Switching selectivity between Pb2+ and Hg2+ ions through variation of substituents at xanthene end; ‘turn-on’ signalling responses by FRET modulation†
Biswonath Biswal and
Bamaprasad Bag*
Colloids and Materials Chemistry Department, Academy of Scientific and Innovative Research, CSIR-Institute of Minerals and Materials Technology, P.O.: R.R.L., Bhubaneswar-751 013, Odisha, India. E-mail: bpbag@immt.res.in; Fax: +91 674 258 1637; Tel: +91 674 237 9254
First published on 1st July 2014
Abstract
Few rhodamine based probes (L1–L4) that consist of a similar 2-(aminoethyl)-pyridine unit at their carboxamide end but vary with substituents attached to the N-atom at their xanthene end were synthesized. Rhodamine G based probes L1–L3 have shown preferential chromogenic and fluorogenic ‘turn-on’ spectral responses in the presence of Pb2+ ions, where one of the two ethyl substituted secondary amino groups attached to the xanthene core either remains un-substituted (as in L1) or is functionalized with a bulky aromatic group (as in L2) or a long alkyl chain (as in L3). On the contrary, the L4 probe that incorporates two ethyl-substituents at both N-atoms attached to the xanthene core has selectively exhibited a dual mode spectral amplification in the presence of Hg2+ ions. The reversible selective dual mode signalling pattern of bifluorophoric L2 in the presence of Pb2+ ions is because of the perturbation of the combined PET (photo-induced electron transfer) inhibition and FRET (fluorescence resonance energy transfer) initiation processes. The observed ratiometric signalling pattern enabled it to detect Pb2+ ions at a low concentration level, even in living organisms such as E. coli. The altered selectivity in the signalling pattern infers a modulated stereo-electronic environment for metal ion coordination, which in turn is caused by induced amine rigidity at the xanthene end.
Introduction
The development of molecular probes for the selective detection of various analytes of environmental and biological importance have attracted significant attention1 in the past couple of decades, relying heavily on methodological approaches, involving various binding sites, signalling subunits and operational principles, which can concurrently display selective changes in photo-physical signatures upon guest binding. The on-site, real time, selective detection and analyses of transition and heavy metal ions, which are such targeted analytes, are highly desirable. Their presence or absence, deviating from the requisite concentration threshold, imparts lethal toxic effects2 to cause serious human health hazards and environmental impacts; although, their existence in the active sites of enzymes and proteins of a living organism is highly essential to operate several biological functions. Toxic lead and mercury ion contamination in the environment, which occur through various natural and human activities are a matter of concern. Intake of ionic lead even in an ultra-trace quantity causes severe health problems3 such as memory loss, anaemia, and slow nerve conduction velocity in children.4 Exposure to mercury leads to cell dysfunctions5 resulting in several physiological disorders6 and other health hazards7 because of its high affinity for proteins/enzymes based thiol-groups. Hence, the selective detection of these two ions at very low concentrations has been indispensable for their impact assessment. Among several feasible and viable solutions, the development of efficient chemosensory probes is promising owing to synthetic and operational advantages. Fluorescent probes are advantageous because of their highly selective, sensitive, quick response and non-destructive nature. At the same time, chromogenic probes facilitate a visual perception of the signalling behaviour as a mode of colour change. Thus, covalent architectures which undergo both fluorogenic and chromogenic signal modulation are preferable for designing molecular chemosensory probes. In this context, the excellent spectroscopic features of xanthene based dyes along with contrast structure–function correlation prompt them to be a prejudiced choice8 as a signalling entity. Several rhodamine-based probes for the selective detection of Hg2+ and Pb2+ ions have already been known9,10 following their metal-induced spiro-cyclic ring-opening. With our quest on metal ion selectivity and signalling pattern of derivatized rhodamine chemosensors, we have recently demonstrated11 that few “amino–alkyl–amino” substituted rhodamine-B based probes selectively exhibit dual mode signalling of Hg2+ ions among various metal ions in an organic–aqueous binary solvent. Such probes quite often lack in efficiency concerning the sensitivity level of detection despite advantages in dual mode signalling pattern because a low signal to noise ratio in spectral amplification at a very low concentration level of the analyte restricts the limit of detection. On the contrary, a bi-fluorophoric ratiometric probe incorporating rhodamine as one of the fluorophores should be promising for addressing sensitivity issues. It would not only allow the measurement of fluorescence intensities at two different wavelengths12 along with an increased dynamic13 range, but would also render a built-in correction for environmental effects for smaller concentration variation. Because the sensitivity and dynamic range of such probes are controlled by the ratio of emission intensities, which lowers the dependency on the signal to noise ratio to achieve a higher sensitivity, the probe–metal ion interaction is crucial for exhibiting high ratiometric signals. For a higher signalling efficiency, such interactions need to overcome various interactive yet detrimental parameters such as probe-solvent/metal ion-solvent interactions and the hydration of metal ions etc. to induce selectivity in coordination. Thus, the efficiency of probe–metal interaction can effectively be tuned through the manipulation of the stereo-electronic environment of spatially disposed donor atoms of the receptor in a probe. To anticipate the variation in electron density over donor atoms that results in altered coordination preferences of the receptor, it is imperative to understand the effect of various substituents attached to rhodamine based probes on its coordination preferences towards metal ions and the ability to open up its spiro-ring for dual mode signalling responses. Previously reported11,14 substituted ‘amino–alkyl–amino’ coupled rhodamine B based probes, although preferentially exhibited selective signalling responses11 for Hg2+ ions in an organic aqueous medium, irrespective of the substitution at the receptor unit, they were hardly observed to alter the selectivity towards metal ions, which coordinated to the ‘amino–alkyl–amide’ segment at the dye's spiro-lactam end. On the other hand, either none or only one alkyl group substitution at each of the N-donor atoms of the xanthene unit, as in the rhodamine-G dye, or even their rigidization with a bulky group attachment, significantly lowers the activation of the internal conversion process.15 Hence, to investigate the stereo-electronic effect of alkyl-/bulky aromatic substitution to mono ethyl-substituted amino groups (as in rhodamine G) on preferential metal ion coordination that initiates a ring-opening process, few such probes (L1–L4) were synthesized (Scheme 1) and their photophysical behaviour was investigated in the presence of various metal ions. These probes consist of a 2-(aminoethyl) pyridine unit attached to their carboxamide end, while they incorporate varied alkyl-substitution to one of the amino groups at the xanthene end. With structural modifications, L1–L3 probes contain a rhodamine G core while L4 consists a rhodamine B moiety to execute the signalling action. Furthermore, the bifluorophoric signalling probe L2 consists of both anthracene and rhodamine G fluorophores in its architecture in a ‘donor(anthracene)–spacer(methylene)–acceptor(rhodamine 6G)’ format to exhibit signalling action in a ratiometric pattern through the modulation of various simultaneously operative processes, predominantly the metal ion induced photo-induced