
Palladacycle-catalyzed Suzuki–Miyaura reaction of aryl/heteroaryl halides with MIDA boronates in EtOH/H2O or H2O†
Yabo Lia,
Jingran Wanga,
Zhiwei Wanga,
Mengmeng Huang*a,
Beiqi Yana,
Xiuling Cuia,
Yusheng Wu*ab and
Yangjie Wu*a
aCollege of Chemistry and Molecular Engineering, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou, 450052, P. R. China. E-mail: wyj@zzu.edu.cn; hmm@zzu.edu.cn; Fax: +86-371-67767993
bTetranov Biopharm, LLC, 75 Daxue Road, Zhengzhou, 450052, P.R. China. E-mail: yusheng.wu@tetranovglobal.com
First published on 7th August 2014
Abstract
With good to excellent yields, a series of mono- or diheteroaryl compounds were synthesized via the palladacycle-catalyzed Suzuki–Miyaura reaction of various N-methyliminodiacetic acid (MIDA) boronates with aryl/heteroaryl halides in EtOH/H2O or H2O.
Introduction
Mono- and diheteroaryl compounds as one of the most prevalent and important motifs are ubiquitous in a wide range of pharmaceutical building blocks, natural products, unnatural nucleotides, molecule probes, advanced materials, and metal-complexing ligands.1 Although these compounds are generally synthesized via typical cross-coupling reactions,2 there are still some limitations for the synthesis of 2-heteroaryls such as the instability of the building blocks,3 use of toxic metals,4 and inefficient couplings with aryl chlorides.5 Recently, the low toxicity and high tolerance of organoboronic acids are widely used as one of the coupling partners in Suzuki reaction. Many progresses have been achieved in this field, the reaction conditions in most cases could not be well suited for the reaction with inherently unstable 2-heteroaryl boronic acids.6 To solve this