methylene chloride) and 2 (hydrogen) amu signals of a mass spectrometer as a function of the sample temperature. The results are displayed in Fig. 1, which shows that the methylene chloride signal decreases (○) at higher temperatures due to the reaction of methylene chloride with the iron. The decrease in the methylene chloride signal is proportional to the rate at which it reacts with the surface allowing an Arrhenius plot of ln(rate) versus 1/T to be made and the result is displayed as an inset to Fig. 1. This results in a measured activation energy for the reaction of methylene chloride with iron of ∼9.5 kcal mol−1. Hydrogen is the only gas-phase product found to form during the reaction, and it is observed that the resulting 2 amu signal (●) increases as the methylene chloride signal decreases, thereby indicating that it arises directly from the decomposition of methylene chloride. An in situ analysis of the sample after reaction reveals the presence of FeCl2.32,41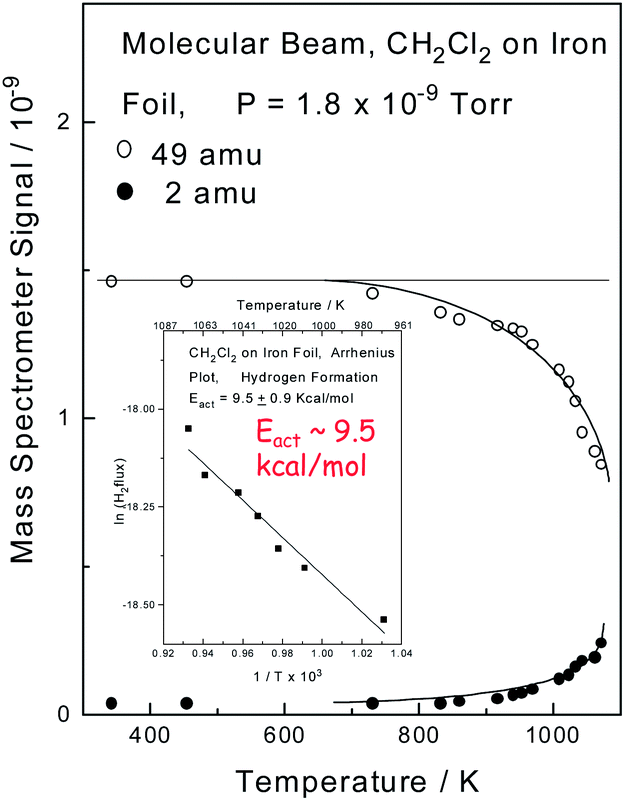 | ||
| Fig. 1 Plot of mass spectrometer signal as a function of sample temperature for a beam of methylene chloride impinging on an iron sample measured in ultrahigh vacuum by monitoring 49 (○, methylene chloride) and 2 (●, hydrogen) amu. Shown as an inset is an Arrhenius plot of the decrease in methylene chloride signal as a function of temperature. Reprinted with permission from ref. 32. Copyright 1998 American Chemical Society. | ||
In order to establish whether similar reactions take place at higher methylene chloride pressures, the film growth rate as a function of temperature was monitored using a microbalance.33 In this case the growth kinetics were measured from the change in the mass of the sample as a function of time by assuming that the film consisted predominantly of ferrous chloride. The results are displayed in Fig. 2A, where the change in mass is plotted as a function of time for various sample temperatures, and where the mass changes have been converted into film thickness. This reveals that the film initially grows rapidly but slows as the film becomes thicker, eventually leading to a plateau. This self-limiting growth is due to an electric-field-assisted Mott–Cabrera mechanism42 and shows that a film of finite thickness is formed by reaction with the chlorinated hydrocarbon. Measuring the initial reaction rate as a function of the sample temperature and plotting the data in Arrhenius form reveals an activation energy of ∼9.5 kcal mol−1, indicating that the kinetics measured at higher pressures are similar to those found in UHV conditions. The film is now sufficiently thick that it can be analysed using Raman spectroscopy33 and the resulting spectrum is displayed in Fig. 2B. The spectrum displays vibrational modes due to ferrous chloride as found in ultrahigh vacuum, with additional peaks that are assigned to the presence of small carbonaceous particles arising from the carbon in the methylene chloride precursor.