electron transfer (PET)16,17 inhibition and the subsequent initiation of intra-molecular fluorescence resonance energy transfer (FRET)18–20 process. Rhodamine structures in a spiro-cyclic conformation have an interrupted π-conjugation restricted within the substituted-aniline of the xanthene core, hence they do not act as energy acceptors. Their metal ion induced ring-opened form facilitates them to be good energy acceptors, which are capable of triggering a FRET process. In an earlier report20 on metal ion induced dual mode signalling of bifluorophoric probes incorporating the rhodamine B and anthracene couple, as fluorophores attached to the respective amino ends of the bis-(amino-ethyl-piperazine) spacer, a weak FRET was observed to be operative because of the poor spectral overlap between both the fluorophores. To induce a higher order of energy transfer efficiency in such a bifluorophoric donor–acceptor ensemble, factors such as their distance and orientation are essentially crucial besides a stronger spectral overlap. The choice of anthracene–rhodamine G couple in L2 is obvious because the excited state emission spectrum of donor (anthracene) largely overlaps with the ground state absorption spectrum of acceptor(rhodamine 6G), which is better than with the spectrum of rhodamine B. In general, earlier design of rhodamine based probes with FRET operational principles for metal ion induced signal perturbation mostly incorporated both fluorophores that are discretely attached to both the ends of the receptor unit, where rhodamine was coupled to the receptor at its spiro-cyclic end. Herein, a different strategy of FRET-based probe design has been followed as fabricated in L2. Both the fluorophores were coupled at amino groups attached to the xanthene core with an anticipated FRET perturbation upon metal ion coordination to the receptor unit at the lactonized end. With all these methodological prejudices, the photophysical investigations of L1–L4 in the presence of metal ions have been envisaged to exhibit altered preferences in their dual mode signalling pattern as a function of the varied stereo-electronic situation. The 2-(aminoethyl)-pyridine derivatized rhodamine G based probe L1 reported here to exhibit selectivity towards Pb2+ ions, which remained unaltered on its derivatization at one of its amino groups attached to the xanthene unit, with either an octyl-substitution (as in L3) or methyl-anthracene substitution (as in L2), continuing to operate through a combined PET-FRET process modulation. However, the L4 probe, which is a rhodamine-B analogue of L1, selectively exhibited dual mode signalling of Hg2+ ions substantiating the effect of structural modification on tuning their preferences towards metal ion coordination. These chemosensors are also expected to possess high sensitivity, selectivity, reversibility and operation under physiological pH for possible applications, such as the fluorescence imaging of metal ions in living cells for their in situ detection, at a low concentration.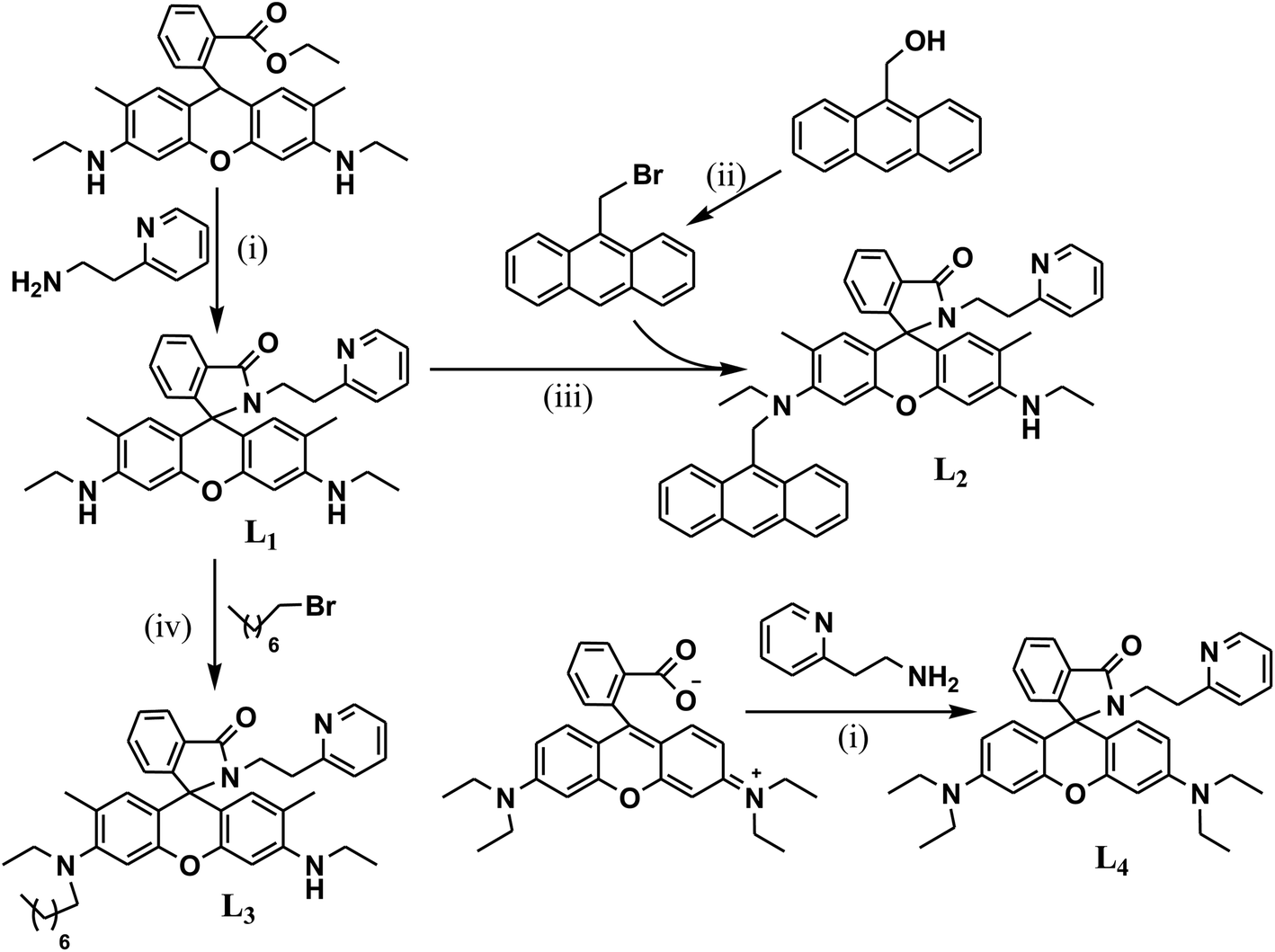 | ||
| Scheme 1 Synthetic route to probes L1, L2, L3 and L4. Reaction conditions: (i) 2-aminoethyl pyridine, EtOH, reflux, 6 h.; (ii) PPh3, Br2, MeCN, RT, 1 h; (iii) 9-bromomethyl anthracene, toluene, Et3N, reflux, 4 h; (iv) 1-bromo-octane, toluene, Et3N, reflux, 8 h. | ||
Results and discussion
The probes (L1–L4) were synthesized as per the synthetic route depicted in Scheme 1. L1 was synthesized through the condensation of rhodamine 6G with 2-aminoethylpyridine. Its subsequent nucleophilic substitution (SN2) reaction with 9-bromomethyl anthracene yielded L2, while it resulted in L3 with 1-bromo-octane. The condensation of 2-aminoethylpyridine with rhodamine B yielded in L4, which further crystallized in ethanol to afford single crystals suitable for X-ray diffraction through the slow evaporation technique. All these probes were characterized by MS, 1H NMR and 13C NMR spectral analyses. The characteristic quaternary carbon peak near ∼65 ppm in their 13C-NMR spectrum in CDCl3 ascertained predominant existence of the spirolactam conformation of rhodamine. Similar to other derivatives, solutions of L1–L4 in various organic solvents also appeared colourless along with a weak fluorescence, inferring the existence of the spirolactam conformation. The single crystal X-ray diffracted structural analysis† for L4 also confirmed the spiro-cyclic conformation of rhodamine (Fig. 1). The non-bonded N3–N4 distance and torsion angle (O2⋯N3⋯C31–N4), which constitute the coordination cavity for metal ion complexation, were found to be 4.436 Å and 118.5°, respectively, in L4.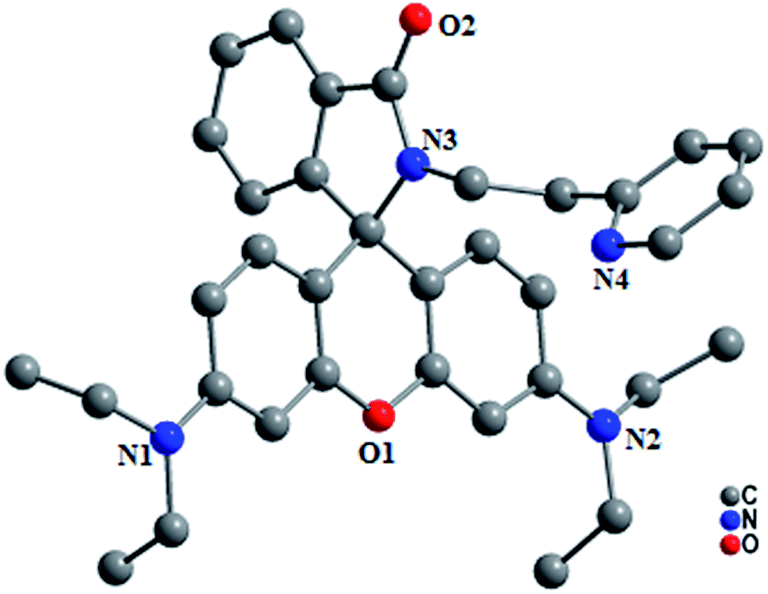 | ||
| Fig. 1 Perspective view of the X-ray crystallographic structure of L4 (H-atoms are omitted for clarity). | ||
The UV-Vis absorption spectral pattern of L1–L4 in various solvents revealed that these probes did not absorb in the 500–600 nm region because of the spirolactam conformation of rhodamine, and exhibited high energy ligand localized absorption transitions in the 250–340 nm region. In addition to this, the bifluorophoric probe L2 incorporating both rhodamine and anthracene fluorophores exhibited a characteristic peak of 9-alkyl substituted anthracene (0, 0) absorption transitions at 388 nm along with its Frank–Condon vibrational structures at 368, 350, and 333 nm. Absence of long range, broad and structured band in its absorption spectra inferred that the lone pair of electrons of the donor N-atom did not interact with anthracene in their ground state, which was commonly observed17 in several ‘anthracene-methyl-amino’ based probes.
Furthermore, colourless solutions of L1–L4 in acetonitrile–aqueous binary mixtures exhibited either none or a weak fluorescence upon the excitation of the rhodamine dye (at 350/550 nm) because of its spirolactam conformation, which facilitated21 an intersystem crossing in its emissive CT state. The fluorescence of L2 was observed to be quenched because of various operative processes such as (a) PET from the xanthene core to the excited anthracene, (b) through bond ET from the donor amino group to the xanthene unit, (c) activated non-radiative pathways because of the spiro-cyclic conformation of rhodamine, etc. The commonly observed16,17 PET from the tert-amino donor to the excited anthracene via the methyl spacer might not be individually contributing here because electron density over the donor N-atom drifted towards the xanthene core, inducing a partial double bond character to preferably prevail a through-bond electron transfer; although, the contributions of such operative processes may not be completely overruled. It is worth mentioning here that the partial derivatization of the xanthene end amino groups of L1 with alkyl-substitution and the induction of amine rigidity has envisaged activating the internal conversion process. As a consequence, lowering in the fluorescence of L2 and L3 was observed in comparison to that of L1. Apart from the non-radiative deactivation through internal conversion, operative photo-physical processes correlating to the predominant spirolactam state of rhodamine have compelled these probes to exhibit an overall quenched fluorescence (ϕFT < 0.001). It was further observed from the spectral responses of L1–L4 in EtOH at different temperatures (30 °C and 70 °C) that their spiro-cyclic conformation has been retained at lower temperatures (at <30 °C). At an elevated temperature (70 °C), probes, such as L1 and L3, exhibited spectral enhancement due to ring-opening, whereas L2 and L4 remained in the spirocyclic state inducing none or negligible spectral changes. This indicates that a through-bond electron transfer is favoured at elevated temperatures in probes (L1–L3) where one of the amino groups attached to the xanthene core is derivatized, which consequently enhances CT character through induced conjugation. However, the deviation of the spectral behaviour of L2 from its other analogues (L1 and L3), at elevated temperatures is more complicated and requires further investigation on various operative processes before rendering any conclusion in this regard.