problem, professor Martin Burke and Gillis6b,7 found that the easily handled and stable MIDA-protected boronates could slowly hydrolyze and release the active boronic acids under mild aqueous basic conditions to take part in Suzuki reaction.8 To improve the existing technology, more recently, Lipshutz and co-workers reported a catalytic system for the reaction of aryl/heteroaryl MIDA boronates with aryl halides in water.9 Their reaction protocol realized the value of environmental conservation, however, unusual polymer phase transfer catalyst TPGS-750-M and high catalytic loading (PdCl2(dtbpf)) were employed. Therefore, the development of effective and environmentally friendly protocols for Suzuki–Miyaura coupling reaction of heteroaryl MIDA boronates with heteroaryl halides is still needed. In the course of our investigation on the catalytic activity of cyclopalladated ferrocenylimines (Scheme 1),10 herein, we report the palladacycle-catalyzed Suzuki–Miyaura coupling reaction involving various MIDA boronates with aryl/heteroaryl halides in EtOH/H2O or H2O.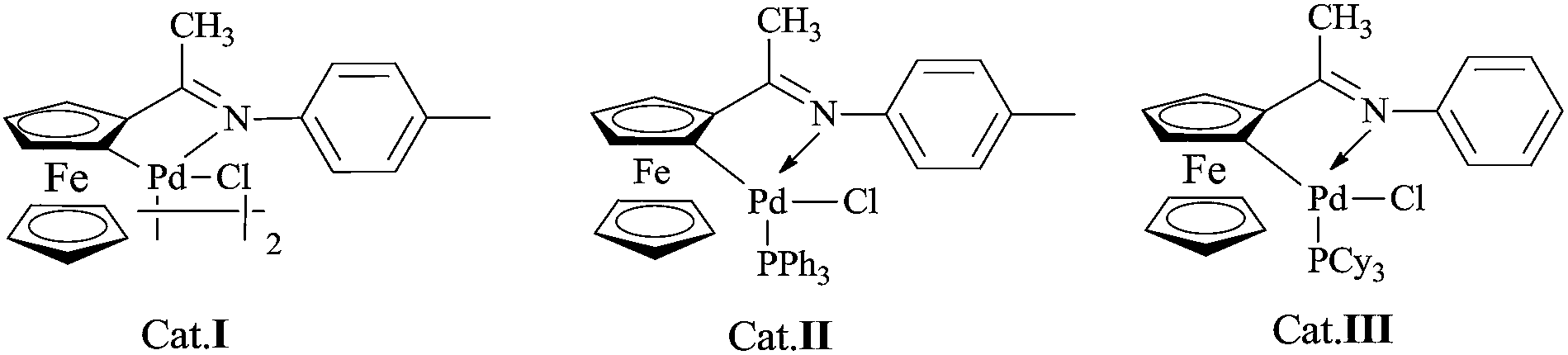 | ||
| Scheme 1 Palladacycle precatalysts used in this study. | ||
Results and discussion
Our initial experiments were focused on identifying reaction conditions with 4-bromoanisole (1a) and phenylboronic acid MIDA ester (2a). Palladacycle catalysts Cat.I-Cat.III (Scheme 1) were selected for the reaction, Cat.II could provide superior yield 76% of 3a (Table 1, entries 1–3). It was found that large amounts of water were necessary to promote hydrolysis of MIDA boronates (Table 1, entries 2 and 4). Some bases (e.g., Cs2CO3, K2CO3, and KOH) were evaluated, and the best choice was K3PO4 (84%; Table 1, entries 4–7). Surprisingly, the discovered reaction was carried out in H2O (with 20% TBAB) or EtOH/H2O (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the excellent yields (Fig. 1). Other factors such as reaction temperature, reaction time, and catalyst loading were respectively investigated in two different solvents EtOH/H2O (6:1) or H2O (with 20% TBAB) (Fig. 2). It was found that the coupling reaction could smoothly proceed at 90 °C to give excellent yield. Although the conversion yield was reduced with decreasing the catalyst loading, it was worth noting that the yield could be given in 71–80% with 0.1 mol% catalyst. In addition, a lower yield was obtained when the reaction was carried out under air atmosphere (Table 1, entry 10–11).
:1) to give the excellent yields (Fig. 1). Other factors such as reaction temperature, reaction time, and catalyst loading were respectively investigated in two different solvents EtOH/H2O (6:1) or H2O (with 20% TBAB) (Fig. 2). It was found that the coupling reaction could smoothly proceed at 90 °C to give excellent yield. Although the conversion yield was reduced with decreasing the catalyst loading, it was worth noting that the yield could be given in 71–80% with 0.1 mol% catalyst. In addition, a lower yield was obtained when the reaction was carried out under air atmosphere (Table 1, entry 10–11).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Base | Solvent (mL) | T (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 4-bromoanisole (0.5 mmol), phenylboronic acid MIDA ester (0.52 mmol), base (2 mmol), solvent (2–3 mL), under a nitrogen atmosphere.b Isolated yield.c Under an air atmosphere. | ||||||
| 1 | I (1) | K3PO4 | DMF:H2O = 9:1 (2) |