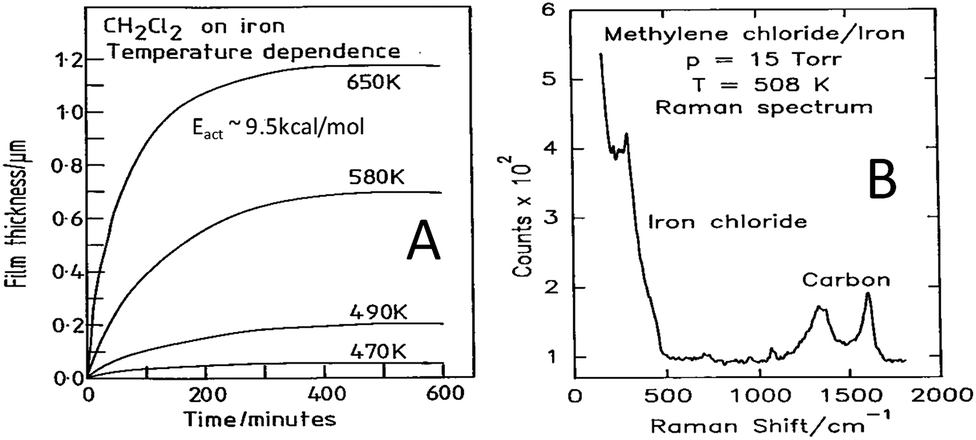 | ||
| Fig. 2 (A) Growth kinetics of 15 Torr of methylene chloride with iron, measured using a microbalance, as a function of sample temperature, where the sample temperatures are marked adjacent to the corresponding trace. (B) Raman spectrum obtained for a film grown by the thermal decomposition of methylene chloride on an iron foil at 508 K using a methylene chloride pressure of 15 Torr. Reprinted with permission from ref. 33 and 41 Copyright 1994 American Chemical Society. | ||
The tribological behaviour of a model EP lubricant consisting of 3.0 wt% of methylene chloride dissolved in a poly α-olefin was measured in a pin and V-block apparatus. This resulted in a relatively low value of friction coefficient (∼0.1). In addition, the load was increased until lubrication failed, as evidenced by a sudden increase in the friction coefficient, designated as the seizure load. This value is plotted as a function of methylene chloride concentration in Fig. 3 (●).41 The results indicate that the tribochemical reaction between methylene chloride and iron forms a film that reduces friction and prevents contact and seizure of the sliding interfaces up to high loads (∼2600 N). The presence of ferrous chloride as the lubricating film is confirmed by measuring the friction coefficient of ferrous chloride films evaporated onto an iron substrate in UHV43 and finding a similarly low value of ∼0.08.
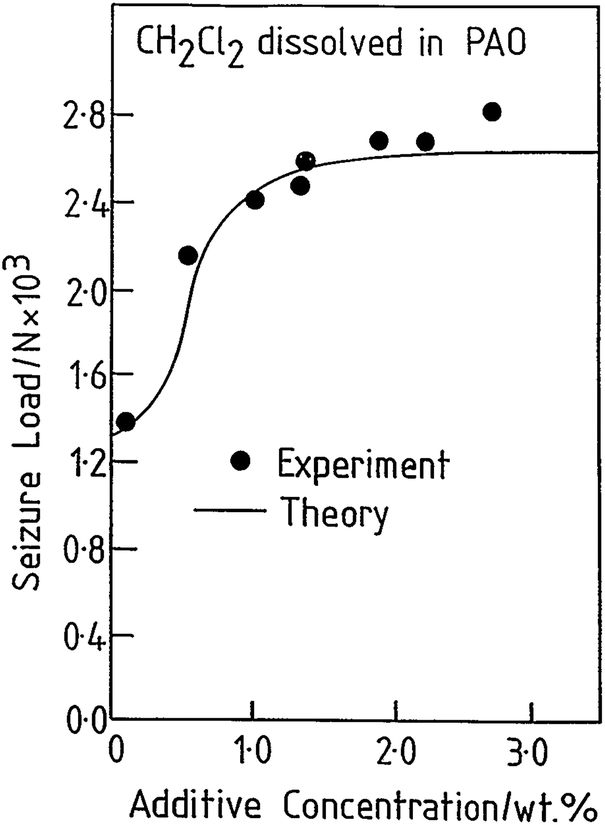 | ||
| Fig. 3 Comparison of a calculation of the seizure load versus additive concentration for methylene chloride (solid line) with experimental results (●) obtained using a pin and V-block apparatus. Reprinted with permission from ref. 41 Copyright 1993 American Chemical Society. | ||
It is clear that methylene chloride reacts with the iron substrate to form a low-friction film of ferrous chloride that incorporates some carbonaceous particles, and evolves hydrogen gas. This film is simultaneously worn from the surface and the wear rate of the ferrous chloride film can be measured from the width of the wear tracks as a function of time. Therefore, the equilibrium film thickness arises from a kinetic balance between the rate at which it is formed on the surface by thermal reaction with the methylene chloride additive, and the rate at which it is worn away. This postulate is tested further by assuming that, if a condition is reached at which the ferrous chloride film is completely removed from the surface, the load at which this occurs corresponds to the seizure load. In this case, the seizure load versus concentration data in Fig. 3 are modelled for a ramped load as in the experiment. This causes the interfacial temperature to increase due to frictional heating and the growth of a solid ferrous chloride lubricant film. However, this film is simultaneously worn from the surface so that the equilibrium film thickness X arises from a balance between these two processes. This variation can be modelled from the film growth kinetics discussed above, and the wear rate. The load at which the film thickness becomes zero is then designated as the seizure load. The results of this calculation are also shown as a solid line in Fig. 3, revealing excellent agreement with the experiment.44,45 The interfacial temperature is proportional to the load40 and the plateau found in the experiment corresponds to the melting temperature of FeCl2 and the presence of such a plateau arises naturally from the model. Increasing the additive concentration results in an initially faster rate of ferrous chloride film formation but, as soon as the interfacial temperature reaches the melting point of the film material, the wear rate becomes asymptotically large and the film is removed. This approach has also been used to explore the EP tribochemistry of other chlorine,32,33,46–56 sulphur,57,58 and phosphorus-containing59,60 lubricant additives.
In the high-temperature limit found under EP conditions, the large frictional heating leads to an interface that is, to a good approximation at thermodynamic equilibrium. This allows Arrhenius rate laws to be used to model the thermal film growth rate as a function of temperature and the thermal chemistry to be used to fully model the EP behaviour of lubricant additives.
