
Original access to 5-aryluracils from 5-iodo-2′-deoxyuridine via a microwave assisted Suzuki-Miyaura cross-coupling/deglycosylation sequence in pure water†
T. Lussier‡
a,
G. Hervéa,
G. Enderlina and
C. Len*ab
aTransformation Intégrée de la Matière renouvelable, UTC-ESCOM, Centre de Recherche de Royallieu, F-60200 Compiègne, BP 20529, France. E-mail: christophe.len@utc.fr; Fax: +33 (0)3 44 97 15 91
bDepartment of Chemistry, University of Hull, Hull, HU6 7RX, England, UK
First published on 17th September 2014
Abstract
A facile and efficient methodology to obtain various 5-aryluracil derivatives was developed through a two steps sequence: a ligand-free Suzuki-Miyaura cross-coupling reaction starting from totally deprotected 5-iodo-(2′-deoxy)uridine followed by a very simple deglycosylation procedure in pure water with assistance of microwave irradiation.
Introduction
5-Aryluracil derivatives are known for their biological activities.1–4 For example, compound 1 is reported as a potent human gonadotropin-releasing hormone receptor (hGnRH-R) antagonist (Fig. 1). | ||
| Fig. 1 Compound 1 as potent human gonadotropin-releasing hormone receptor. | ||
Different pathways to those compounds are described in the literature. They can, for example, be obtained by SRN1 Aromatic Nucleophilic Substitution,5 or by tin- or palladium-catalyzed intramolecular arylation of pyrimidines,6,7 or by heating several 2-aryl-3-hydroxypropenoic acid esters with urea followed by base-catalyzed cyclization.8 More often, 5-aryluracils are prepared by cross-coupling reactions such as Suzuki-Miyaura, Stille or also Negishi.9–17 More recently, direct 5-arylations of N1,N3-methylated or benzylated uracils have been reported by Hocek's group18 and Kim's group.19 In 2014, Wnuk described a palladium-catalyzed disrect arylation of monoprotected 5-halouracils with arene promoted by TBAF.20 All the above reported methodologies shared the fact that they require organic solvents and/or protected starting material which is not compatible with the development of green chemistry.
The most common way to prepare 5-arylnucleoside derivatives consisted in palladium cross-coupling reactions.21–26 Unfortunately, despite all our efforts, application of these methodologies to the direct coupling of unprotected 5-iodouracil with various boronic acids failed.