To investigate their metal ion induced signalling responses, various transition and heavy metal ions were added to the solution of L1–L4 in aqueous–acetonitrile binary mixture. The choice of the solvent is prejudiced because the presence of an aqueous component in a medium is known to render functional selectivity in the metal ion induced dual mode signalling in rhodamine-B based probes,11 correlating through competitive parameters such as probe–metal ion interaction and hydration energy of metal ions. Addition of various metals ions to the colourless solution of L1 in CH3CN–H2O (9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 v/v) has resulted in the appearance of an absorption peak at 527 nm with subsequent change in colour to orange in the presence of Pb2+ (ε = 40768 dm3 mol−1 cm−1, ε/ε0 = 59.2 fold), although a few other metal ions have also induced appreciable changes (Ni2+, ε/ε0 = 18.6 fold) to a lesser extent in comparison to that by Pb2+ ions. Metal ion induced change in the fluorescence (I555) spectral pattern followed its absorption spectral behaviour, exhibiting optimal fluorescence enhancement (ϕFT = 0.632) in the presence of Pb2+ ions. These spectral amplifications along with the subsequent appearance of colour in L1 is attributed to the complexation-induced transformation of the spiro-ring of rhodamine to its ring-opened carboxamide conformation, and the extent of dual mode signalling was observed to be maximum in the preferential presence of Pb2+ ions over other metal ions under investigation.
:0.5 v/v) has resulted in the appearance of an absorption peak at 527 nm with subsequent change in colour to orange in the presence of Pb2+ (ε = 40768 dm3 mol−1 cm−1, ε/ε0 = 59.2 fold), although a few other metal ions have also induced appreciable changes (Ni2+, ε/ε0 = 18.6 fold) to a lesser extent in comparison to that by Pb2+ ions. Metal ion induced change in the fluorescence (I555) spectral pattern followed its absorption spectral behaviour, exhibiting optimal fluorescence enhancement (ϕFT = 0.632) in the presence of Pb2+ ions. These spectral amplifications along with the subsequent appearance of colour in L1 is attributed to the complexation-induced transformation of the spiro-ring of rhodamine to its ring-opened carboxamide conformation, and the extent of dual mode signalling was observed to be maximum in the preferential presence of Pb2+ ions over other metal ions under investigation.
The absorption and emission spectral pattern of L2 (1 μM) in CH3CN–H2O (9.5:0.5 v/v) is shown in Fig. 2. None of these metal ions were observed to induce any changes in the colour of the solution or modulate the absorption transitions, except Pb2+, which upon addition turned the colourless solution to orange along with the appearance of an absorption transition at ∼530 nm corresponding to the ring-opened rhodamine (Fig. 2a). When excited at 350 nm, its quenched fluorescence (ϕRho < 0.001) also simultaneously enhanced (ϕRho(L2+Pb(II)) = 0.583) with a fluorescence transition at ∼555 nm upon the addition of Pb2+ (10 eq.). The Pb2+-induced fluorescence enhancement is attributed to the perturbation of various interactive processes occurring simultaneously such as its coordination at the carboxamide–pyridine receptor cavity enabling the spiro-lactam to open, the consequent suppression of operative PET favouring a charge transfer conjugated channel involving the xanthene core and the subsequent initiation of a FRET from the excited anthracene to the ring-opened rhodamine. When monitored at the anthracene emission window (I417), Pb2+ ions did not induce substantial spectral enhancement in comparison to that of the rhodamine moiety; its fluorescence from the excited anthracene component hardly resulted in a 2-fold enhancement in the presence of Pb2+ ions through an anticipated PET suppression and still remained lower because of initiated FRET pathways. In the context of anthracene monomer fluorescence, maximum enhancement was observed in the presence of Cu2+ ions (ϕAn = 0.159) among all the metal ions added. The Pb2+ selective fluorescence enhancement in L2 as a consequence of combined PET inhibition-FRET initiation processes could not be observed for other metal ions, complementing inferences from its absorption spectral analysis.
 | ||
| Fig. 2 Absorption (a) and fluorescence (b) spectra of L2 in the presence of various metal ions in CH3CN–H2O (9.5:0.5 v/v), corresponding photographs showing colour change (c) and fluorescence on excitation at 350 nm (d) [L2] = 1 μM. | ||
The fluorescence titration of L2 (0.1 μM) with Pb2+ in MeCN–H2O (9.5:0.5 v/v) revealed that it exhibited metal ion induced fluorescence enhancement in a ratiometric pattern with an isobestic point at ∼515 nm (Fig. 3). The fluorescence spectra of metal free L2 represented a structured band centred at the 350–500 nm region corresponding to the weak fluorescence (ϕAn = 0.0002) of excited anthracene and did not contain any long range transition corresponding to the lactonized rhodamine G component. Upon the gradual addition of Pb2+ ions to that solution, fluorescence intensity (I417) corresponding to anthracene enhanced (up to 2-fold), indicating inhibition in the PET process; the increasing trend was observed up to an addition of a 50 nM concentration. Further the addition up to 5 μM of Pb2+ resulted in a decrease in anthracene fluorescence. However, the gradual appearance of a new red-shifted emission peak at 555 nm (Fig. 3a) corresponding to that of the ring-opened rhodamine was observed with concomitant colour change from colourless to orange/green (under illumination at 350 nm). The decrease in I417 and increase in I555 fluorescence was observed in a ratiometric manner in the 50 nM–10 μM concentration range of added Pb2+ (Fig. 3b), which logistically supplemented the initiation of the FRET process.
 | ||
| Fig. 3 (a) Fluorescence titration spectra of L2 (0.1 μM) upon the gradual addition of Pb2+ ions in CH3CN–H2O (9.5:0.5 v/v), λex = 350 nm, em. and ex. bp. = 5 nm, RT; (b) plot of its fluorescence intensities at 417 nm and 555 nm as a function of added Pb2+ ion concentration. | ||
The plot of the absorbance of L2 as a function of the mole fractions of added Pb2+ ion (Job's plot) inferred the complexation stoichiometry to be in a 1:1 (L2–Pb2+) ratio. The nonlinear regression of its fluorescence titration data with Pb2+ ions has determined22 the association constant [logKa = 6.22 ± 0.24] of the complex. When estimated from its absorption titration spectral data, it was found (Kabsa = 2.312 × 104 M−1) to be consistent with that obtained through the fluorescence titration profile and it was comparable with a correlation factor [log{ka(fluorescence)/ka(absorption)}] of 1.42.
The appreciable spectral overlap between the emission of excited anthracene and the absorption of the L2–Pb2+ complex with the ring-opened rhodamine 6G conformation enabled these two fluorophores to be a donor (DAn)–acceptor (ARh) pair for a FRET process. The efficiency of singlet–singlet excitation energy transfer (ηEET) between DAn to ARh evaluated23 from its steady-state fluorescence data was found to be 90.35%, which is in good agreement with other rhodamine based systems.24 The energy transfer efficiency depends upon Förster critical radius25 (R0) and on the interchromophoric distances (r) between DAn and ARh, where the energy transfer is effective over a distance of R0 ± 0.5R0. Förster's critical distance (R0) was calculated to be 41.16 Å by assuming rapid relative orientation of DAn and ARh with a dynamic isotropic average of the orientation factor 〈κ2〉 as 2/3.
The quenched fluorescence of the metal free L1, as mentioned, was observed to further decrease upon partial substitution to one of the amino groups attached to its xanthene core with an octyl chain (as in L3). This inferred that the amino rigidity through partial derivatization induced the activation of the internal conversion process in these probes leading to the further quenching of fluorescence. In a mixed medium containing various metal ions under investigation in CH3CN–H2O (9.5:0.5 v/v), Pb2+ selectively rendered the appearance of an absorption transition at ∼525 nm (ε = 120171 dm3 mol−1 cm−1) along with the enhancement of fluorescence at ∼550 nm (λex = 525 nm) in L3 while other metal ions rendered no or negligible change. Its colourless solution also selectively turned orange which appeared green on illumination (at 350 nm) in the presence of Pb2+ ions. Fluorescence enhancement in the presence of Pb2+ was observed to be higher in L1 (325 fold) in comparison to those in L2 (28 fold) and L3 (24 fold).
