
Enalos InSilicoNano platform: an online decision support tool for the design and virtual screening of nanoparticles
Georgia Melagraki* and
Antreas Afantitis*
Novamechanics Ltd, Nicosia, Cyprus. E-mail: melagraki@novamechanics.com; afantitis@novamechanics.com
First published on 23rd September 2014
Abstract
Engineered nanoparticles (ENPs) are being extensively used in a great variety of applications with a pace that is increasingly growing. The evaluation of the biological effects of ENPs is of utmost importance and for that experimental and most recently computational methods have been suggested. In an effort to computationally explore available datasets that will lead to ready-to-use applications we have developed and validated a QNAR model for the prediction of the cellular uptake of nanoparticles in pancreatic cancer cells. Our insilico workflow was made available online through the Enalos InSilicoNano platform (http://enalos.insilicotox.com/QNAR_PaCa2/), a web service based solely on open source and freely available software that was developed with the purpose of making our model available to the interested user wishing to generate evidence on potential biological effects in the decision making framework. This web service will facilitate the computer aided nanoparticle design as it can serve as a source of activity prediction for novel nano-structures. To demonstrate the usefulness of the web service we have exploited the whole PubChem database within a virtual screening framework and then used the Enalos InSilicoNano platform to identify novel potent nanoparticles from a prioritized list of compounds.
1. Introduction
Nanotechnology has already contributed a wide range of significant products in several areas of application such as medicine, environment, electronics, cosmetics, defense etc.1–4 Due to the unique physical and chemical properties that particles possess in the nano scale, the research in the field is swiftly progressing and many more promising applications are rapidly being developed. As a result, nanoparticles (NPs) are increasingly used in our everyday life followed by concerns that have now been raised for their safety that is still to be explored.5–10 The bioactivity profile and risk assessment of NPs, including exposure and hazard assessment, is now gaining greater concern by academia, government and industry and many initiatives worldwide are working on defining the strategies and setting the priorities towards this goal (i.e. NNI National Nanotechnology Initiative, NanoSafety Cluster).The evaluation of NPs biological activity and toxicity by in vitro and in vivo studies is costly and time consuming and therefore alternative novel techniques that are fast, inexpensive and reduce the animal testing are required.11–17 To date a great number of Quantitative Structure Activity (QSAR) models have been proposed in literature. These models usually cover the biological profile of small organic molecules and have been proven accurate in predicting the biological effect for a wide range of molecular scaffolds. This is not the case for NPs that have recently emerged as important chemical structures with a wide range of significant properties that find applications in different areas of interest. Although ‘classic’ QSAR models own a great proportion of their success in the presence of organized databases, no such databases are available for NPs. Experimental data are scarce and produced by different groups of scientists following different protocols and it is often difficult to select and combine the available information from different sources. On top of that, the structural characteristics of NPs cannot be encoded by the “conventional” widely used 2D and 3D molecular descriptors. NPs include organic as well as inorganic elements with sometimes unknown composition and highly complex structures that demand new approaches for developing molecular descriptors. These hurdles have already been recognized and now international efforts are being organized towards the development of large datasets for NPs and the computational exploration of these results.
The potential of computational methods for advancing risk assessment of NPs is commonly accepted and a few computational attempts to predict the toxicity of NPs are reported in the literature the last few years.18–24 As mentioned, although “classic” Quantitative Structure Activity Relationship (QSAR) models have been for long proposed in the literature to assess different properties of compounds, Quantitative Nanostructure Activity (QNAR) models have not yet been extensively studied and limited examples have been published.1,5,7,9 Many factors have contributed in this including major hurdles such as lack of organized datasets and inadequate descriptors for NPs. On top of that attempts on the computational exploration of the activity of NPs and the produced QNAR models in principal are not made directly available to the community to be further used as useful tools for the risk assessment of novel NPs. Thus their utility is quite limited, whereas an online version of the model could spread the knowledge gained and generate more advancement in the field.
One of the few organized datasets on NPs that has been presented in literature includes the cellular uptake of 109 NPs in pancreatic cancer cells (PaCa2). Each NP within this dataset includes the same metal core (iron oxide/NH2 cores) but different surface modifiers which are organic small molecules conjugated to the NP surface.25 Different computational approaches have been proposed in literature for the exploitation of this dataset with interesting results in model development. Recent models presented in the literature are briefly discussed below.