We now report an original methodology leading to various 5-aryluracil derivatives in sole water as solvent, under microwave irradiation, without any protection/deprotection step starting from 5-iodo-2′-deoxyuridine.
Results and discussion
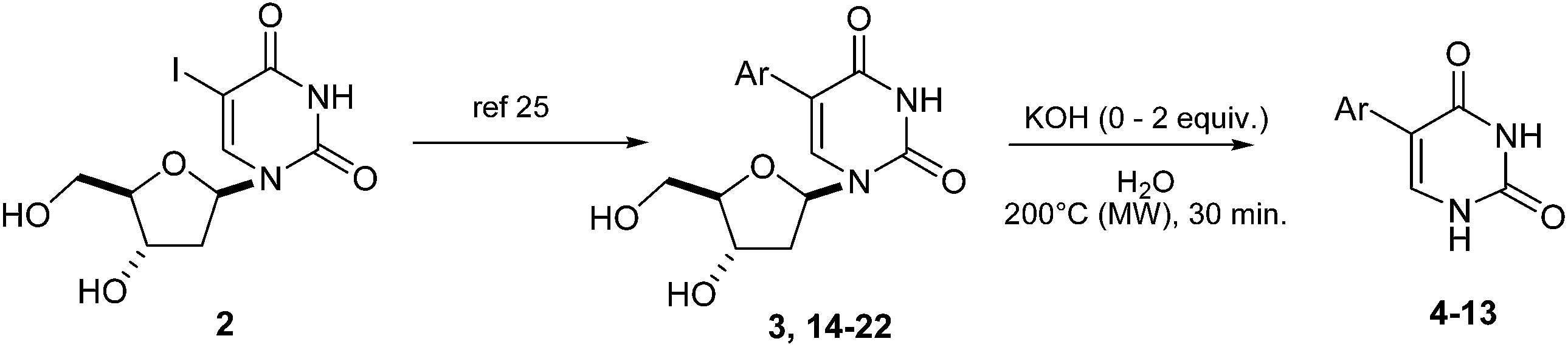
Following one of our most recent work, in an attempt to improve reaction efficiency of our cross-coupling model reaction (5-iodo-2′-deoxyuridine (2) and phenylboronic acid) (Scheme 1), we tested the effect of an increase of the temperature (from 100 °C to 200 °C progressively). | ||
| Scheme 1 Synthesis of 5-phenyl-2′-deoxyuridine (3). | ||
We gradually observed formation of side-products mainly due to dehalogenation of the starting-material and also deglycosylation of both the dehalogenated substrate and the cross-coupled product. Moreover it was noticed that at 200 °C only uracil was obtained indicating that, at this temperature, dehalogenation of the starting-material 2 was faster than cross-coupling reaction. Based on this finding, an original route to 5-phenyluracil (4) consisting in a “one-pot, two-steps” way could be considered. To be effective, it must first take into account the optimum time and temperature previously reported for the cross-coupling reaction step. For this purpose, 5-iodo-2′-deoxyuridine (2) and phenylboronic acid were engaged in a Suzuki-Miyaura cross-coupling reaction in the presence of a small amount of PdII (0.1 mol%) and KOH (2 equiv.) in pure water, first at 100 °C using microwave irradiation (monowave 850W) for 5 minutes, then at 200 °C for 30 minutes (Scheme 2).
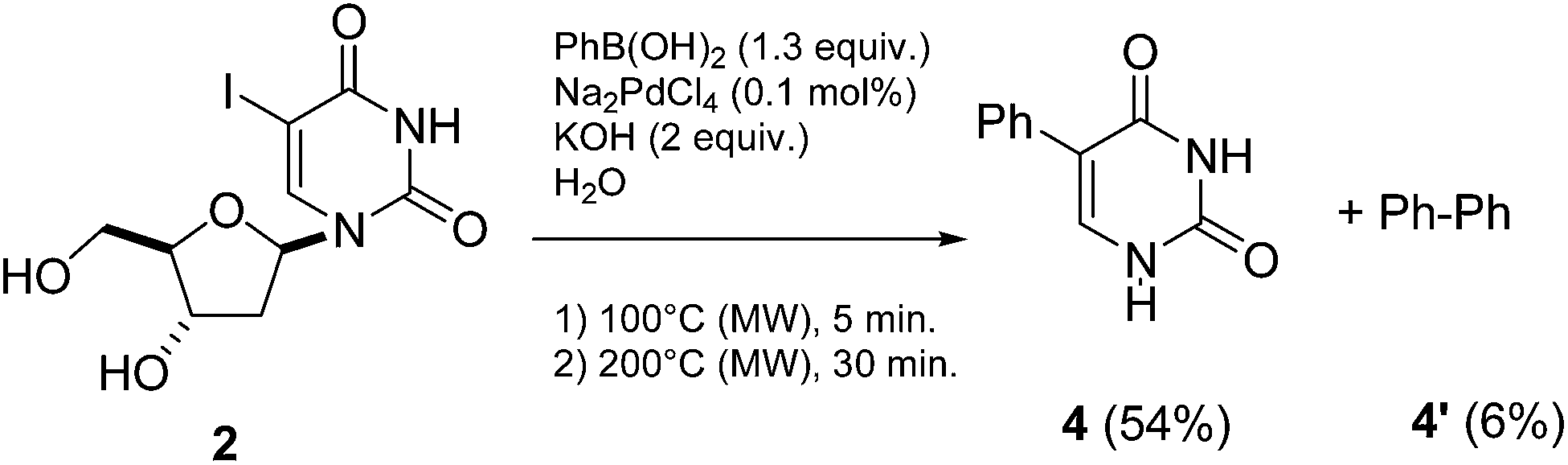 | ||
| Scheme 2 “One-pot, two-steps” synthesis of 5-phenyluracile (4). | ||
After reaction, analyses of the crude mixture showed that 5-phenyluracil (4) was predominantly formed beside the homocoupled compound 4′. The latter was not observed in our previous works23–26 so one could imagine that it might be formed during the deglycosilation step maybe due to the residual presence of palladium, boronic acid and effect of the higher temperature. 5-Phenyluracil (4) and the homocoupled product were readily separated from the glycone degradation by-products by extraction. Ultimately, 5-phenyluracil (4) represented 90% of the final mixture. The applicability of this methodology was then studied and various 5-aryluracils have been obtained (Table 1).