As noted above, the interface is also expected to be close to thermodynamic equilibrium when the sliding conditions (speed and load) are mild, resulting in a negligible interfacial temperature rise. However, in the majority of such cases, the low surface temperature (∼300 K) results in film growth rates through a Mott–Cabrera mechanism and rates of thermal decomposition of adsorbates being so low as not to lead to film formation. Thus, other shear-induced film formation pathways must be sought. However, the insights into the film formation pathways in the EP regime discussed above will guide our exploration of shear-induced effects. Based on the above discussion, the elementary steps consist of the following: (i) a reaction of the additive or gas-phase lubricant with the surface to form an adsorbed precursor, (ii) a sliding-induced decomposition of the adsorbate, (iii) a shear-induced process that causes the formation of a tribofilm that (iv) regenerates a clean surface that allows this tribochemical cycle to continue to form a thicker film. These processes will be explored for mild sliding conditions in the next section using a model system comprising DMDS as a gas-phase lubricant for copper.
4. Ambient-temperature tribochemistry of dimethyl disulfide on copper
Shear effects in the formation of a tribofilm were explored using DMDS as a gas-phase lubricant for copper where the friction was measured using a UHV tribometer. This approach ensures that the copper sample is atomically clean and that the surface does not become contaminated after a tribological experiment. Thus, the surface composition measured after a tribological experiment is expected to resemble that formed while rubbing. While lubrication of copper is less technologically important than iron, there are a number of applications, such as brushes in electric motors or generators that involve sliding copper–copper contacts. In order to first establish whether DMDS is indeed a gas-phase lubricant for copper, the friction was measured while exposing the copper sample to DMDS in UHV. This experiment was carried out by exposing the sample to DMDS through a dosing tube directed towards the sample to minimize background contamination of the vacuum chamber.The results of this experiment are displayed in Fig. 4, which shows the plot of friction coefficient as a function of the number of times that it is rubbed. The experiment was initiated by first rubbing the clean copper surface for ∼70 cycles.61 This created an initial wear track (about 100 μm wide) and resulted in a run-in period during which the friction decreased from an initial value of ∼0.8 to a lower and steady value of ∼0.52. Thus, any further changes in friction coefficient are due to DMDS exposure and not to any evolution in the nature of the contact. DMDS exposure (at a background pressure of 8 × 10−8 Torr, where the local pressure at the sample will be higher than this value) results in a rapid decrease in friction coefficient to ∼0.3. Continued rubbing in the presence of a beam of DMDS causes a further but rather small decrease. In order to ensure that the UHV data are relevant to more conventional liquid lubricants, the friction of a model lubricant consisting of 5 wt% of DMDS in a poly α-olefin was measured in a pin and V-block apparatus and a value of friction coefficient of ∼0.04 was found. A profile of the composition across the wear track was measured by Auger spectroscopy62 using an electron beam spot size of ∼70 μm. The resulting profile of the sulphur KLL signal across the wear track collected after ∼100 rubbing cycles is shown as an inset to Fig. 4, revealing that a significant amount of sulphur has been deposited in the wear track. Therefore DMDS does indeed act as a gas-phase lubricant for copper and shear effects cause sulphur to be deposited on the surface. The continual decrease in friction coefficient as rubbing proceeds, as well as the growth in the amount of sulphur in the wear track, indicates that this tribofilm is continually being formed while rubbing. The shear-induced mechanisms by which this occurs are described in the context of the four elementary steps discussed above.
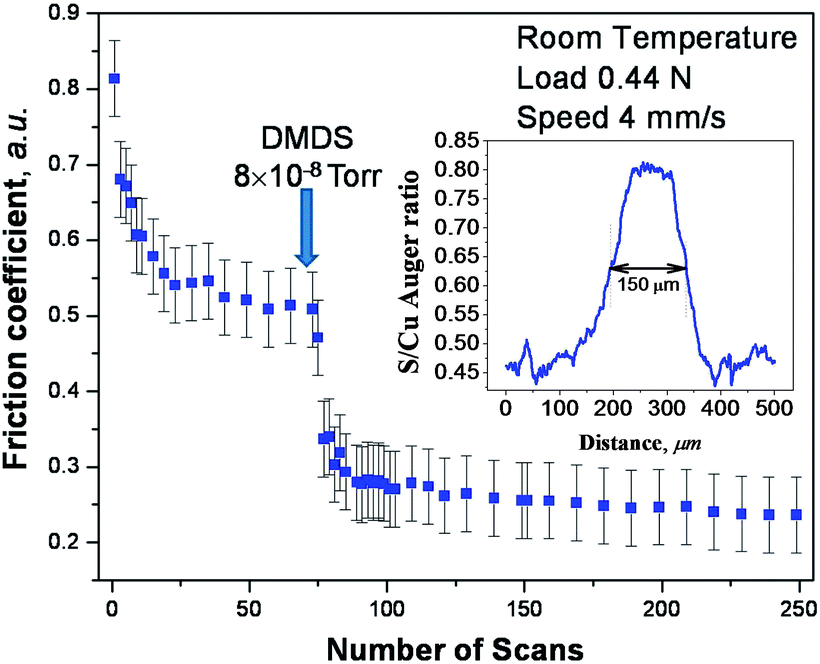 | ||
| Fig. 4 Plot of friction coefficient versus number of rubbing cycles measured in ultrahigh vacuum at a sliding speed of 4 × 10−3 m s−1 and a normal load of 0.44 N. The clean surface was initially rubbed 70 times to reach a steady-state value of friction coefficient, and then dimethyl disulfide was introduced via a dosing tube at a background pressure of ∼5 × 10−8 Torr and the friction coefficient then recorded in the presence of gas-phase DMDS. The inset shows an elemental profile of sulphur across the wear track measured by Auger spectroscopy. Adapted with permission from ref. 61. Copyright 2011 American Chemical Society. | ||