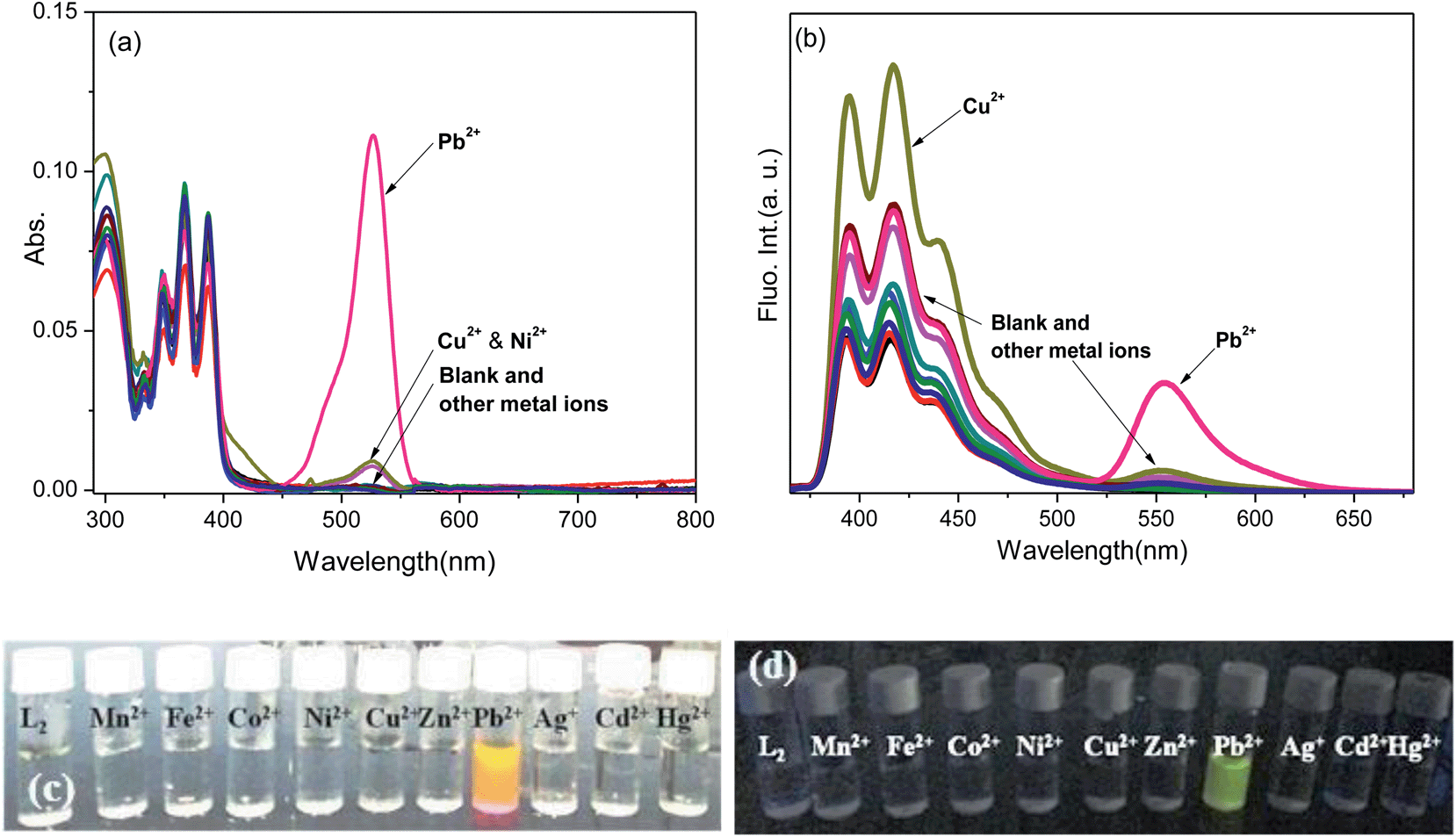
The L4 probe, a rhodamine B analogue of L1, was anticipated to show longer wavelength absorption and emission in comparison to the latter. Its fluorescence was also observed to be lowered (ϕF < 0.001) owing to the activated internal conversion process because of ethyl substitutions to both the amino groups attached to the xanthene core of L1. Contrary to the metal ion induced spectral modulation in L1, its photo-physical properties in the absence and presence of various metal ions in the MeCN–H2O (9.5:0.5 v/v) medium revealed that the selective addition of Hg2+ ions resulted in absorption (ε555 = 28125 dm3 mol−1 cm−1) and fluorescence (ϕF = 0.591) ‘turn-on’ signalling responses, while the presence of other metal ions did not induce any appreciable change in its absorption and fluorescence intensity (Fig. 4). The Hg2+ ion selective dual mode spectral amplification in L4 is consistent with those observed for other rhodamine B based probes in an aqueous–acetonitrile mixture solvent and attributed to a prevailing Hg2+–L4 interaction over the hydration of Hg2+. Other metal ions failed to exhibit such changes because their hydration was favoured over probe–metal ion interaction under the investigating conditions. The plot of absorbance against the mole fractions of Hg2+ added to L4 inferred a 1:1 complexation stoichiometry and the association constant of complexation was estimated to be 2.6 × 104 M−1 from its absorption titration spectra, which is comparable to that of the L2–Pb2+ complex (Fig. 5). However, the fluorescence titration profile of L4 with Hg2+ in MeCN–H2O (9.5:0.5 v/v) resulted in a higher association constant for the complex (logKflua = 11.64) and is correlated with that obtained from the absorption titration through a factor [log{ka(fluo.)/ka(abs.)}] of 2.63.
 | ||
| Fig. 4 (a) Absorption and (b) fluorescence spectra of L4 in the absence and presence of various metal ions in CH3CN–H2O (9.5:0.5 v/v), λex = 500 nm, em. and ex. bp. = 5 nm, RT, [L4] = 10 μM (abs.), 1 μM (fluo.), [M2+] = 50 μM. | ||
 | ||
| Fig. 5 Plot of the absorption transition of (a) L2 (A527) and (b) L4 (A555) as a function of equivalents of added metal ions in CH3CN–H2O (9.5:0.5 v/v). [L2] = 1 × 10−5 M, [L4] = 1 × 10−4 M (inset a and b): double reciprocal plot of absorption {1/(A − A0)} against the concentration of respective metal ions. | ||
To comprehend the impact of solvents on the preferences of these rhodamine G based probes towards metal ion coordination that induces dual mode signalling, the photo-physical behaviour of L2 (10 μM) was also investigated in the presence of various metal ions in pure CH3CN. Its absorption and emission spectral pattern revealed that the probe does not have any preferences towards metal ions in their coordination induced signalling module in pure MeCN. Upon excitation at 350 nm, it resulted in the appearance of an absorption spectral transition at 525 nm because of the spiro-ring opening of rhodamine-G moiety, simultaneously rendered the colourless → orange/green colour transition and triggered a FRET to exhibit fluorescence at 550 nm in the presence of a few metal ions such as Fe2+ (ϕF = 0.91), Ni2+ (ϕF = 0.83), and Pb2+ (ϕF = 0.82), while other metal ions induced none or negligible spectral changes. A comparison in the metal ion induced spectral modulation of L2 in pure CH3CN with that in aqueous CH3CN inferred that the aqueous component in the solvent promoted its preferential coordination to Pb2+ ion over the hydration of metal ion. On the contrary, the presence of an aqueous component in the medium endorsed a preferential coordination of L4 to Hg2+ ions. These observations inferred that the aqueous component in solvent composition plays a vital role in inducing the functional selectivity of the metal ion in coordination mediated dual mode signalling because of competitive parameters such as metal–probe interaction against the hydration of metal ions. Furthermore, from the fluorescence titration data of L2 with Fe2+ in dry CH3CN, the association constant (Ka) of complexation for 1:1 stoichiometry (as determined through Job's plot) was estimated to be 1.13 × 1012 M−1. Its comparison with that obtained for the L2–Pb2+ complex (logKa = 6.22 ± 0.24) in the CH3CN–H2O (9.5:0.5 v/v) medium revealed that the presence of an aqueous component in the medium lowers the extent of the coordination affinity of the probe for metal ions. Aqueous environment promotes the hydration of metal ions while probe–metal interaction is the driving force for complexation through an entropy-effect resulting from the extra dehydration in inner-sphere complexes. Nevertheless, the aqueous component has provided a suitable coordination environment to L2 for preferential complexation with Pb2+ ions while restricting other metal ions through their hydration to render selectivity in the signalling pattern of the probe.
In a controlled experiment, spectral changes of L2 were monitored in the presence of Pb2+ in varying compositions of aqueous (pH = 7.02, HEPES buffer) and CH3CN components as solvent (Fig. 6) to determine appropriate aqueous–organic binary composition of the solvent medium that facilitates the optimal spectral perturbations of the probe and to explore the effect of the aqueous component in promoting selectivity. The preferential Pb2+ induced absorption (A525) and fluorescence (I555) enhancements pertaining to the ring-opened conformation of rhodamine were observed to be optimal in dry CH3CN. Among binary mixtures, the CH3CN–H2O (9.5:0.5 v/v) composition exhibited higher Pb2+ induced spectral amplifications because strong hydration ability of the metal ion preferred its hydration in composition with a higher aqueous component over the probe–metal interaction to induce none or negligible changes. However being a bifluorophoric probe, the ratiometric signalling pattern of the FRET mediated I555 fluorescence enhancement upon the excitation of the L2–Pb2+ complex at 350 nm revealed that the fluorescence signal (I555/I417) ratio was optimal in the CH3CN–H2O (9.5:0.5 v/v) composition (inset, Fig. 6b), and even higher than in dry CH3CN, which establishes the role of the aqueous component and justifies the appropriateness of the composition in binary mixture for obtaining optimal spectroscopic changes.