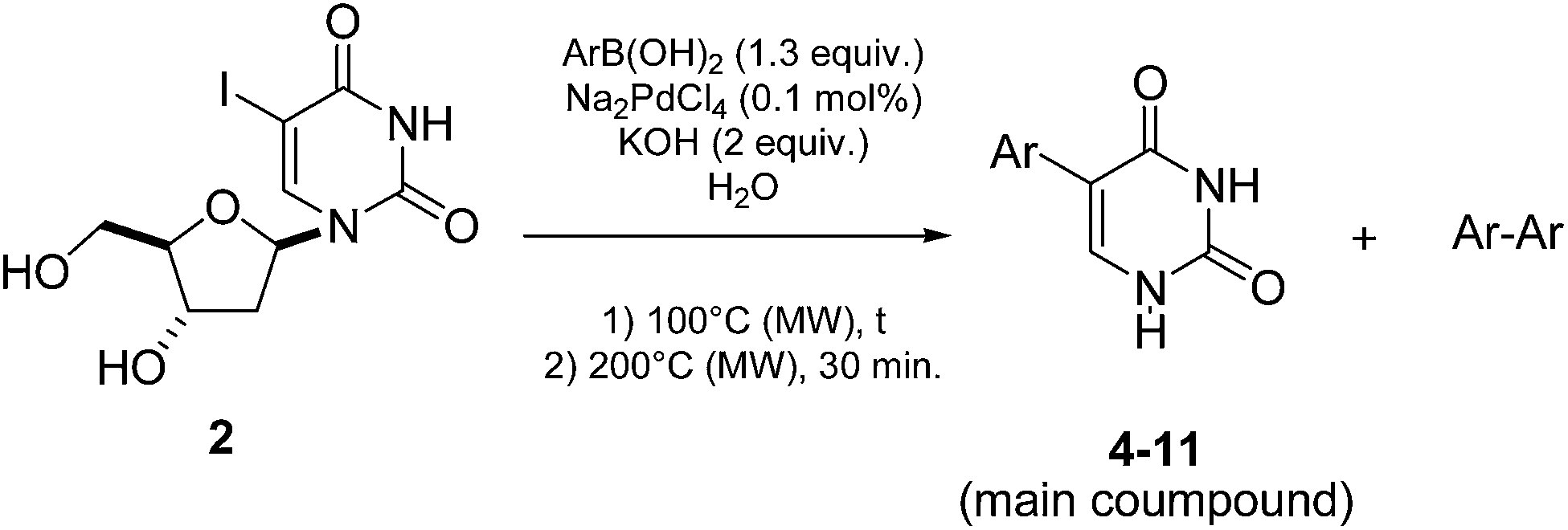


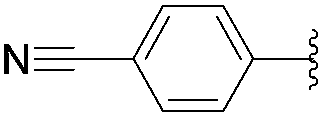


This methodology provided seven 5-aryluracil derivatives in yields (two steps) comprised between 30% and 60%. One must note that the already described compounds 48,9,18a and 518b were all obtained in purely organic medium either in better or lower yields. Uracil derivatives bearing electron-donating group on the phenyl moiety (Table 1, entries 1–4) were isolated in good yields (45–60%) while uracils with electron-withdrawing groups (Table 1, entries 6 and 7) were obtained in quite lower yields (30–50%). Only one deglycosylation could not be achieved. Indeed in the case of the cyano derivative 8 only degradation was obtained from which no product could actually been extracted or characterized (Table 1, entry 5). This might be due to hydrolysis of the nitrile function in presence of KOH at 200 °C.
Nevertheless, even if this “one-pot, two steps” obtention of 5-aryluracil derivatives seemed original and encouraging, it had a drawback. As determined by NMR experiments, all the resulting 5-aryluracils 4–11 were obtained in mixture with the corresponding homocoupled compound in a proportion of around 10% in accordance with the excess of boronic acid used in the cross-coupling step.