4.1 Surface reactions of dimethyl disulfide with copper
The surface chemistry of DMDS on copper was studied in UHV using a number of surface-sensitive techniques, including temperature-programmed desorption (TPD),63 X-ray photoelectron spectroscopy,62 and reflection–absorption infrared spectroscopy (RAIRS).34 These experiments reveal that DMDS adsorbs molecularly on copper at ∼80 K, but heating to ∼200 K causes S–S bond scission to form methyl thiolate (CH3–S–) species on the surface. The methyl thiolate species are stable up to ∼426 K, where they thermally decompose to desorb methane along with some ethylene and ethane. This leaves a surface covered with atomic sulphur but with very little carbon since this is removed as small hydrocarbons. The resulting adsorbed sulphur is thermodynamically more stable on the surface than in the bulk and is still present when the copper sample is heated to ∼550 K. The resulting surface reaction pathways of DMDS on copper are shown in Scheme 1. Thus, room-temperature exposure of copper to DMDS results in the formation of a methyl thiolate-covered surface and the thiolates are thermally stable to ∼420 K. This system is therefore ideal for exploring shear effects since the stability window for adsorbed methyl thiolates formed at ∼300 K is ∼120 K. This is almost three orders of magnitude larger than the interfacial temperature rise during sliding under the applied rubbing conditions (∼0.34 K). Thus, while the disparity between different estimates of flash temperature rise during sliding has been pointed out,64 such estimates are not orders of magnitude in error, so that any changes found during rubbing must be ascribed to shear effects and not frictional heating. This therefore sets the stage for experiments to explore changes that occur while rubbing.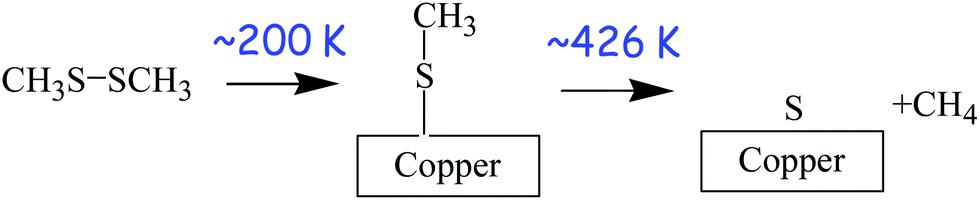 | ||
| Scheme 1 The reaction pathways for dimethyl disulphide on copper. | ||
4.2 Shear-induced tribofilm formation
In the case of EP lubrication, the formation of a tribofilm consisting of FeCl2 containing carbonaceous particles is thermally driven due to the high surface temperatures caused by frictional heating. Clearly, in the case of the gas-phase lubrication of copper by DMDS, the interfacial temperature rise during sliding is sufficiently small to allow such thermal effects to be excluded.A possible mechanism for shear-induced surface-to-bulk transport of adsorbed layers comes from molecular dynamics simulations65–72 where Kelvin-Helmholtz instabilities at the sliding metal–metal interface cause atoms that were originally on the surface to be transported into the subsurface region, and provide a possible mechanism for shear-induced tribofilm formation. In order to explore whether shear causes a loss of surface species, the amount of sulphur in the wear track of an initially thiolate-covered surface was measured as a function of the number of rubbing cycles using Auger spectroscopy. The results of this experiment are displayed in Fig. 5,73 which plots the relative sulphur coverage in the wear track as a function of the number of rubbing cycles. This clearly shows a rapid decrease in surface sulphur signal, which eventually decreases to zero. According to the above molecular dynamics simulations, this is proposed to be a result of shear-induced surface-to-bulk transport. An alternative possibility is that the sulphur could have been worn from the surface. In order to establish whether the sulphur has penetrated into the bulk, we take advantage of the observation made above that sulphur is thermodynamically stable on the surface so that any subsurface sulphur is metastable. Thus, heating the sample should cause the subsurface sulphur to diffuse to the surface of the sample. However, since the whole surface was dosed with DMDS, but only that within the wear track was removed, there is a large inventory of sulphur outside the wear track, which could potentially diffuse into the worn region when the sample is heated. Therefore the remaining thiolate species (outside the wear track) were carefully removed by Argon ion bombardment, and their absence was confirmed using Auger spectroscopy. Subsequently heating the copper sample revealed that sulphur is indeed found in the previously worn region of the sample, confirming that it had been transported into the subsurface region.61 Note that the removal of sulphur from the surface via this process regenerates a clean surface, thereby allowing the adsorption and film formation cycles to continue in the rubbed region.