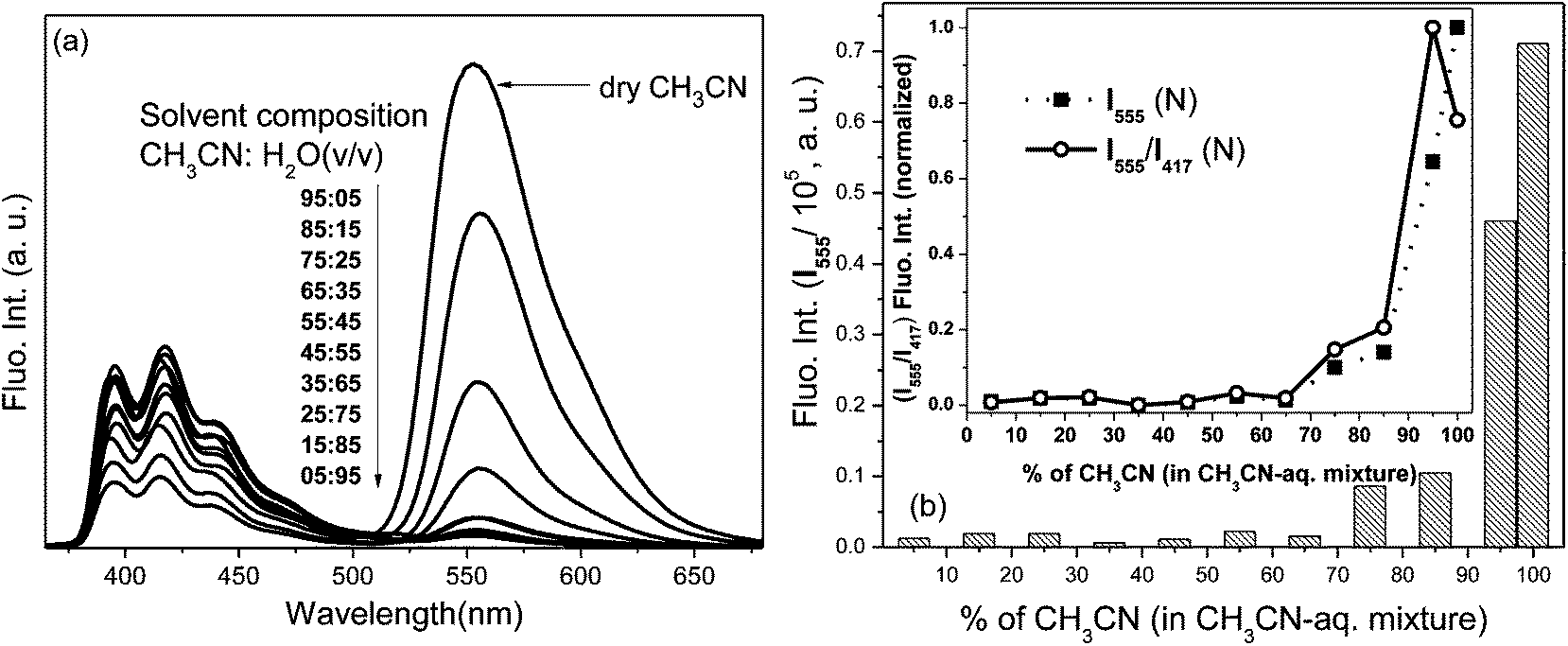 | ||
| Fig. 6 (a) Fluorescence spectra and (b) corresponding intensity (I555) of L2–Pb2+ complex in various compositions of CH3CN–H2O mixture, HEPES buffer, pH = 7.02, λex = 350 nm, em. and ex. bp. = 5 nm, RT, [L2] = 1 μM, [Pb2+] = 5 μM; (inset b): normalized fluorescence intensity ratio (I555/I417) showing optimal spectral enhancement in CH3CN–H2O: 9.5:0.5 v/v composition. | ||
The alteration in the selectivity of metal ion induced dual mode signalling responses of these probes incorporating the same receptor unit at the carboxamide end, i.e. Hg2+ selectivity for L4 in comparison to that of Pb2+ for L1 under similar (aq.–organic) solvent conditions, was attributed to stereo-electronic perturbation at the receptor site originating from ethyl substitution at amino groups attached to the xanthene core. Despite having a 2-aminoethyl pyridine substitution at the carboxamide end in these probes, the electron density drift to the spirolactam carbonyl group from a tertiary amino group of rhodamine B (as in L4) is presumed to be higher than that from a secondary one of rhodamine G (as in L1). Consequently, spatial disposition and coordination abilities of lactamide donors in the receptor unit get reoriented in L4, modulating binding preferences towards metal ions to exhibit altered signalling responses when compared with those of L1. Partial substitution at one of the –N(H)Et aniline segments of the xanthene core in L1 functionalized with either bulky aromatics (L2) or flexible long alkyl chains (L2) might not have effected a profound stereo-electronic modulation in comparison to L1 for such changes in binding preferences at the receptor unit to exhibit subsequent altered metal ion selective spectral responses.
The reversibility in the fluorescence signal responses of L2 because of a metal ion induced FRET process was evaluated with the subsequent addition of ammonium salts of various counter anions in a CH3CN–H2O (9.5:0.5 v/v) medium. The Pb2+-induced enhancement in absorption (A527) and emission (I550) of L2 decreased almost to that of its metal free state and the orange/green coloured solution turned colourless within 1 min after addition of the AcO− anion. Addition of other counter anions exhibited a time dependent decrease in spectral responses and a decolourization of its solution. Apart from anions, its spectral responses revealed that the initial spirolactam state was regenerated upon the subsequent addition of chelating agents such as EDTA and ethylene diamine to the L2–Pb2+ complex in solution. Subsequent addition of Pb2+ ions to the decolourized solution of anion (AcO−), mediated Pb2+-decomplexed L2 (ϕFT ≤ 0.002), resulted in its colourization with the re-appearance of enhanced A527 and I550 spectral transitions almost to the same extent as those upon initial Pb2+ addition to L2. This establishes its reusability as a probe for selective Pb2+ ion detection.
The ability of a probe to selectively detect a metal ion in the presence of various competitive ones enables it to be a chemosensor for that particular metal ion. Hence, competitive experiments (Fig. 7a) were carried out for L2 in aq.–organic medium where the addition of metal ions (5 equiv.) other than Pb2+ to the solution containing L2 and Pb2+ could not generate appreciable changes to its Pb2+ induced FRET mediated fluorescence emission (I550) upon excitation at 350 nm. However, solutions containing L2 and metal ions other than Pb2+, which exhibited none or negligible I550, profoundly triggered FRET to enhance I550 when Pb2+ was added. Similar competitive experiments have established the ability of L4 for the selective detection of Hg2+ in aqueous–organic medium. The limit26 of selective Pb2+ detection by L2 (LOD = 2.1 × 10−7 M) and Hg2+ detection by L4 (2.6 × 10−8 M) were estimated to be very low.
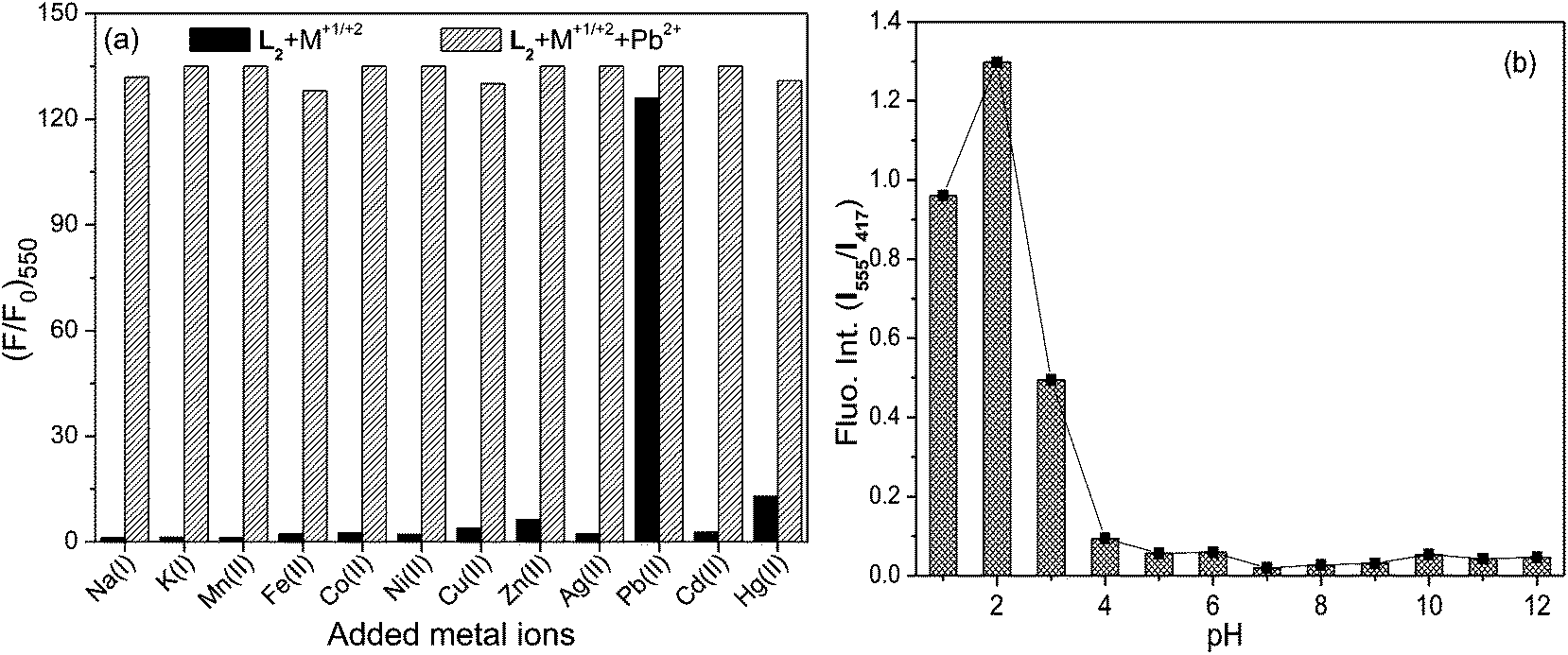 | ||
| Fig. 7 (a) Fluorescence intensities of L2–Pb2+ upon the addition of various metal ions in CH3CN–H2O (9.5:0.5 v/v) followed by the subsequent addition of Pb2+ (5 μM) ion, λex = 350 nm, [L2] = 1 μM, [M+1/+2] = 5 μM. (b) Ratiometric fluorescence signals of L2 in various pH showing its stability over a wide pH range. | ||
A faster response time for executing the signalling action enables a probe to overcome its associated implications for practical implementation in real time monitoring. Hence, the fluorescence spectral responses (I550) of L2 upon excitation at 350 nm were monitored as a function of time in the presence of varying concentrations of Pb2+. Its Pb2+ induced FRET triggered I550 signals were observed to appear <1 min of the addition of metal ions. Similarly, a faster response time (<1 min) of signalling was also obtained for L4 with Hg2+ ion. The spectroscopic signals corresponding to the Pb2+ complexes of L1–L3 and Hg2+ complex of L4 were found to be photo-stable because spectral patterns of solutions containing respective complexes remained enhanced for at least up to 6 h after irradiation at 350 nm, retaining the metal ion induced ring-opened conformation in these probes.