The next task was to consider a more conventional way to obtain the 5-aryluracil derivatives consisting in two distinguished stages: (i) the Suzuki-Miyaura cross-coupling of those same boronic acids with 5-iodo-2′-deoxyuridine (2) and then (ii) the deglycosylation. This time, each resulting 5-aryl-2′-deoxyuridine derivative will be purified as previously reported25 before being engaged in the deglycosylation step. As no more palladium and boronic acid would be present in the second stage, the homocoupled product formation should be avoided. Nevertheless a question remains: what about KOH? Indeed presence of a base was necessary for the Suzuki-Miyaura cross-coupling but perhaps not for the deglycosylation. Shapiro reported that alkaline medium was not appropriate27 and to the best of our knowledge most of glycosides or nucleosides hydrolysis were done in acidic or neutral conditions.28–30 Therefore in order to examine KOH influence on the deglycosylation, two routes were developed (Table 2): one in presence of KOH (2 equiv.) and one without any trace of base.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ar | Suzuki-Miyaura cross-coupled compound (yield %) | Product | Yielda (%) | Total yield (%) | |
| a Deglycosylation step. Isolated yields.b Deglycosylation in presence of KOH (2 equiv.).c Deglycosylation without any KOH. | ||||||
| 1 |  |
3 (85) |  |
4 | 71b | 60b |
| 78c | 66c | |||||
| 2 |  |
14 (79) | 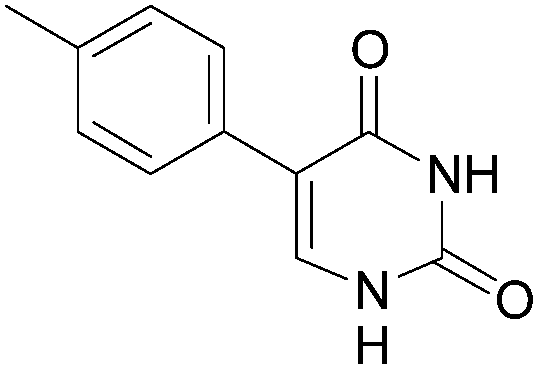 |
5 | 76b | 60b |
| 78c | 63c | |||||
| 3 | 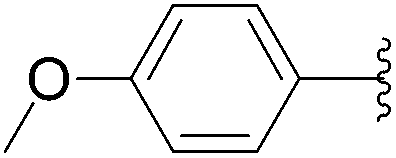 |
15 (84) | 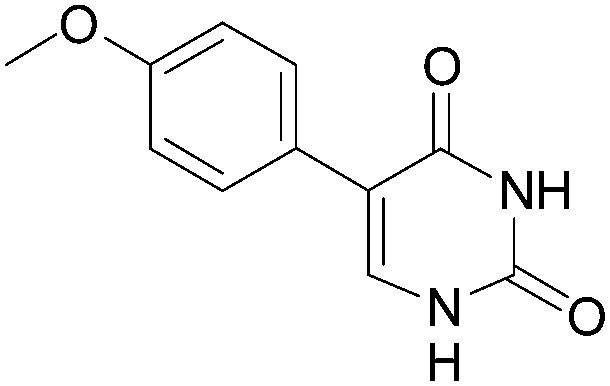 |
6 | 66b | 55b |
| 75c | 63c | |||||
| 4 | 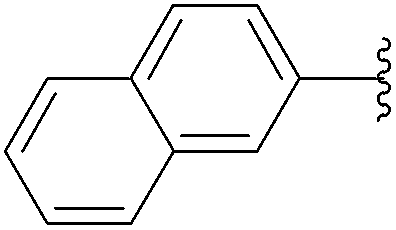 |
16 (84) | 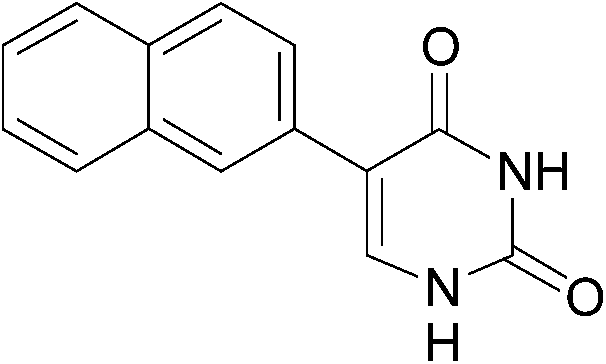 |
7 | 71b | 59b |
| 80c | 67c | |||||
| 5 | 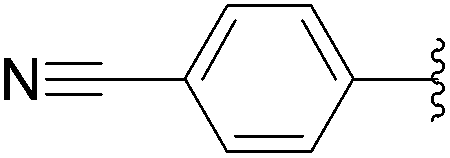 |