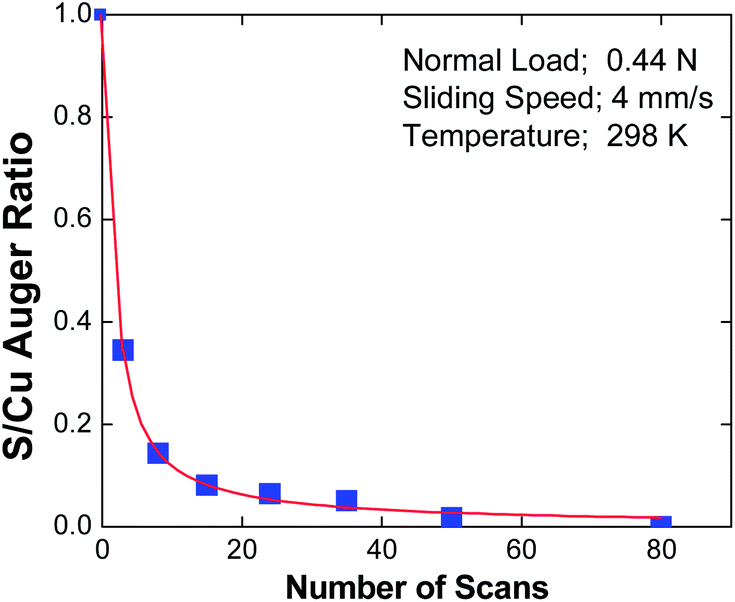 | ||
| Fig. 5 Plot of the peak-to-peak intensity of the sulphur LMM Auger feature within the wear track, ratioed to the peak-to-peak intensity of the Cu LMM feature as a function of the number of passes over the wear track.73 With kind permission from Springer Science and Business Media. | ||
4.3 Shear-induced chemistry of thiolate overlayer
Clearly the surface methyl thiolate species decompose as the surface is sheared to allow atomic sulphur to penetrate the subsurface region as described in the previous section. This regenerates a clean surface to allow the reaction to continue, eventually accumulating a relatively large amount of sulphur on the surface as shown in Fig. 1. Further insights into the amount of sulphur and carbon in the subsurface regions can be gained by heating the sample after 170 cycles of rubbing while dosing DMDS after it had been carefully Argon ion bombarded to remove any methyl thiolates from the unrubbed region. The resulting Auger scans across the wear track after heating to various temperatures are displayed in Fig. 6. Here a large amount of sulphur is detected after heating to ∼780 K, but only a relatively small amount of carbon. This indicates that the thiolate species have undergone some shear-induced reaction that removes carbon from the surface, while the sulphur penetrates the subsurface region to lower friction (Scheme 1).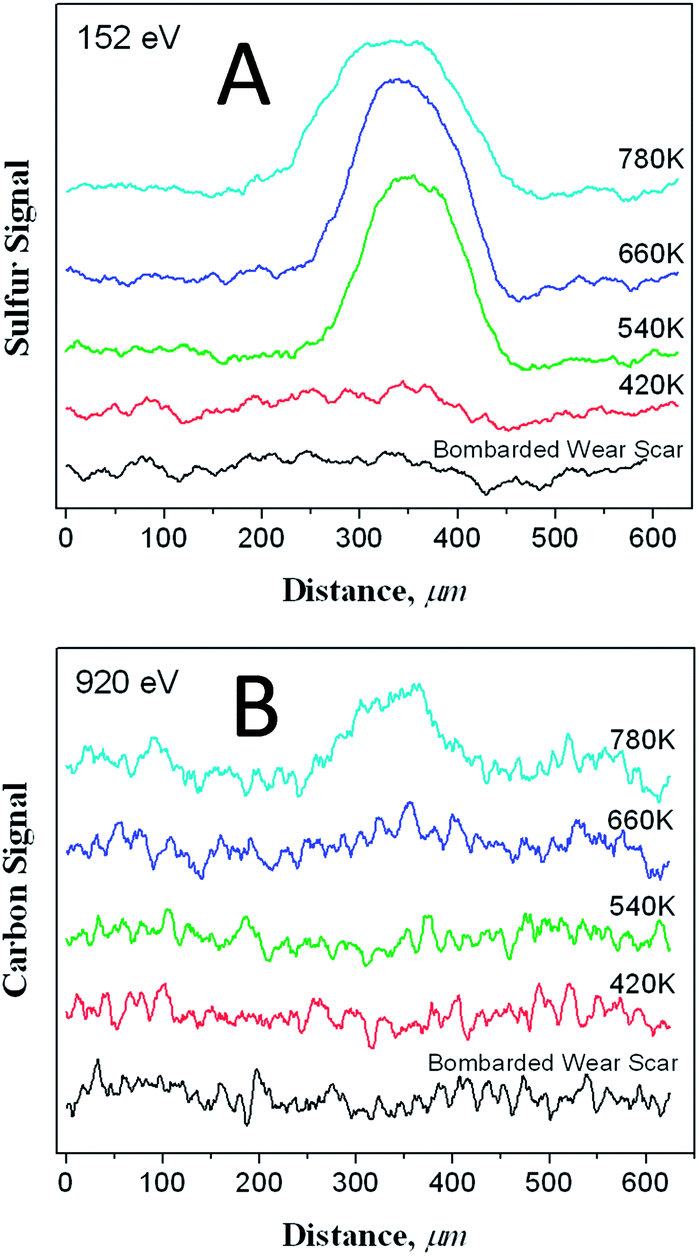 | ||
| Fig. 6 Peak to-peak amplitude of A the sulphur LMM Auger feature and B the carbon KLL transition along a line scan across a wear track that had been formed by rubbing the surface for 170 scans while exposing it to dimethyl disulphide via a dosing tube with a background pressure of ∼5 × 10−8 Torr, and then ion bombarded until no sulphur was detected within or outside of the wear scar, after annealing the sample to various temperatures. The annealing temperatures are indicated adjacent to the corresponding profile. Reproduced with permission from ref. 61 Copyright 2011 American Chemical Society. | ||
5. Comparison of shear and thermal effects in tribofilm formation
The above discussions identify the film formation pathways that occur at a sliding interface under conditions where the system is expected to be at, or close to thermodynamic equilibrium. While the physicochemical processes that occur for each of the elementary steps are different, they can each be classified into the four elementary steps as discussed above. In the case of EP lubrication, these processes are dominated by the high temperatures (∼1000 K) that are generated by frictional heating. Thus, the initial adsorption and reaction of the additive molecules is very rapid at these high temperatures and invariably results in the complete thermal decomposition of the precursor additive molecules into their elements. The film subsequently forms by a self-limiting, electric-field-assisted Mott–Cabrera growth mechanism that forms a friction-reducing film consisting of FeCl2 when using chlorinated hydrocarbon additives. This mechanism inherently limits the thickness to which the film can grow. This process regenerates a surface on which the additive can react further, and the equilibrium film thickness arises from a balance between the rate that it forms and the rate at which it is worn from the surfaces; such a model can be used to rationalize the observed tribological behaviour.In the case in which the temperature is only very slightly affected by frictional heating, film formation occurs by similar elementary steps, but is now driven by interfacial shear rather than the surface temperature rise. Here, initial (still thermally assisted) adsorption occurs to form methyl thiolate species from DMDS, which decompose under shear to remove carbon, and the resulting sulphur is transported into the subsurface region by interfacial shear to regenerate a clean surface to continue the cycle.