The operational pH range of any probe for their metal ion induced optical spectral modulation is crucial in their practical implementation as chemosensors. Hence, absorption responses of these probes in the absence and presence of Pb2+/Hg2+ ions were monitored at varying pH values. I555 fluorescence in L2 was not observed in the 4–12 pH range (Fig. 7b) when excited at 350 nm, suggesting its stability over a wider pH domain. However, under highly acidic conditions (pH < 4), the colour of L2 solution turned orange along with the appearance of A528 absorption and I550 fluorescence transitions as a consequence of proton induced opening of its spiro-ring, apart from fluorescence enhancement at 393 nm along with its vibrational structures originating from anthracene because of proton induced PET inhibition. In the 5–10 pH range, the addition of Pb2+ led to its chromogenic and fluorogenic dual mode spectral amplifications through metal ion induced ring-opening and triggered FRET process, enabling these probes to be suitable for the signalling operation under physiological conditions. Similarly, L4 was observed to exhibit signalling responses in the presence of Hg2+ over a wider operational pH (4–12) range.
The effectiveness and utility assay of L2 for the detection of Pb2+ ions was evaluated through monitoring its fluorescence signals in Pb2+ contaminated E. coli (ESI†). The microorganisms were incubated for 30 min with L2 followed by the addition of Pb2+ and vice versa under physiological conditions. Their fluorescence imaging revealed that the non-fluorescent microorganism did not exhibit any rhodamine based emission when incubated with either only the probe or the metal ion. However, incubation with L2 followed by the addition of Pb2+ led to fluorescence upon excitation at 350 nm. Hence, these observations demonstrated that L2 has the potential for the detection and quantification of Pb2+ ion accumulation in microorganisms and other biological functionalities.
Conclusion
In summary, the photo-physical behaviour of L1–L4 probes, which incorporate the same receptor unit at their spirocyclic end but structurally vary as a function of substituents at one of the amino-groups attached to their xanthene unit, were carried out to investigate the effect of alkyl substitution on selectivity in their metal ion induced signalling pattern. The rhodamine G based probes (L1–L3) selectively exhibited both chromogenic and fluorogenic signalling in the presence of Pb2+ ions in an organic–aqueous medium through metal ion induced structural equilibration of their spiro-conformation. On the contrary, rhodamine B based probe L4 exhibited similar dual mode signalling selectively in the presence of Hg2+ ions under similar operational conditions of the solvent medium, which is in good agreement with that of the other rhodamine based probes incorporating ‘amino alkyl’-substitution at their spiro-cyclic end. The switching in selectivity between Pb2+ and Hg2+ ions in these probes, as reflected in their signalling pattern, is attributed to a perturbed stereo-electronic situation at the receptor unit because of the partial substitution of various alkyl groups at the amino N-atoms attached to the xanthene core, which in turn modulated the induced amine rigidity to perturb the activation of internal conversion processes to various extents. Among rhodamine G based probes (L1–L3), the extent of selective fluorescence enhancement in the presence of Pb2+ was observed in the order of L1 > L2 ≥ L3, which is in concomitant correlation with the extent of induced rigidity at amino groups. Interestingly, among those is L2 that selectively executed dual mode transduction in the presence of Pb2+ following FRET modulation pathways. The distinctive design of L2 incorporating other fluorophore at the aniline –N(H)Et segment of the xanthene core differs from commonly designed FRET mediated rhodamine chemosensors19a–d,20 where the second fluorophore mostly attached at the lactonized end. The ratiometric fluorescent spectral module of L2 with Pb2+ enabled it to have a significantly lower detection limit, fulfilling the pre-requisite criteria of subsequent chemosensing applications. Its extent of resonance energy transfer and fluorescence decay time was observed to be in alignment with its metal ion induced FRET modulated fluorescence enhancement. The signalling pattern in L2 responsive to multiple ions in acetonitrile in comparison with that of highly selective Pb2+ in aq.–acetonitrile medium inferred the role of aqueous component in the mixture medium for inducing selectivity. However, this requires further investigation before ascertaining a concluding justification. Nevertheless, structural modifications at xanthene amino-groups in these probes effectively modulated the selectivity for metal ion coordination at the receptor unit to subsequently alter the chromogenic and fluorogenic signalling pattern for their detection at the desired low concentration level.Experimental section
Synthesis
Yield: 0.383 g (73%); mixed m.p. = 94–96 °C; ESI-MS (C33H34N4O2): m/z+ (%), 519.25 [L1 + 1]+ (100), 1H-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 8.54 (1H, d, J = 3.99 Hz), 8.35 (1H, d, J = 7.99 Hz), 7.93 (1H, d, J = 7.99 Hz), 7.59 (1H, dd, J1 = 7.99 Hz, J2 = 3.99 Hz), 7.43 (2H, t, J = 3.99 Hz), 7.13 (2H, dd, J1 = 5.99 Hz, J2 = 3.99 Hz), 6.98 (1H, d, J = 7.99 Hz), 6.35 (2H, s), 6.23 (1H, s), 3.48 (2H, dt, J1 = 7.99 Hz, J2 = 1.99 Hz), 3.19 (2H, dd, J1 = 5.99 Hz, J2 = 3.99 Hz), 3.11 (2H, t, J = 3.99 Hz), 2.92 (2H, t, J = 7.99 Hz), 2.68 (2H, d, J = 5.99 Hz), 1.84 (4H, d, J = 7.99 Hz), 1.30 (6H, t, J = 6.99 Hz); 13C-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 160.04, 151.70, 149.35, 148.91, 147.33, 136.37, 136.04, 132.36, 131.06, 128.50, 127.91, 123.71, 123.39, 123.15, 122.72, 121.28, 120.99, 117.81, 106.04, 96.63, 65.10, 41.92, 40.23, 38.34, 36.57, 16.69, 14.79. Anal calcd for C33H34N4O2 (Mw = 518.65), %: C, 76.42, H, 6.61, N, 10.80; found: C, 75.97, H, 6.79, N, 10.93.
:1 v/v) as the eluent.Yield: 0.604 g (84%); mixed m.p. = 123–126 °C; ESI-MS (C48H44N4O2): m/z+ (%), 709.24 [L2 + 1]+ (25%); 1H-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 8.60 (1H, d, J = 3.99 Hz), 8.43 (1H, s), 8.36 (1H, d, J = 7.99 Hz), 8.20 (1H, d, J = 7.99 Hz), 7.93 (1H, d, J = 7.99 Hz), 7.54 (2H, s), 7.40 (4H, q, J = 3.99 Hz), 7.36 (2H, d, J = 7.99 Hz), 7.28 (2H, br s), 6.98 (1H, d, J = 7.99 Hz), 6.40 (2H, s), 6.35 (1H, br s), 6.23 (1H, br s), 5.66 (1H, s), 4.65 (1H, s), 3.50 (2H, t, J = 3.99 Hz), 3.19 (2H, d, J = 3.99 Hz), 2.72 (2H, t, J = 3.99 Hz), 1.86 (4H, d, J = 7.99 Hz), 1.78 (2H, s), 1.32 (2H, d, J = 3.99 Hz), 1.29 (2H, d, J = 7.99 Hz), 0.88 (6H, br s); 13C-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 168.39, 151.71, 149.81, 148.93, 147.34, 136.04, 132.38, 131.47, 131.31, 129.08, 128.87, 128.81, 128.51, 127.93, 127.52, 126.36, 125.54, 125.34, 125.30, 125.05, 124.82, 124.76, 124.72, 124.01, 123.78, 123.16, 122.73, 120.99, 65.11, 40.22, 38.35, 36.59, 16.72, 14.74; anal calcd for C48H44N4O2 (Mw = 708.89), %: C, 81.33, H, 6.26, N, 7.90; found: C, 81.19, H, 6.10, N, 8.07.