17 (33) | 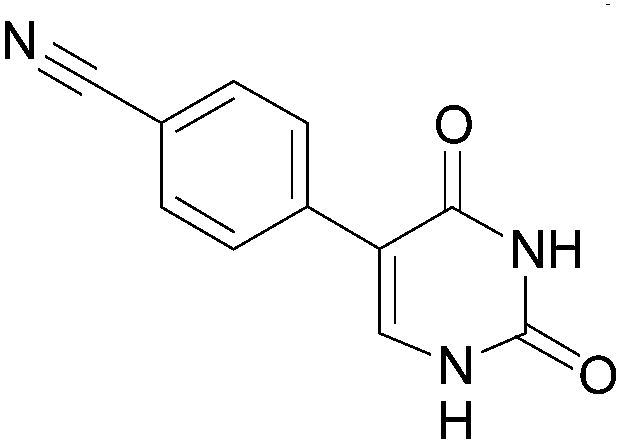 |
8 | 0b | 0b |
| 0c | 0c | |||||
| 6 | 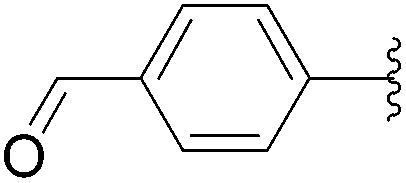 |
18 (66) | 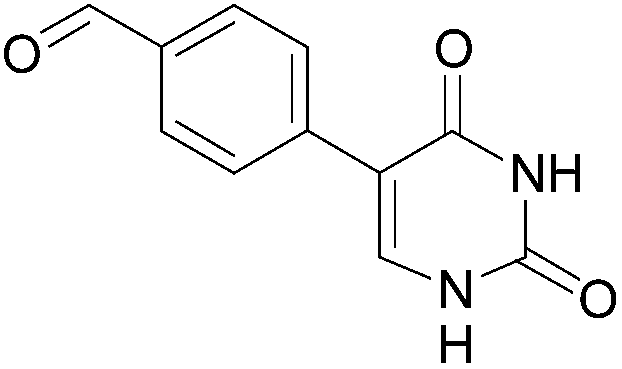 |
9 | 60b | 40b |
| 64c | 42c | |||||
| 7 | 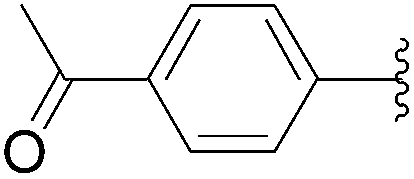 |
19 (78) |  |
10 | 55b | 43b |
| 66c | 52c | |||||
| 8 | 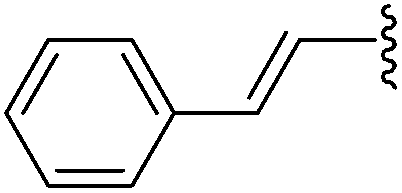 |
20 (85) | 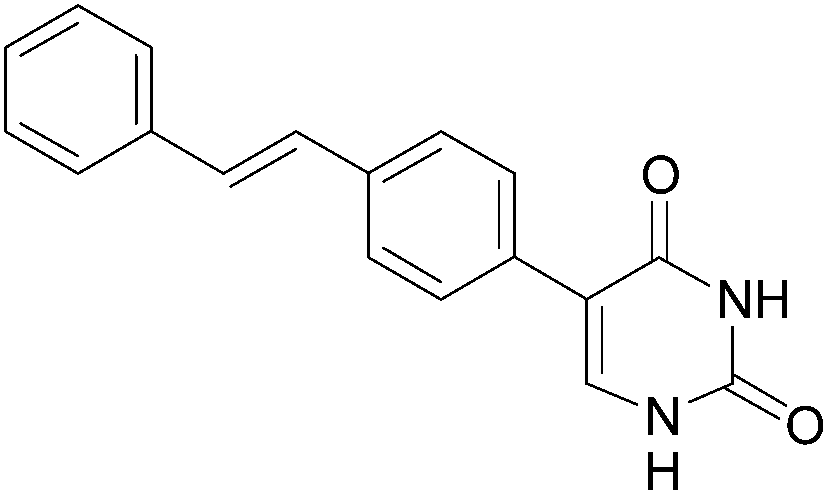 |
11 | 52b | 43b |
| 53c | 44c | |||||
| 9 | 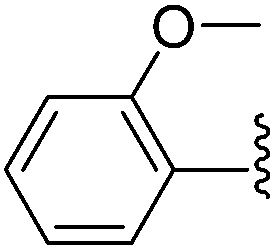 |
21 (30) |  |
12 | 75b | 22b |
| 79c | 24c | |||||
| 10 | 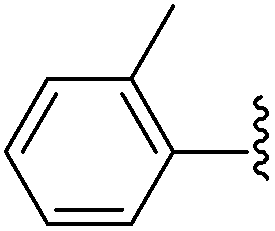 |
22 (45) | 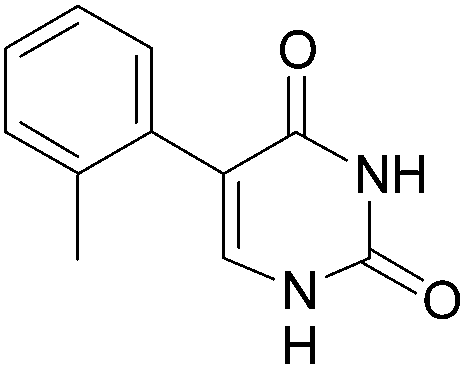 |
13 | 68b | 31b |
| 75c | 34c | |||||
When the intermediates 5-aryl-2′-deoxyuridine 3 and 14–22 were purified prior to being engaged, the deglycosylation step was more effective whatever the electronic effect on the phenyl core. Indeed the 5-aryl uracil derivatives 4–11 were obtained in similar to higher yields (Table 2, entries 1–8). Furthermore using this new methodology no trace of any homocoupled product was detected, the deglycosylated product being the sole final isolated compound. The intermediate purification (after the Suzuki-Miyaura step) allowed us to test the deglycosilation on two more compounds 21 and 22 (Table 2, entries 9 and 10). The deglycosylation step was performed in high yield for both those compounds, respectively 75% and 68%. After the Suzuki cross-coupling those compounds were obtained in mixture with the iodated starting-material 2 and could not, de facto, be engaged in a “one step, two pots” reaction. The total yields of compounds 4–13 seemed to be more dependants on the cross-coupling steps than on the deglycosylation's one, excepted for compound 8 (Table 2, entry 5).