At intervening temperatures between mild lubrication, discussed above, and EP lubrication, which likely occur, for example, in the formation of antiwear films from zinc dialkyl dithiophosphates (ZDDP),74–85 a common antiwear additive to engine oils, both thermal and shear effects are likely to occur simultaneously. While, in such cases, the surface conditions (particularly temperature) are less well defined, the processes will likely be a combination of the effects observed at the extrema. Recent molecular dynamics simulations have suggested that the reaction is driven by interfacial shear that causes it to become thermodynamically favoured because of an increase in entropy of the reaction.86
The nature of the tribochemical reactions will therefore depend critically on the interfacial conditions, which dictate the processes by which the films are formed, on the nature of the surface, which dictates the initial adsorption of the precursor molecule, as well as the tribological conditions, and on the nature of the reactant molecule itself. For example, it has been shown that the surface chemistry of model borate esters profoundly influences the nature of the tribofilm that is formed and the frictional behaviour.87 Indeed, the most common strategy for synthesizing lubricants for a particular application is to tune the nature of the additive molecule. However, the dependence of the tribochemistry on the nature of the surface suggests an alternative approach of modifying the surface to tune its chemical properties. Indeed, recent work has shown that poly α-olefins react to form diamond-like carbon films on appropriately modified surfaces, to provide low friction without the use of additives.88
Acknowledgements
We gratefully acknowledge support by the National Science Foundation under grant numbers CHE-0654276 and CMMI-1265742.Notes and references
- J. O. M. Bockris, Electrochemistry, Butterworths, University Park Press, London, Baltimore, 1973 Search PubMed.
- D. C. S. Bryce-Smith, Photochemistry, Chemical Society, London, England, 1970 Search PubMed.
- E. Barraud, S. Bégin-Colin, G. Le Caër, O. Barres and F. Villieras, J. Alloys Compd., 2008, 456, 224–233 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- F. K. Urakaev and V. V. Boldyrev, Powder Technol., 2000, 107, 93–107 CrossRef CAS.
- F. K. Urakaev and V. V. Boldyrev, Powder Technol., 2000, 107, 197–206 CrossRef CAS.
- V. V. Boldyrev and K. Tkáčová, J. Mater. Synth. Process., 2000, 8, 121–132 CrossRef CAS.
- P. G. Fox, J. Mater. Sci., 1975, 10, 340–360 CrossRef CAS.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS PubMed.
- S. Kipp, V. Šepelák and K. D. Becker, Chem. Unserer Zeit, 2005, 39, 384–392 CrossRef CAS.
- V. I. Levitas, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 184118 CrossRef.
- B. Rodríguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213–2233 CrossRef.
- B. M. Rosen and V. Percec, Nature, 2007, 446, 381–382 CrossRef CAS PubMed.
- Z. V. Todres, Organic mechanochemistry and its practical applications, Taylor & Francis, Boca Raton, FL, 2006 Search PubMed.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- H. J. Theophrastus, Theophrastus's history of stones: with an English version, and critical and philosophical notes, including the modern history of the gems, &c., described by that author, and of many other of the native fossils, London, 1774 Search PubMed.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, Nat. Chem., 2013, 5, 110–114 CrossRef CAS PubMed.
- S. L. Craig, Nature, 2012, 487, 176–177 CrossRef CAS PubMed.