100 | 12 | 38 |
| 2 | II (1) | K3PO4 | DMF:H2O = 9:1 (2) |
100 | 12 | 76 |
| 3 | III (1) | K3PO4 | DMF:H2O = 9:1 (2) |
100 | 12 | 70 |
| 4 | II (1) | K3PO4 | DMF:H2O = 9:1 (3) |
100 | 12 | 84 |
| 5 | II (1) | Cs2CO3 | DMF:H2O = 9:1 (3) |
100 | 12 | trace |
| 6 | II (1) | K2CO3 | DMF:H2O = 9:1 (3) |
100 | 12 | trace |
| 7 | II (1) | KOH | DMF:H2O = 9:1(3) |
100 | 12 | 40 |
| 8 | II (1) | K3PO4 | EtOH:H2O = 6:1 (2.1) |
90 | 4 | 94 |
| 9 | II (1) | K3PO4 | H2O (20% TBAB) (2.5) | 90 | 4 | 95 |
| 10c | II (1) | K3PO4 | EtOH:H2O = 6:1 (2.1) |
90 | 4 | 90 |
| 11c | II (1) | K3PO4 | H2O (20%TBAB) (2.5) | 90 | 4 | 87 |
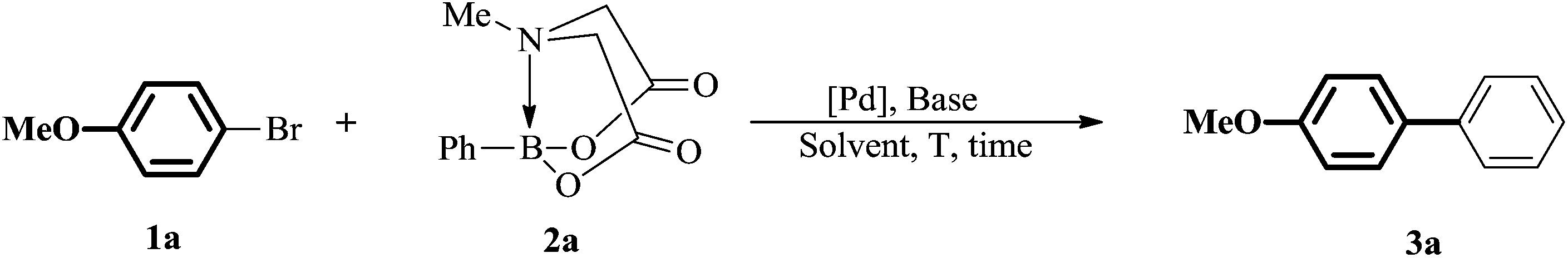
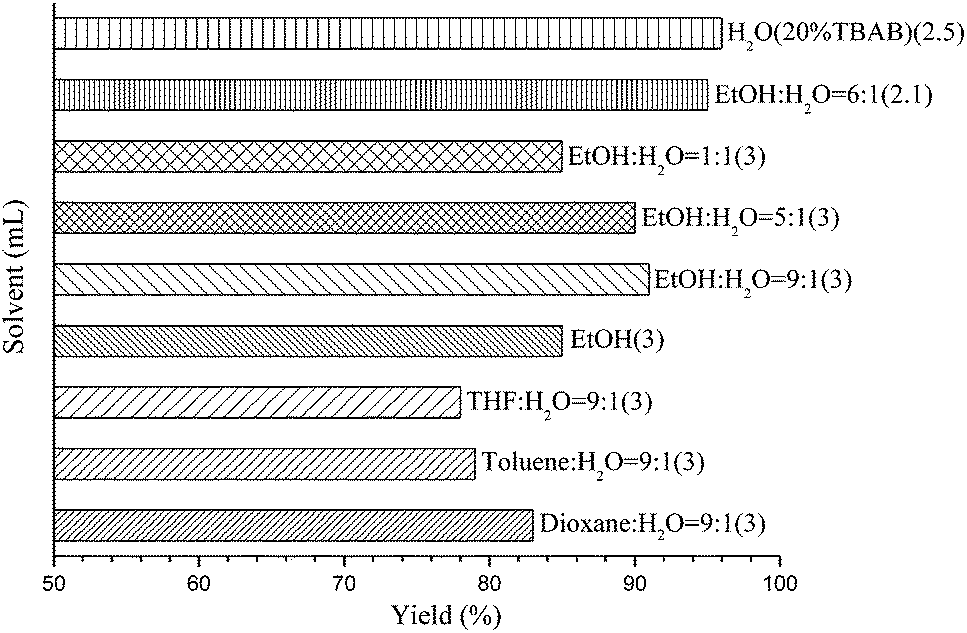 | ||
| Fig. 1 Influence of solvents. Reaction conditions: 4-bromoanisole (0.5 mmol), phenyl boronic acid MIDA ester (0.52 mmol), K3PO4 (2 mmol), Cat.II (1 mol%), solvent (2–3 mL) under nitrogen at 100 °C in 12 h. | ||
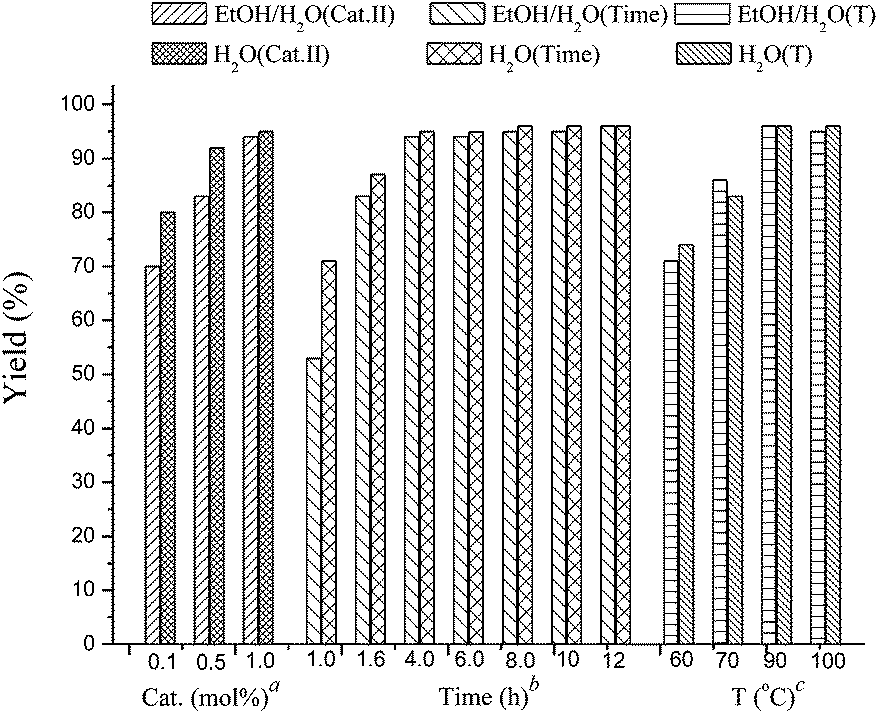 | ||
| Fig. 2 Influence of catalyst loading, reaction time and temperature. a The catalyst loading of Cat.II was optimized at 90 °C in 4 h. b Reaction time was optimized with 1 mol% Cat.II at 90 °C. c The reaction temperature was optimized with 1 mol% Cat.II in 12 h. | ||
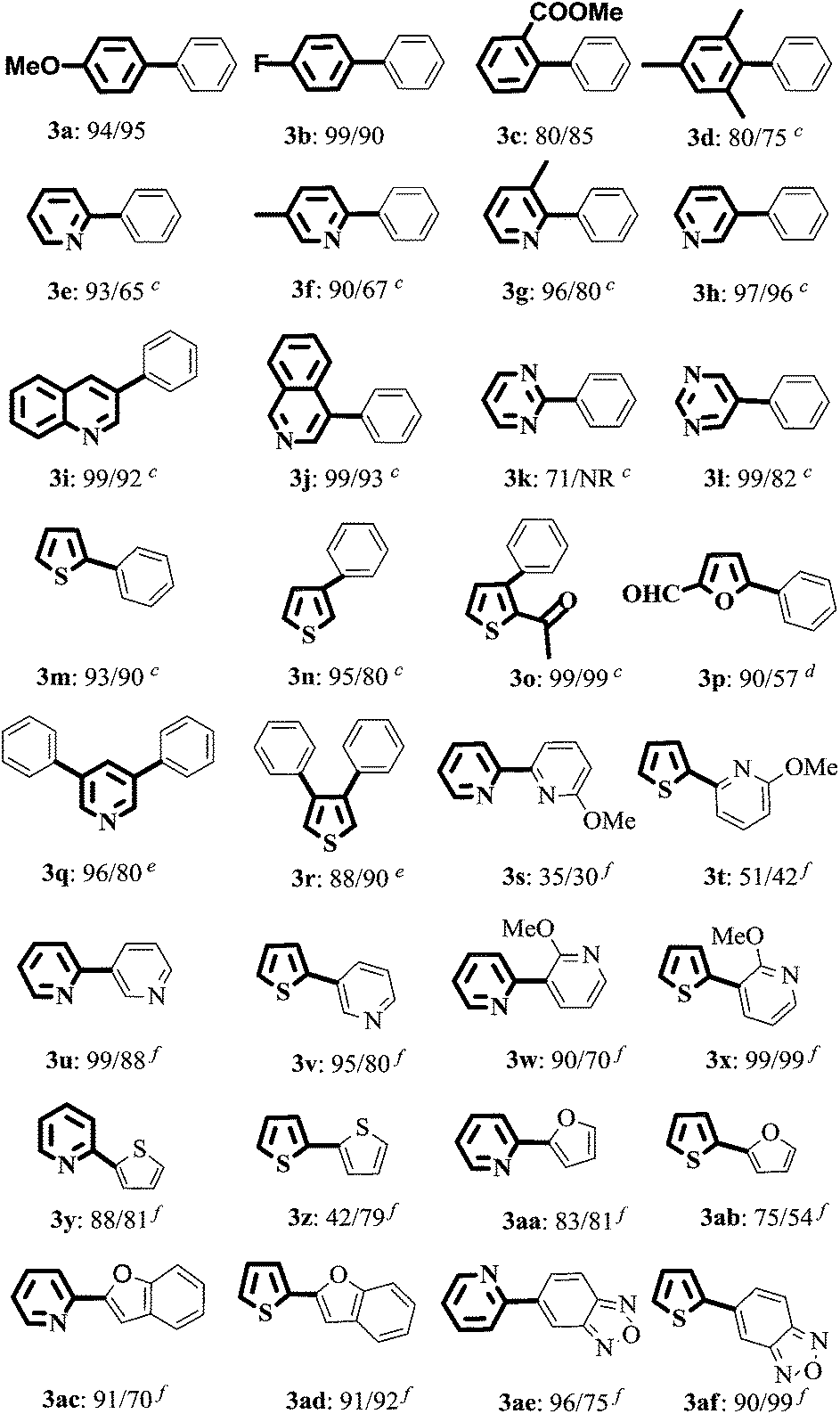
With the optimized reaction conditions, the substrate scope of both catalytic systems (EtOH/H2O or H2O with 20% TBAB) was investigated with a series of aryl/heteroaryl MIDA boronates and aryl/heteroaryl bromides (Table 2). The desired products 3a and 3b were obtained in excellent yields regardless of the electronic effect of functional groups on aryl bromides. Sterically hindered ortho-substituted aryl bromides