Concerning the necessity of KOH for the deglycosylation step, our results showed that its presence was not required. Indeed, deglycosylations were achieved in better yields (Table 2, entries 1, 3, 4, 6, 7 and 9) or at least with the same efficiency (Table 2, entries 2 and 8). Unfortunately, the cyano derivative, once again, could not be isolated even without KOH (Table 2, entry 5).
Conclusions
In summary, a new and very simple procedure for 5-aryl-2′-deoxyuridine deglycosylation in neat water has been developed using microwave irradiation at 200 °C furnishing nine 5-aryluracil analogues in 66–78% yields, among which sterically hindered compounds 12 and 13. The sugar part of the starting nucleoside analogue might be seen as the first totally green protecting group. To the best of our knowledge, this is the first time that such green and easy conditions for deglycosylation of nucleosides are reported.Experimental
1 General methods
All products were purchased either to Acros or Sigma Aldrich depending on their availability. All Solvents were purchased to Carlo Erba. All reactions were monitored by TLC (Kieselgel 60F254 MERCK aluminium sheet) with detection by UV light and/or with sulfuric acid in ethanol (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v) and by HPLC (Shimadzu). The column used is a GRACE Prevail C18. The detectors used are SPD-M20A photo diode array detector (Shimadzu), LCMS-2020 mass spectrometer (Shimadzu) and ELSD-LTII (Shimadzu). The mobile phase is a mixture of water and MeOH (50:50). Mass spectrometry analyses were performed on a Shimadzu LCMS-2020 mass spectrometer equipped with an electrospray source (ESCI). 1H and 13C NMR spectra were recorded on a 400 MHz Bruker UltraShield 400 MHz/54 mm Ultra long hold. Chemical shifts (δ) are quoted in ppm and are referenced to TMS as an internal standard. Coupling constants (J) are quoted in Hz. Melting points are recorded on a Stuart SMP 10 and are uncorrected.