In 2010 Fourches et al.26 presented a QNAR model based on MOE descriptors calculated for the organic molecules conjugated to the NP surface and k-nearest neighbors (kNN) methodology. The proposed model was proven robust and accurate as indicated by external predictions, cross validation and Y randomization. Winkler and coworkers27,28 also studied this dataset and generated quantitative, predictive and informative models of cellular uptake using a pool of molecular descriptors. In a recent publication, Y. T. Chau and C. W. Yap29 used four different modeling methods, namely Naive Bayes, logistic regression, k nearest neighbor and support vector machine, to develop candidate models. A consensus model was developed using the top 5 candidate models and validated by repeating the entire model development process five times using different combinations of training and validation sets. The final consensus model had a sensitivity of 86.7 to 98.2% and a specificity of 67.3 to 76.6%. In a different publication Toropov et al.30 used CORAL software to build a QSAR model for the prediction of cellular uptake of this dataset. The software gave satisfactory and stable predictions of the cellular uptake of NPs in PaCa2 cancer cells for five random splits. Another attempt was made by Ghorbanzadeh et al.31 who presented an artificial neural network that was built based on descriptors calculated with Hyperchem program and Dragon. The results revealed the accuracy and reliability of the proposed model and moreover a sensitivity analysis indicated that the number of hydrogen-bond donor sites in the organic coating of a NP is the predominant factor responsible for cellular uptake. Moreover, Liu et al.32 proposed a robust Relevance Vector Machine (RVM) model built with nine descriptors, which demonstrated prediction accuracy as quantified by a 5-fold cross-validated squared correlation coefficient. Ensemble learning based QNAR models for predicting the biological effects of this dataset were also constructed by Singh et al.33 based on simple structural descriptors and various statistical parameters suggested robustness of the model. Finally a recent attempt for the modeling of this dataset was reported by Kar et al.34 in their publication were a statistically significant regression – based QNAR model was developed using a PLS method and a small number of interpretable descriptors.
In this work we present a fully validated and predictive QNAR model that was developed based on Mold2 descriptors and the kNN algorithm. Our model was made publicly available through Enalos InSilicoNano platform (http://enalos.insilicotox.com/QNAR_PaCa2/), a web service developed with the aim to facilitate NPs design and evaluation. The user can draw a new structure, enter a SMILES notation or upload many structures in an sdf file. By the click of a button a prediction is made available together with a value that indicates if the structure can be tolerated by the model in terms of its domain of applicability. We have used our web service in a virtual screening framework mining PubChem database. We have successfully retrieved several potent inhibitors with the aim to prioritize compounds for screening. This online tool could be a useful aid for the decision making of both research groups and regulatory bodies interested in NPs' design and screening.
2. Results and discussion
2.1 Building a KNIME QNAR workflow
In this work we have tried to address the need of robust and predictive QNAR models for the assessment of the biological profile of ENPs and on top of that the proposed model has been made available online through Enalos InSilicoNano platform. The platform was used in a virtual screening framework to identify promising compounds within PubChem. For our study we have worked with 109 ENPs with the same metal core and different organic coating.25 The model was built based on a KNIME workflow that was developed for this purpose. KNIME is a freely available tool and has an extended community of users and developers and is increasingly gaining more attention for solving cheminformatics problems.35 Our efforts to address the lack of ready-to-use applications based on QSAR models were facilitated by the use of this open source platform.Our overall strategy is targeting the development of a validated QNAR model and the release of this model to the wider community through a web service. For the model development a KNIME35 workflow was developed that executes the following procedures: (i) data preprocessing, (ii) descriptors calculation, (iii) variable selection and model development, (iv) model validation, (v) domain of applicability determination. In the proposed workflow all these computational steps were incorporated and this complete line of operations was made feasible with the invaluable help of our in house made Enalos KNIME nodes, namely Enalos Mold2 node, Enalos Model Acceptability Criteria node and Enalos Domain – Similarity node.36 These nodes have been developed by Novamechanics Ltd and are publicly available through the KNIME Community and the company's website.37
The CfsSubset variable selection with BestFirst evaluator method was then applied on the training data to select the most significant descriptors.40,41 Among the available descriptors, nine have emerged as the most critical in capturing the significant structural characteristics that affect the biological profile of the studied NPs as proposed by the variable selection algorithm. These descriptors include:
Geary topological structure autocorrelation length-7 weighted by atomic van der Waals volumes (D461), Geary topological structure autocorrelation length-5 weighted by atomic Sanderson electronegativities (D467), number of total quaternary C-sp3 (D599), number of group secondary amines (aliphatic) (D649), number of group donor atoms for H-bonds (with N and O) (D712), number of group CH3R and CH4 (D714), number of group phenol or enol or carboxyl OH (D753), number of group Al2–NH (D758) and hydrophilic factor index (D775). Their physical meaning is briefly described below.
Descriptors D461 and D467 encode information as described by Geary topological structure autocorrelation length-7 weighted by atomic van der Waals volumes and length-8 weighted by atomic Sanderson electronegativities. Geary index is a general index of spatial autocorrelation and is a distance-type function varying from zero to infinite. In each descriptor the index is either weighted by atomic van der Waals volumes or atomic Sanderson electronegativities.42 The hydrophilic factor index (D775) accounts for the hydrophilicity of each of the structures described. All other descriptors included are counting for the number of different important features present in the structure such as total quaternary C-sp3 (D599), secondary amines (D649), donor atoms for H-bonds (D712), the presence of CH3R and CH4 (D714), phenol or enol or carboxyl OH (D753) and Al2–NH (D758).