could be also transformed into target products in good yields (Table 2, 3c and 3d). When the reaction of methyl 2-bromobenzoate with 2a was carried out in EtOH/H2O, an ester-exchange reaction product of ethyl 2-biphenylcarboxylate was found in 20% yield by NMR spectroscopy (Table 2 and 3c, ESI,† NMR spectra). 3-Bromopyridine, 3-bromoquinoline, 4-bromoisoquinoline, 2-bromothiophene, and 2-acetyl-3-bromothiophene could be converted to the desired products in excellent yields in EtOH/H2O and neat water (Table 2, 3h–3j, 3m, and 3o). 2-Phenylpyridinederivatives (3e, 3f, and 3g) were obtained in excellent yields (93%, 90%, and 96%) in EtOH/H2O and moderate yields in H2O (65%, 67%, and 80%). This analogous phenomenon also occurred in the synthesis of 3k, 3l, 3n, and 3p. Heteroaryl dibromides gave triaryl products 3q and 3r in good to excellent yields in both solvents. Subsequently, 2-bromopyridine and 2-bromothiophene were chosen to evaluate the reactivity of various heteroaryl substituted MIDA boronates. The reaction of 3-pyridyl-, 2-thiophenyl-, 2-furanyl-, 2-benzofuranyl-, or 5-bebzofurazanboronic acid MIDA esters with 2-bromoheteroaryls afforded 3u–3af in good to excellent yields in both green solvents, except the coupling of 2-thiophenylboronic acid MIDA ester with 2-bromothiopene in EtOH/H2O (3z). However, the reaction of 2-pyridyl MIDA boronate with 2-bromopyridine gave only a trace amount of product 2,2′-bipyridine by GC analysis. But changing 2-pyridine MIDA boronate to 6-methoxy-2-pyridylboronic acid MIDA ester, surprisingly, heterobiaryls 3s and 3t were isolated in 30–51% yields in EtOH/H2O and H2O.The structure of 3af was determined by X-ray diffraction (ESI, Fig. S1†).