:1 v/v) as the eluent.Yield: 0.538 g (85%); mixed m. p. = 113–116 °C; ESI-MS (C41H50N4O2): m/z+ (%), 631.27 [L3 + 1]+ (63%); 1H-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 8.35 (1H, s), 8.28 (1H, s), 7.85 (1H, dd, J1 = 3.79 Hz, J2 = 1.59 Hz), 7.55 (1H, dt, J1 = 3.79 Hz, J2 = 1.59 Hz), 7.35 (1H, td, J1 = 3.99 Hz, J2 = 1.59 Hz), 7.17 (1H, d, J = 5.59 Hz), 7.10 (1H, d, J = 3.19 Hz), 6.95 (1H, dd, J1 = 3.99 Hz, J2 = 1.19 Hz), 6.92 (1H, d, J = 1.19 Hz), 6.86 (1H, d, J = 7.59 Hz), 6.29 (1H, s), 6.14 (1H, s), 5.72 (1H, br s), 3.50 (2H, t, J = 4.79 Hz), 3.40 (2H, s), 3.32 (4H, dt, J1 = 7.99 Hz, J2 = 1.59 Hz), 3.09 (4H, br s), 2.94 (4H, t, J = 7.19 Hz), 2.68 (2H, d, J = 5.59 Hz), 2.57 (2H, t, J = 7.59 Hz), 1.80 (6H, s), 1.29 (2H, d, J = 5.59 Hz), 1.21 (6H, td, J1 = 7.19 Hz, J2 = 1.59 Hz), 0.80 (3H, br s); 13C-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 168.17, 159.06, 158.87, 157.96, 153.74, 151.68, 149.07, 148.93, 137.34, 136.66, 132.43, 130.96, 128.36, 127.97, 123.80, 122.65, 121.59, 117.85, 105.75, 96.50, 65.18, 53.48, 47.94, 46.98, 40.30, 38.27, 36.49, 34.82, 33.86, 32.75, 31.69, 29.24, 28.93, 27.16, 26.68, 25.34, 22.54, 16.72, 14.66, 14.03; anal calcd for C41H50N4O2 (Mw = 630.35), %: C, 78.06, H, 7.99, N, 8.88, found: C, 77.91, H, 8.11, N, 8.99.
Yield: 0.710 g (69%); mixed m.p. = 103–106 °C; ESI-MS (C35H38N4O2) m/z+ (%): 583.15 [L4·HCl]+ (100); 1H-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 8.53 (1H, d, J = 3.99 Hz), 8.48 (1H, d, J = 5.99 Hz), 7.92 (1H, d, J = 7.99 Hz), 7.61 (1H, d, J = 1.99 Hz), 7.41 (1H, d, J = 7.99 Hz), 7.12 (2H, td, J1 = 7.99 Hz, J2 = 3.59 Hz), 6.99 (1H, s), 6.43 (2H, d, J = 7.99 Hz), 6.38 (2H, d, J = 3.99 Hz), 6.23 (2H, d, J = 7.99 Hz), 3.30 (2H, t, J = 7.99 Hz), 3.12 (2H, t, J = 7.99 Hz), 2.95 (4H, d, J = 7.99 Hz), 2.92 (4H, d, J = 7.99 Hz), 1.18 (12H, t, J = 7.99 Hz); 13C-NMR (400 MHz, CDCl3, 25 °C, TMS, δ): 168.21, 159.6(d), 153.81(d), 149.38(d), 148.92(m), 137.26, 136.73(m), 132.33, 131.09, 128.82, 127.94, 126.05, 123.72(m), 122.70(d), 121.68(m), 108.03, 105.48, 97.77, 65.05, 44.32, 40.65(d), 39.91(d), 36.60, 12.60; anal. calcd for C35H38N4O2 (Mw = 546.70), %: C, 76.89, H, 7.01, N, 10.25, found: C, 76.43, H, 7.16, N, 10.15.
Acknowledgements
BPB wishes to thank DST, New Delhi for financial support (SB/EMEQ-226/2013) for this work; UGC New Delhi for a senior research fellowship to BB; Director of CSIR-IMMT for infrastructural support through CSIR-CSC-0101.Notes and references
- For reviews, see: (a) J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780 CrossRef CAS PubMed; (b) B. Bag and P. K. Bharadwaj, in Photo, Electrochemistry & Photobiology in the Environment, Energy and Fuel, ed. S. Kaneco, 2007, p. 201 Search PubMed; (c) V. Amendola, L. Fabbrizzi, F. Forti, M. Licchelli, C. Mangano, P. Pallavicini, A. Poggi, D. Sacchi and A. Taglieti, Coord. Chem. Rev., 2006, 250, 273 CrossRef CAS PubMed; (d) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551 CrossRef CAS PubMed; (e) B. Valeur and I. Leroy, Coord. Chem. Rev., 2000, 205, 3 CrossRef CAS; (f) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlauggsson, A. J. M. Huxley, C. P. McCoy, J. T. Radmacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- (a) Heavy Metals in the Environment, ed. B. Sarkar, Marcel Dekker Inc., New York, 2002 Search PubMed; (b) D. Beyersmann, in Metals and Their Compounds in the Environment, ed. E. Merian, Wiley-VCH, Weinheim, 1990 Search PubMed; (c) W. F. Fitzgerald, C. H. Lamborg and C. R. Hammerschmidt, Chem. Rev., 2007, 107, 641 CrossRef CAS PubMed; (d) T. W. Clarkson and L. Magos, Crit. Rev. Toxicol., 2006, 36, 609 CrossRef CAS PubMed; (e) O. Malm, Environ. Res., 1998, 77, 73 CrossRef CAS PubMed; (f) P. Grandjean, P. Weihe, R. F. White and F. Debes, Environ. Res., 1998, 77, 165 CrossRef CAS PubMed.
- (a) H. L. Needleman, Human Lead Exposure, CRC Press, Boca Raton, FL, 1992 Search PubMed; (b) J. D. Cremin, Jr, M. L. Luck, N. K. Laughlin and D. R. Smith, Toxicol. Appl. Pharmacol., 1999, 161, 283 CrossRef PubMed; (c) Y. Finkelstein, M. E. Markowitz and J. F. Rosen, Brain Res. Rev., 1998, 27, 168 CrossRef CAS.
- (a) T. I. Lidsky and J. S. Schneider, Brain, 2003, 126, 5 CrossRef PubMed; (b) H.-W. Tang, G. Huel, D. Campagna, G. Hellier, C. Boissinot and P. Blot, J. Appl. Toxicol., 1999, 19, 167 CrossRef CAS; (c) L. Stokes, R. Letz, F. Gerr, M. Kolczak, F. E. McNeill, D. R. Chettle and W. E. Kaye, Occup. Environ. Med., 1998, 55, 507 CrossRef CAS; (d) K. M. Stiles and D. C. Bellinger, Neurotoxicol. Teratol., 1993, 15, 27 CrossRef CAS.
- E. K. Silbergeld, I. A. Silva and J. F. Nyland, Toxicol. Appl. Pharmacol., 2005, 207, S282 CrossRef CAS PubMed.
- (a) P. Kaur, M. Aschner and T. Syversen, Neurotoxicology, 2006, 27, 492 CrossRef CAS PubMed; (b) E. R. Milaeva, J. Inorg. Biochem., 2006, 100, 905 CrossRef CAS PubMed; (c) R. K. Zalups and S. Ahmad, J. Am. Soc. Nephrol., 2004, 15, 2023 CrossRef CAS PubMed; (d) R. K. Zalups and L. H. Lash, Toxicol. Appl. Pharmacol., 2006, 214, 88 CrossRef CAS PubMed; (e) G. Shanker, L. A. Mutkus, S. J. Walker and M. Aschner, Mol. Brain Res., 2002, 106, 1 CrossRef CAS.
- (a) I. Oniyido, A. R. Norris and E. Buncel, Chem. Rev., 2004, 104, 5911 CrossRef PubMed; (b) H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed; (c) D. W. Boening, Chemosphere, 2000, 40, 1335 CrossRef CAS.
- For selected reviews, see: (a) L. Yuan, W. Lin, K. Zheng, L. He and W. Hyang, Chem. Soc. Rev., 2013, 42, 622 RSC; (b) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef CAS PubMed; (c) M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC; (d) J. F. Jhang and J. S. Kim, Anal. Sci., 2009, 25, 1271 CrossRef; (e) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC.
- Few recent examples for Hg2+ ions: (a) X. Chen, X. Meng, S. Wang, Y. Cai, Y. Wu, Y. Feng, M. Zhu and Q. Guo, Dalton Trans., 2013, 42, 14819 RSC; (b) W. Wang, Y. Li, M. Sun, C. Zhou, Y. Zhang, Y. Li and Q. Yang, Chem. Commun., 2012, 48, 6040 RSC; (c) K. Ghosh, T. Sarkar and A. Sammader, Org. Biomol. Chem., 2012, 10, 3236 RSC; (d) F. Wang, S.-W. Nam, Z. Guo, S. Park and J. Yoon, Sens. Actuators, B, 2012, 161, 948 CrossRef CAS PubMed; (e) S. Saha, P. Mahato, G. U. Reddy, E. Suresh, A. Chakrabarty, M. Baidya, S. K. Ghosh and A. Das, Inorg. Chem., 2012, 51, 336 CrossRef CAS PubMed; (f) N. Vasimalai and S. A. John, Analyst, 2012, 137, 3349 RSC.
- Pb2+ ions: (a) Z.-Q. Hu, C.-S. Lin, X.-M. Wang, L. Ding, C.-L. Cui, S.-F. Liu and H. Y. Lu, Chem. Commun., 2010, 46, 3765 RSC; (b) X. Zhang, Y. Shiraishi and T. Hirai, Tetrahedron Lett., 2007, 48, 5455 CrossRef CAS PubMed; (c) J. Y. Kwon, Y. J. Jang, Y. J. Lee, K. M. Kim, M. S. Seo, W. Nam and J. Yoon, J. Am. Chem. Soc., 2005, 127, 10107 CrossRef CAS PubMed.