- V. Sepelak, S. Begin-Colin and G. Le Caer, Dalton Trans., 2012, 41, 11927–11948 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- A.-S. Duwez, S. Cuenot, C. Jerome, S. Gabriel, R. Jerome, S. Rapino and F. Zerbetto, Nat. Nanotechnol., 2006, 1, 122–125 CrossRef CAS PubMed.
- J. P. Joule, Philos. Trans. R. Soc. London, 1850, 140, 61–82 CrossRef.
- G. W. Stachowiak and A. W. Batchelor, Engineering tribology, Elsevier Butterworth-Heinemann, Amsterdam, Boston, 2005 Search PubMed.
- H. Blok, Wear, 1963, 6, 483–494 CrossRef.
- S. Suzuki and F. E. Kennedy, Jr, IEEE Trans. Magn., 1989, 25, 3728–3730 CrossRef.
- R. Holm, J. Appl. Phys., 1948, 19, 361–366 CrossRef PubMed.
- O. A. Mazyar, H. Xie and W. L. Hase, J. Chem. Phys., 2005, 122, 094713 CrossRef PubMed.
- R. Y. Rubinstein, Simulation and the monte carlo method, Wiley, New York, 1981 Search PubMed.
- F. P. Bowden and K. E. W. Ridler, Proc. R. Soc. London, Ser. A, 1936, 154, 640–656 CrossRef.
- B. D. Dawson, S. M. Lee and J. Krim, Phys. Rev. Lett., 2009, 103, 205502 CrossRef CAS.
- J. F. Archard and W. Hirst, Proc. R. Soc. London, Ser. A, 1956, 236, 397–410 CrossRef.
- J. Lara and W. T. Tysoe, Langmuir, 1998, 14, 307–312 CrossRef CAS.
- L. Huezo, C. Crumer, C. Soto and W. T. Tysoe, Langmuir, 1994, 10, 3571–3576 CrossRef CAS.
- O. J. Furlong, B. P. Miller, Z. Li, J. Walker, L. Burkholder and W. T. Tysoe, Langmuir, 2010, 26, 16375–16380 CrossRef CAS PubMed.
- G. Wu, F. Gao, M. Kaltchev, J. Gutow, J. K. Mowlem, W. C. Schramm, P. V. Kotvis and W. T. Tysoe, Wear, 2002, 252, 595–606 CrossRef CAS.
- J. C. Jaeger, J. Proc. R. Soc. N. S. W., 1943, 76, 21 Search PubMed.
- D. G. Bansal and J. L. Streator, Wear, 2009, 266, 721–732 CrossRef CAS PubMed.
- M. F. Ashby, J. Abulawi and H. S. Kong, Tribol. Trans., 1991, 34, 577–587 CrossRef.
- E. H. Smith and R. D. Arnell, Tribol. Lett., 2013, 52, 407–414 CrossRef PubMed.
- T. J. Blunt, P. V. Kotvis and W. T. Tysoe, Tribol. Lett., 1996, 2, 221–230 CrossRef CAS.
- P. V. Kotvis, L. A. Huezo and W. T. Tysoe, Langmuir, 1993, 9, 467–474 CrossRef CAS.
- N. Cabrera and N. F. Mott, Rep. Prog. Phys., 1949, 12, 163 CAS.
- F. Gao, P. V. Kotvis and W. T. Tysoe, Wear, 2004, 256, 1005–1017 CrossRef CAS PubMed.
- P. V. Kotvis and W. T. Tysoe, Tribol. Trans., 1998, 41, 117–123 CrossRef CAS.
- T. J. Blunt, P. V. Kotvis and W. T. Tysoe, Tribol. Trans., 1998, 41, 129–139 CrossRef CAS.
- P. V. Kotvis, J. Lara, K. Surerus and W. T. Tysoe, Wear, 1996, 201, 10–14 CrossRef CAS.
- L. A. Huezo, P. V. Kotvis, C. Crumer, C. Soto and W. T. Tysoe, Appl. Surf. Sci., 1994, 78, 113–122 CrossRef CAS.
- P. V. Kotvis and W. T. Tysoe, Tribol. Trans., 1998, 41, 117–123 CrossRef CAS.
- T. J. Blunt, P. V. Kotvis and W. T. Tysoe, Tribol. Trans., 1998, 41, 129–139 CrossRef CAS.
- J. Lara, P. V. Kotvis and W. T. Tysoe, Tribol. Lett., 1997, 3, 303–309 CrossRef CAS.
- W. T. Tysoe, K. Surerus, J. Lara, T. J. Blunt and P. V. Kotvis, Tribol. Lett., 1995, 1, 39–46 CrossRef CAS.
- P. V. Kotvis, L. A. Huezo and W. T. Tysoe, Langmuir, 1993, 9, 467–474 CrossRef CAS.
- F. Gao, O. Furlong, P. Kotvis and W. Tysoe, Tribol. Lett., 2005, 20, 171–176 CrossRef CAS.
- M. Kaltchev, G. Celichowski, J. Lara and W. T. Tysoe, Tribol. Lett., 2001, 9, 161–165 CrossRef.
- J. Lara, H. Molero, A. Ramirez-Cuesta and W. T. Tysoe, Langmuir, 1996, 12, 2488–2494 CrossRef CAS.
- J. Lara and W. T. Tysoe, Tribol. Lett., 1999, 6, 195–198 CrossRef CAS.