|
|---|
| a Reaction conditions: aryl or heteroaryl bromide (0.5 mmol), boronic acid MIDA ester (0.52 mmol), base (2 mmol), Cat.II (1 mol%), EtOH/H2O (6:1, 2.1 mL) or H2O (2.5 mL) with TBAB (20 mol%), under nitrogen at 90 °C in 4 h.b Isolated yield for EtOH/H2O and H2O.c Reaction time 12 h.d 20 h.e Heteroaryl bromide (0.5 mmol), boronic acid MIDA ester (1.1 mmol), base (4 mmol), Cat.II (2 mol%), EtOH/H2O (6:1, 4.2 mL) or H2O (3 mL), TBAB (40 mol%).f 100 °C in 24 h. |
 |
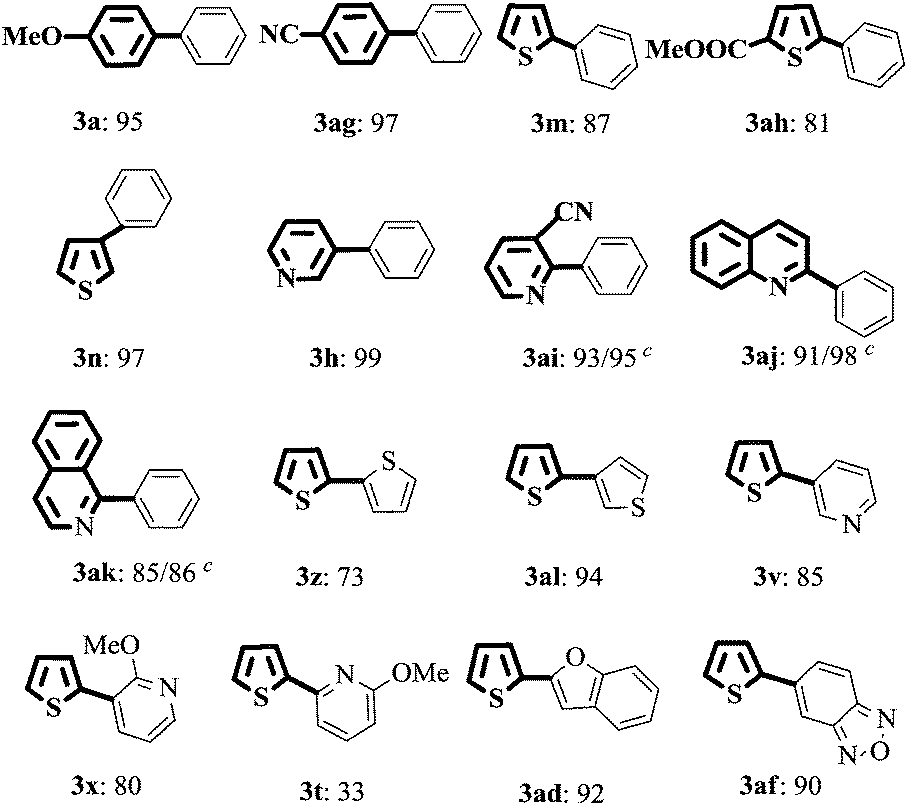
Encouraged by the above results, aryl/heteroaryl chlorides were then investigated with Cat.I (1 mol%) and 4 mol% X-Phos in H2O at 90 °C (Table 3). It was found that the yields of 3ai, 3ak, and 3aj in EtOH/H2O were similar with their reaction in H2O. Thus, considering the economic factor, our subsequent research was carried out in water. The modified catalytic system was suitable for aryl/heteroaryl chlorides substituted with electron-rich, electron-deficient or sterically hindered groups to afford the desired products 3a–3ak in good to excellent yields. Under the standard reaction conditions, 2-chlorothiophene could also react with a series of heteroaryl MIDA boronates to form the desired products 3z–3af (Table 3) in moderate to excellent yields except 3t.
Conclusion
In conclusion, mono- and diheteroaryls were synthesized via the palladacycle-catalyzed Suzuki–Miyaura reaction of various MIDA boronates with aryl/heteroaryl halides in EtOH/H2O or H2O. The desired cross-coupling products were obtained in good to excellent yields. This modified catalytic system was suitable not only for aryl or heteroaryl halides substituted with electron-rich, electron-deficient or sterically hindered groups, but also for aryl/heteroaryl MIDA boronates.Experimental section
General
All reactions were run under nitrogen in Schlenk tubes using vacuum lines. Palladacycle precatalysts,10 phenyl, 2-thiophene and 3-pyridine boronic acid MIDA ester11 were prepared according to the literature procedures. Unless otherwise noted, chemical reagents used in experiments were purchased from commercial suppliers without further purification. 1H NMR and 13C NMR spectra were recorded on a 400 and 100 MHz instrument using CDCl3 as the solvent at room temperature. High resolution mass spectrometry data of the products were collected on an LC/MS instrument. Melting points were measured using a WC-1 microscopic apparatus and were uncorrected. Thin-layer chromatography was visualized with UV light (254 and 365 nm). Flash chromatography was performed on silica gel (200–300 mesh).General procedures of Suzuki–Miyaura reaction with aryl/heteroaryl bromides
In a typical experiment, Cat.II (3.7 mg, 1 mol%), aryl/heteroaryl MIDA boronate (0.52 mmol), aryl/heteroaryl bromide (0.5 mmol) and K3PO4 (425 mg, 2 mmol) were dissolved in EtOH/H2O (6:1, 2.1 mL) or H2O (2.5 mL with 20 mol% TBAB, 32 mg) under nitrogen atmosphere. Unless otherwise noted, the reaction mixture was stirred at 90 °C for 24 hours. Then, the suspension was cooled to room temperature and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried with Na2SO4. After evaporation of the solvents, the residue was purified by silica gel column chromatography affording the desired product.