:10, v/v) and by HPLC (Shimadzu). The column used is a GRACE Prevail C18. The detectors used are SPD-M20A photo diode array detector (Shimadzu), LCMS-2020 mass spectrometer (Shimadzu) and ELSD-LTII (Shimadzu). The mobile phase is a mixture of water and MeOH (50:50). Mass spectrometry analyses were performed on a Shimadzu LCMS-2020 mass spectrometer equipped with an electrospray source (ESCI). 1H and 13C NMR spectra were recorded on a 400 MHz Bruker UltraShield 400 MHz/54 mm Ultra long hold. Chemical shifts (δ) are quoted in ppm and are referenced to TMS as an internal standard. Coupling constants (J) are quoted in Hz. Melting points are recorded on a Stuart SMP 10 and are uncorrected.
2 General procedures
:MeOH 95:5 to 5:95). The fractions were combined and methanol was removed under reduce pression. The remaining solution was frozen with liquid nitrogen and lyophilisated. Pure 5-aryl-2′-deoxyuridine (50 mg, 1 equiv.), KOH (2 equiv. if used) and 2 mL of nitrogen flushed water were introduced in a 10 mL vial. The mixture was stirred under microwave irradiation heating at 200 °C for 30 min. The crude residue was then diluted in sodium hydroxyde 1 M (10 mL). Aqueous phase was then extracted with AcOEt (3 × 20 mL). Organic phases were dried over MgSO4 and concentrated under reduce pressure to give pure deglycosydated compounds.3 Spectroscopic data
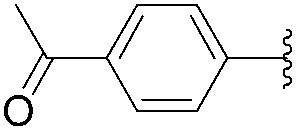
1H NMR (400 MHz, DMSO-d6) δ = 11.33 (s, 2H, NH), 7.93 (d, 2H, J = 8.4 Hz, Har), 7.80 (s, 1H, H6), 7.74 (d, 2H, J = 8.4 Hz, Har), 2.57 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ = 192.1 (COCH3), 163.04 (C4), 154.08 (C2), 140.90 (C6), 133.41 (Car), 129.00 (Car), 127.2 (Car), 126.9 (Car), 125.8 (Car), 125.8 (Car), 108.77 (C5). One signal is missing (behind DMSO). MS (ESI): 229.19 [M − H]−.
Acknowledgements
TL thanks Dr G. Santini and ESCOM for their financial support.Notes and references
- C. F. Regan, Z. Guo, Y. Chen, C. Q. Huang, M. Chen, W. Jiang, J. K. Rueter, T. Coon, C. Chen, J. Saunders, M. S. Brown, S. F. Betz, R. S. Struthers, C. Yang, J. Wen, A. Madan and Y.-F. Zhu, Bioorg. Med. Chem. Lett., 2008, 18, 4503 CrossRef CAS PubMed.
- C. Chen, D. Wu, Z. Guo, Q. Xie, G. J. Reinhart, A. Madan, J. Wen, T. Chen, C. Q. Huang, M. Chen, Y. Chen, F. C. Tucci, M. Rowbottom, J. Pontillo, Y.-F. Zhu, W. Wade, J. Saunders, H. Bozigian and R. S. Struthers, J. Med. Chem., 2008, 51, 7478 CrossRef CAS PubMed.
- Z. Guo, Y. Chen, C. Q. Huang, T. D. Gross, J. Pontillo, M. W. Rowbottom, J. Saunders, S. Struthers, F. C. Tucci, Q. Xie, W. Wade, Y.-F. Zhu, D. Wu and C. Chen, Bioorg. Med. Chem. Lett., 2005, 15, 2519 CrossRef CAS PubMed.
- C. Chen, Y. Chen, J. Pontillo, Z. Guo, C. Q. Huang, D. Wu, A. Madan, T. Chen, J. Wen, Q. Xie, F. C. Tucci, M. Rowbottom, Y.-F. Zhu, W. Wade, J. Saunders, H. Bozigian and R. S. Struthers, Bioorg. Med. Chem. Lett., 2008, 18, 3301 CrossRef CAS.