- (a) B. Bag and B. Biswal, Org. Biomol. Chem., 2012, 10, 2733 RSC; (b) A. Pal and B. Bag, J. Photochem. Photobiol., A, 2012, 240, 42 CrossRef CAS PubMed; (c) B. Bag and A. Pal, Org. Biomol. Chem., 2011, 9, 4467 RSC.
- (a) X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025 CrossRef CAS PubMed; (b) J. V. Mello and N. S. Finney, Angew. Chem., Int. Ed., 2001, 40, 1536 CrossRef CAS; (c) H. Takakusa, K. Kikuchi, Y. Urano, H. Kojima and T. Nagano, Chem.–Eur. J., 2003, 9, 1479 CrossRef CAS PubMed; (d) A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2005, 127, 10464 CrossRef CAS PubMed; (e) W. Lin, L. Yuan, L. Long, C. Guo and J. Feng, Adv. Funct. Mater., 2008, 18, 2366 CrossRef CAS; (f) Z. Xu, X. Qian and J. Cui, Org. Lett., 2005, 7, 3029 CrossRef CAS PubMed.
- (a) F. D. Lewis, Y. Zhang and R. L. Letsinger, J. Am. Chem. Soc., 1997, 119, 5451 CrossRef CAS; (b) J. Lou, T. A. Hatton and P. E. Laibinis, Anal. Chem., 1997, 69, 1262 CrossRef CAS; (c) A. T. Reise Sousa, E. M. S. Castanheira, A. Fedorov and J. M. G. Martinho, J. Phys. Chem. A, 1998, 102, 6406 CrossRef; (d) H. Nohta, H. Satozono, K. Koiso, H. Yoshida, J. Ishida and M. Yamaguchi, Anal. Chem., 2000, 72, 4199 CrossRef CAS; (e) A. Okamoto, T. Ichiba and I. Saito, J. Am. Chem. Soc., 2004, 126, 8364 CrossRef CAS PubMed.
- (a) C. Kaewtong, B. Wanno, Y. Uppa, N. Morakot, B. Pulpoka and T. Tuntulani, Dalton Trans., 2011, 40, 12578 RSC; (b) Y. H. Lee, M. H. Lee, J. F. Zhang and J. S. Kim, J. Org. Chem., 2010, 75, 7159 CrossRef CAS PubMed; (c) S. K. Kim, K. M. K. Swamy, S.-Y. Chung, H. N. Kim, M. J. Kim, Y. Jeong and J. Yoon, Tetrahedron Lett., 2010, 51, 3286 CrossRef CAS PubMed; (d) J. Mao, L. Wang, W. Dou, X. Tang, Y. Yan and W. Liu, Org. Lett., 2007, 9, 4567 CrossRef CAS PubMed.
- (a) T. Lopez Arbeloa, F. Lopez Arbeloa, M. J. Estevez and F. Lopez Arbeloa, J. Lumin., 1994, 59, 369 CrossRef CAS; (b) M. Vogel, W. Rettig, R. Sens and K. H. Drexhage, Chem. Phys. Lett., 1988, 147, 452 CrossRef CAS; (c) M. Vogel, W. Rettig, R. Sens and K. H. Drexhage, Chem. Phys. Lett., 1988, 147, 461 CrossRef CAS; (d) T. Karstens and K. Kobs, J. Phys. Chem., 1980, 84, 1871 CrossRef CAS.
- (a) B. Bag and P. K. Bharadwaj, J. Phys. Chem. B, 2005, 109, 4377 CrossRef CAS PubMed; (b) J. S. Kim, K. H. Noh, S. H. Lee, S. K. Kim and J. Yoon, J. Org. Chem., 2003, 68, 597 CrossRef CAS PubMed; (c) L. Aoki, T. Sakaki and S. Shinkai, J. Chem. Soc., Chem. Commun., 1992, 730 RSC.
- (a) Advanced Concepts in Fluorescence Spectroscopy: Small Molecule Sensing, ed. C. D. Geddes and J. R. Lakowicz, Springer-Sciences, New York, USA, 2005 Search PubMed; (b) G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, VCH, Weinheim, 1993 Search PubMed; (c) K. Bhattacharyya and M. Chowdhury, Chem. Rev., 1993, 93, 507 CrossRef CAS; (d) F. Fages, J.-P. Desvergne and H. Bouas-Laurent, J. Am. Chem. Soc., 1989, 111, 96 CrossRef CAS; (e) C. D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CrossRef.
- (a) K. Kikuchi, Chem. Soc. Rev., 2010, 39, 2048 RSC; (b) K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS PubMed; (c) J. N. Miller, Analyst, 2005, 130, 265 RSC; (d) K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS PubMed; (e) Topics in Fluorescence Spectroscopy, ed. J. R. Lakowicz, Plenum press, New York, 1999 Search PubMed; (f) T. Förster, Z. Naturforsch., A: Phys. Sci., 1949, 49, 321 Search PubMed.
- (a) N. R. Chereddy, S. Thennarasu and A. B. Mandal, Analyst, 2013, 138, 1334 RSC; (b) B. Liu, F. Zeng, Y. Liua and S. Wu, Analyst, 2012, 137, 1698 RSC; (c) V. Luxami, M. Verma, R. Rani, K. Paul and S. Kumar, Org. Biomol. Chem., 2012, 10, 8076 RSC; (d) X. Lv, J. Liu, Y. Liu, Y. Zhao, M. Chen, P. Wang and W. Guo, Org. Biomol. Chem., 2011, 9, 4954 RSC; (e) R. Ferreira, P. Remón and U. Pischel, J. Phys. Chem. C, 2009, 113, 5805 CrossRef CAS; (f) E. S. Barrett, T. J. Dale and J. Rebek Jr, J. Am. Chem. Soc., 2007, 129, 3818 CrossRef CAS PubMed; (g) K. K. Sadhu, B. Bag and P. K. Bharadwaj, Inorg. Chem., 2007, 46, 8051 CrossRef CAS PubMed; (h) A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640 CrossRef CAS PubMed; (i) A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300 CrossRef CAS PubMed.
- B. Biswal and B. Bag, Org. Biomol. Chem., 2013, 11, 4975 CAS.
- (a) R. Menzel, R. Bornemann and E. Thiel, Phys. Chem. Chem. Phys., 1999, 1, 2435 RSC; (b) R. Menzel and E. Thiel, Chem. Phys. Lett., 1998, 291, 237 CrossRef CAS; (c) J. Karpiuk, Z. R. Grabowski and F. C. De Schryver, J. Phys. Chem., 1994, 98, 3247 CrossRef CAS.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC; (b) Y. Shiraishi, S. Sumiya, Y. Kohno and T. Hirai, J. Org. Chem., 2008, 73, 8571 CrossRef CAS PubMed; (c) S. Fery-Forgues, M.-T. Le-Bris, J.-P. Guette and B. Valeur, J. Phys. Chem., 1988, 92, 6233 CrossRef CAS; (d) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS.
- (a) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, 2002, p. 247 Search PubMed; (b) K. Kils, J. Kajanus, J. Mrtensson and B. Albinsson, J. Phys. Chem. B, 1999, 103, 7329 CrossRef CAS; (c) K. K. Jensen, S. B. van Berlekom, J. Kajanus, J. Mrtensson and B. Albinsson, J. Phys. Chem. A, 1997, 101, 2218 CrossRef CAS.
- (a) P. Mahato, S. Saha, E. Suresh, R. Di Liddo, P. P. Parnigotto, M. T. Conconi, M. K. Kesharwani, B. Ganguly and A. Das, Inorg. Chem., 2012, 51, 1769 CrossRef CAS PubMed; (b) C. Ma, F. Zeng, L. Huang and S. Wu, J. Phys. Chem. B, 2011, 115, 874 CrossRef CAS PubMed; (c) B. Ma, S. Wu, Y. Luo, J. Zhao and Z. Tong, Nanotechnology, 2010, 21, 195501 CrossRef PubMed.
- C. G. dos Remedios and P. D. Moens, J. Struct. Biol., 1995, 115, 175 CrossRef CAS PubMed.
- (a) C. R. Lohani, J.-M. Kim, S.-Y. Chung, J. Yoon and K.-H. Lee, Analyst, 2010, 135, 2079 RSC; (b) B. P. Joshi, C. R. Lohani and K.-H. Lee, Org. Biomol. Chem., 2010, 8, 3220 RSC.
- M. Bullpit, W. Kitching, D. Doddrell and W. J. Adcock, Org. Chem., 1976, 41, 760 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for L4: C35H38N4O2; Mw = 546.69; block-shaped; colourless, orthorhombic, space group Pna21, a = 13.399(2) Å, b = 15.319(7) Å, c = 13.902(9) Å, α = β = γ = 90.00, U = 2853.1(6) Å3, T = 100(2) K, Z = 4, μ(Mo Kα) = 0.080 mm−1, F(000) = 1168, ρcalc = 1.273 mg m−3, 7051 reflection data with 370 parameters, 3823 [I ≥ 2 σ(I)] unique reflections used in calculations. Final R1 = 0.0882, wR2 = 0.1827, S = 0.997. CCDC 963835. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra04152f |
| This journal is © The Royal Society of Chemistry 2014 |