General procedures of Suzuki–Miyaura reaction with aryl/heteroaryl chlorides
In a typical experiment, Cat.I (4.6 mg, 1 mol%), X-Phos (9.5 mg, 4 mol%), aryl/heteroaryl MIDA boronate (0.52 mmol), aryl/heteroaryl chloride (0.5 mmol) and K3PO4 (425 mg, 2 mmol) were dissolved in H2O (20 mol% TBAB, 32 mg, 2.5 mL) under nitrogen atmosphere. Unless otherwise noted, the reaction mixture was stirred at 90 °C. Then, the suspension was cooled to room temperature and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried with Na2SO4. After evaporation of the solvents the residue was purified by silica gel column chromatography affording the desired product.Acknowledgements
We are grateful to the National Natural Science Foundation of China (no. 21172200 and 21302172) for financial support to this research.Notes and references
- (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed; (b) I. Shinkai, A. O. King and R. D. Larsen, Pure Appl. Chem., 1994, 66, 1551 CrossRef CAS; (c) A. B. Smith III, T. J. Beauchamp, M. J. LaMarche, M. D.Kaufman, Y. Qiu, H. Arimoto, D. R. Jones and K. Kobayashi, J. Am. Chem. Soc., 2000, 122, 8654 CrossRef; (d) D. Camp, C. F. Matthews, S. T. Neville, M. Rouns, R. W. Scott and Y. Truong, Org. Process Res. Dev., 2006, 10, 814 CrossRef CAS; (e) P. D. de Koning, D. McAndrew, R. Moore, I. B. Moses, D. C. Boyles, K. Kissick, C. L. Stanchina, T. Cuthbertson, A. Kamatani, L. Rahman, R. Rodriguez, A. Urbina, A. Sandoval (née Accacia) and P. R. Rose, Org. Process Res. Dev., 2011, 15, 1011 Search PubMed; (f) T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS.
- For reviews, see: (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (b) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (c) A. Molnar, Chem. Rev., 2011, 111, 2251 CrossRef CAS PubMed; (d) J. K. Stille, Angew. Chem., 1986, 98, 504 CrossRef CAS PubMed; (e) T. Hiyama, J. Organomet. Chem., 2002, 653, 58 CrossRef CAS; (f) E.-i. Negishi, Q. Hu, Z. Huang, M. Qian and G. Wang, Aldrichimica Acta, 2005, 38, 71 CAS; (g) D. S. Surry and S. L. Buchwald, Angew. Chem., 2008, 120, 6438 CrossRef PubMed.
- (a) K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358 CrossRef CAS PubMed; (b) C. A. Fleckenstein and H. J. Plenio, Org. Chem., 2008, 73, 3236 CrossRef CAS PubMed; (c) J.-H. Li, Q.-M. Zhu and Y.-X. Xie, Tetrahedron, 2006, 62, 10888 CrossRef CAS PubMed; (d) S. E. Denmark and C. R. Bulter, Chem. Commun., 2009, 20 RSC.
- (a) J. Milstein and K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef; (b) T. R. Bailey, Tetrahedron Lett., 1986, 27, 4407 CrossRef CAS; (c) A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343 CrossRef CAS PubMed; (d) H. G. Seiler, H. Sigel and A. Sigel in Handbook on Toxicity of Inorganic Compounds, New York, 1988, ch. 37 Search PubMed.