- M. Médebielle, M. A. Oturan, J. Pinson and J.-M. Savéant, Tetrahedron Lett., 1993, 34, 3409 CrossRef.
- K. C. Majumbar, P. Debnath, A. Taher and A. K. Pal, Can. J. Chem., 2008, 86, 325 CrossRef.
- K. C. Majumdar, B. Sinha, P. K. Maji and S. K. Chattopadhyay, Tetrahedron, 2009, 65, 2751 CrossRef CAS.
- M. M. Morshed, Q. Wang, S. Islam and M. M. Hossain, Synth. Commun., 2007, 37, 4173 CrossRef CAS.
- D. Villemin, M. J. Gómez-Escalonilla and J.-F. Saint-Clair, Tetrahedron Lett., 2001, 42, 635 CrossRef CAS.
- K. Pomeisl, A. Holý and R. Pohl, Tetrahedron Lett., 2007, 48, 3065 CrossRef CAS.
- K. Pomeisl, A. Holý and K. Horská, Tetrahedron, 2009, 65, 8486 CrossRef CAS.
- J. M. Sadler, O. Ojewoye and K. L. Seley-Radtke, Nucleic Acids Symp. Ser., 2008, 52, 571 CrossRef CAS.
- U. Wellmar, A.-B. Hörnfeldt and S. Gronowitz, J. Heterocycl. Chem., 1996, 33, 409 CrossRef CAS.
- M. Mosrin, N. Boudet and P. Knochel, Org. Biomol. Chem., 2008, 6, 3237 CAS.
- R. Nencka, I. Votruba, H. Hřebabecký, P. Jansa, E. Tloušt’ová, K. Horská and M. Masojidková, J. Med. Chem., 2007, 50, 6016 CrossRef CAS PubMed.
- A. Coelho and E. Sotelo, J. Comb. Chem., 2005, 7, 526 CrossRef CAS PubMed.
- V. Farina and S. T. Hauck, Synlett, 1991, 3, 157 CrossRef.
- (a) M. Čerňová, R. Pohl and M. Hocek, Eur. J. Org. Chem., 2009, 3698 CrossRef; (b) M. Čerňová, I. Černá, R. Pohl and M. Hocek, J. Org. Chem., 2011, 76, 5309 CrossRef PubMed.
- K. H. Kim, H. S. Lee and J. N. Kim, Tetrahedron Lett., 2011, 52, 6228 CrossRef CAS.
- Y. Liang, J. Gloudeman and S. F. Wnuk, J. Org. Chem., 2014, 79, 4094 CrossRef CAS PubMed.
- L. A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103, 1875 CrossRef CAS PubMed.
- G. Hervé, G. Sartori, G. Enderlin, G. Mackenzie and C. Len, RSC Adv., 2014, 4, 18558 RSC.
- G. Sartori, G. Enderlin, G. Hervé and C. Len, Synthesis, 2012, 44, 767 CrossRef CAS.
- G. Sartori, G. Hervé, G. Enderlin and C. Len, Synthesis, 2013, 45, 330 CrossRef CAS.
- S. Gallagher-Duval, G. Hervé, G. Sartori, G. Enderlin and C. Len, New J. Chem., 2013, 37, 1989 RSC.
- G. Enderlin, G. Sartori, G. Hervé and C. Len, Tetrahedron Lett., 2013, 54, 3374 CrossRef CAS.
- R. Shapiro and S. Kang, Biochemistry, 1969, 8, 1806 CrossRef CAS.
- R. Wolfenden, X. Lu and G. Young, J. Am. Chem. Soc., 1998, 120, 6814 CrossRef CAS.
- G. K. Schroeder and R. Wolfenden, Biochemistry, 2007, 46, 13638 CrossRef CAS PubMed.
- J. L. Przybylski and S. D. Wetmore, J. Phys. Chem. B, 2010, 114, 1104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04814h |
| ‡ Present address: Université de Sherbrooke, 2500 bd de l'Université, Sherbrooke J1K 2R1, Canada. |
| This journal is © The Royal Society of Chemistry 2014 |