- M. Kaltchev, P. V. Kotvis, T. J. Blunt, J. Lara and W. T. Tysoe, Tribol. Lett., 2001, 10, 45–50 CrossRef CAS.
- J. Lara, K. K. Surerus, P. V. Kotvis, M. E. Contreras, J. L. Rico and W. T. Tysoe, Wear, 2000, 239, 77–82 CrossRef CAS.
- F. Gao, P. V. Kotvis, D. Stacchiola and W. T. Tysoe, Tribol. Lett., 2005, 18, 377–384 CrossRef CAS PubMed.
- F. Gao, O. Furlong, P. V. Kotvis and W. T. Tysoe, Langmuir, 2004, 20, 7557–7568 CrossRef CAS PubMed.
- O. J. Furlong, B. P. Miller, P. Kotvis and W. T. Tysoe, ACS Appl. Mater. Interfaces, 2011, 3, 795–800 CAS.
- B. Briggs and M. P. Seah, Practical Surface Analysis: Auger and X-ray Photoelectron Spectroscopy, John Wiley and Sons, New York, 1996 Search PubMed.
- P. A. Redhead, Vacuum, 1962, 12, 203–211 CrossRef CAS.
- M. Kalin and J. Vižintin, Tribol. Int., 2001, 34, 831–839 CrossRef CAS.
- D. A. Rigney, Wear, 2000, 245, 1–9 CrossRef CAS.
- S. Karthikeyan, H. J. Kim and D. A. Rigney, Phys. Rev. Lett., 2005, 95, 106001 CrossRef CAS.
- A. Emge, S. Karthikeyan, H. J. Kim and D. A. Rigney, Wear, 2007, 263, 614–618 CrossRef CAS PubMed.
- H. Kim, W. Kim, M. Falk and D. Rigney, Tribol. Lett., 2007, 28, 299–306 CrossRef.
- A. Emge, S. Karthikeyan and D. A. Rigney, Wear, 2009, 267, 562–567 CrossRef CAS PubMed.
- S. Karthikeyan, A. Agrawal and D. A. Rigney, Wear, 2009, 267, 1166–1176 CrossRef CAS PubMed.
- H. J. Kim, S. Karthikeyan and D. Rigney, Wear, 2009, 267, 1130–1136 CrossRef CAS PubMed.
- D. Rigney and S. Karthikeyan, Tribol. Lett., 2010, 39, 3–7 CrossRef.
- O. Furlong, B. Miller and W. Tysoe, Tribol. Lett., 2011, 41, 257–261 CrossRef CAS.
- Z. Zhang, E. S. Yamaguchi, L. Yu, M. Kasrai and G. M. Bancroft, Tribol. Trans., 2007, 50, 58–67 CrossRef CAS.
- Z. Zhang, E. S. Yamaguchi, M. Kasrai and G. M. Bancroft, Tribol. Trans., 2004, 47, 527–536 CrossRef CAS.
- N. J. Mosey, T. K. Woo, M. Kasrai, P. R. Norton, G. M. Bancroft and M. H. Müser, Tribol. Lett., 2006, 24, 105–114 CrossRef CAS.
- G. W. Canning, M. L. S. Fuller, G. M. Bancroft, M. Kasrai, J. N. Cutler, G. De Stasio and B. Gilbert, Tribol. Lett., 1999, 6, 159–169 CrossRef CAS.
- Z. Zhang, M. Kasrai, G. M. Bancroft and E. S. Yamaguchi, Tribol. Lett., 2003, 15, 377–384 CrossRef CAS.
- E. S. Yamaguchi, Z. Zhang, M. Kasrai and G. M. Bancroft, Tribol. Lett., 2003, 15, 385–394 CrossRef CAS.
- Z. Zhang, E. S. Yamaguchi, M. Kasrai and G. M. Bancroft, Tribol. Lett., 2005, 19, 211–220 CrossRef CAS.
- Z. Zhang, E. S. Yamaguchi, M. Kasrai, G. M. Bancroft, X. Liu and M. E. Fleet, Tribol. Lett., 2005, 19, 221–229 CrossRef CAS.
- M. D. Pauli, T. S. Rufael, J. K. Mowlem, M. Weinert, D. K. Saldin and W. T. Tysoe, Tribol. Int., 2005, 38, 195–204 CrossRef CAS PubMed.
- H. Spikes, Tribol. Lett., 2004, 17, 469–489 CrossRef CAS.
- M. I. de Barros'Bouchet, J. M. Martin, T. Le-Mogne and B. Vacher, Tribol. Int., 2005, 38, 257–264 CrossRef PubMed.
- J. M. Martin, M. Belin, J. L. Mansot, H. Dexpert and P. Lagarde, ASLE Trans., 1986, 29, 523–531 CrossRef CAS.
- J. M. Martin, T. Onodera, C. Minfray, F. Dassenoy and A. Miyamoto, Faraday Discuss., 2012, 156, 311–323 RSC.
- B. Miller, P. Kotvis, O. Furlong and W. Tysoe, Tribol. Lett., 2013, 49, 21–29 CrossRef CAS PubMed.
- A. Erdemir, O. L. Eryilmaz, M. Urgen and K. Kazmanli, Google Patents, 2013.
| This journal is © The Royal Society of Chemistry 2014 |