- (a) J. M. Lovell and J. A. Joule, Synth. Commun., 1997, 27, 1209 CrossRef CAS; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- (a) D. G. Hall, Boronic Acids, Wiley-VCH, Weinheim, Germany, 2005, pp. 3–14 Search PubMed; (b) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed; (c) Y.-B. Li, X. Mi, M.-M. H, R.-R. Cai and Y.-Y. Wu, Tetrahedron, 2012, 68, 8502 CrossRef CAS PubMed; (d) V. I. Potkin, N. A. Bumagin and S. K. Petkevich, et al., Synthesis, 2012, 44, 151 CrossRef CAS PubMed; (e) N. A. Bumagin, S. K. Petkevich and A. V. Kletskov, et al., Chem. Heterocycl. Compd., 2014, 49, 1515 CrossRef CAS PubMed; (f) N. A. Bumagin, I. S. Veselov and D. S. Belov, Chem. Heterocycl. Compd., 2014, 50, 19 CrossRef CAS PubMed.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed; (b) B. E. Uno, E. P. Gillis and M. D. Burke, Tetrahedron, 2009, 65, 3130 CrossRef CAS PubMed; (c) S. J. Lee, T. M. Anderson and M. D. Burke, Angew. Chem., Int. Ed., 2010, 49, 8860 CrossRef CAS PubMed; (d) G. R. Dick, D. M. Knapp, E. P. Gillis and M. D. Burke, Org. Lett., 2010, 12, 2314 CrossRef CAS PubMed.
- (a) S. J. Lee, K. C. Gray and J. S. Paek, J. Am. Chem. Soc., 2008, 130, 466 CrossRef CAS PubMed; (b) J. E. Grob, J. Nunez, M. A. Dechantsreiter and L. G. Hamann, J. Org. Chem., 2011, 76, 4930 CrossRef CAS PubMed; (c) J. E. Grob, M. A. Dechantsreiter, R. B. Tichkule, M. K. Connolly, A. Honda, R. C. Ronald and L. G. Harmann, Org. Lett., 2012, 14, 5578 CrossRef CAS PubMed; (d) E. M. Woerly, A. H. Cherney, E. K. Davis and M. D. Burke, J. Am. Chem. Soc., 2010, 132, 6941 CrossRef CAS PubMed; (e) T. Delaunay, M. E. Sayed, J. P. Vors, N. Monteiro and G. Blame, Chem. Lett., 2011, 40, 1434 CrossRef CAS; (f) J. D. St. Denis, C. C. G. Scully, C. F. Lee and A. K. Yudin, Org. Lett., 2014, 16, 1338 CrossRef CAS PubMed; (g) M. A. J. Duncton and R. Singh, Org. Lett., 2013, 15, 4284 CrossRef CAS PubMed.
- N. A. Isley, F. Gallou and B. H. Lipshutz, J. Am. Chem. Soc., 2013, 135, 17707 CrossRef CAS PubMed.
- (a) S. Q. Huo, Y. J. Wu, C. X. Du, Y. Zhu, H. Z. Yuan and X. A. Mao, J. Organomet. Chem., 1994, 483, 139 CrossRef CAS; (b) Y. J. Wu, S. Q. Huo, J. F. Gong, X. L. Cui, L. Ding, K. L. Ding, C. X. Du, Y. H. Liu and M. P. Song, J. Organomet. Chem., 2001, 27, 637 Search PubMed; (c) Y. J. Wu, L. R. Yang, J. L. Zhang, M. Wang, Li. Zhao, M. P. Song and J. f. Gong, ARKIVOC, 2004, ix, 111 CrossRef; (d) J. Ma, X. L. Cui, B. Zhang, M. P. Song and Y. J. Wu, Tetrahedron, 2007, 63, 5529 CrossRef CAS PubMed.
- (a) Synthesis of phenyl boronic acid MIDA ester: T. Mancilla, M. A. C. Romo and L. A. Delgado, Polyhedron, 2007, 26, 1023 CrossRef CAS PubMed; (b) Synthesis of 2-thiophene boronic acid MIDA ester: D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed; (c) Synthesis of 3-pyridine boronic acid MIDA ester: J. L. Gustafson, D. Lim, K. T. Barrett and S. J. Miller, Angew. Chem., Int. Ed., 2011, 50, 5125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 965688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra07486f |
| This journal is © The Royal Society of Chemistry 2014 |