The proposed KNIME workflow gave us the opportunity to test the performance of various algorithms included in the WEKA suite of programs and select the combination that best describes our data. The kNN algorithm was selected to describe the significant correlation among the selected descriptors and the cellular uptake in PaCa2. This algorithm outperformed various different algorithms that were also tested. The kNN methodology was applied on our training data with an optimized value of k equal to 2.43 Euclidean distance was used with all nine descriptors and contributions of neighbors weighted by the inverse of distance.
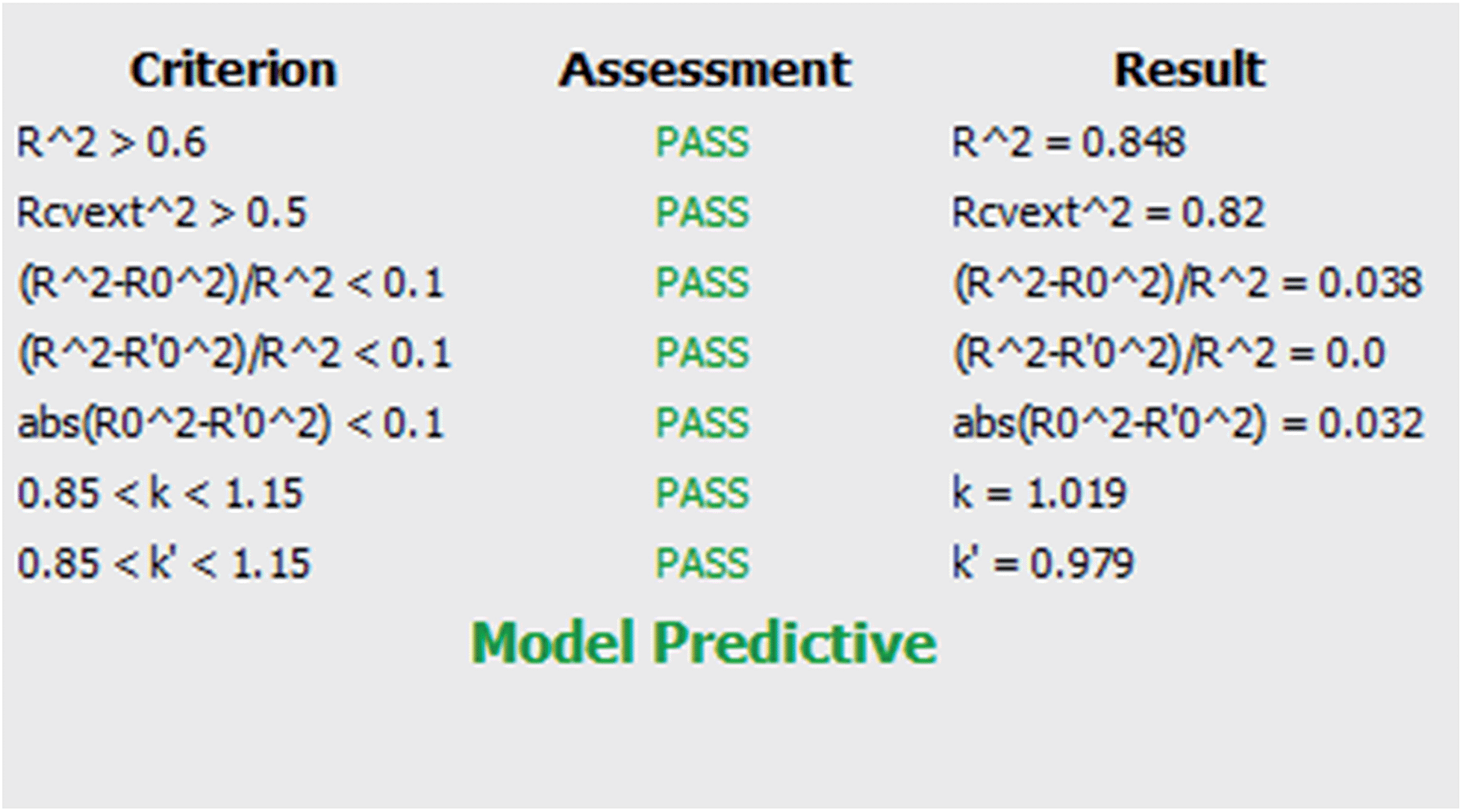 | ||
| Scheme 1 Model evaluation summary results. | ||
R2 is the coefficient of determination between experimental values and model prediction on the test set (R2pred). Mathematical calculations of R2o, R′2o, k, and k′ are based on regression of the observed activities against the predicted activities and vice versa using the equations described in Materials and methods section.
The model was also quite stable to the inclusion–exclusion of compounds measured by the ten-fold cross validation procedure. The R2L10O was calculated equal to 0.74. In addition the Y-randomization test was used as a method for testing the robustness and statistical significance of the model. Since low values of the correlation coefficient were measured we can eliminate the possibility of chance correlation.
The values of all the above statistical tests illustrate the accuracy, significance and robustness of the proposed model.
It is important that the limitations of the model are also described via the domain of applicability. This gives an important indication as the user can freely and creatively design novel molecules but will be warned for the reliability of the prediction when the structural characteristics cannot be tolerated by the model. After model validation, the domain of applicability of our model was also defined to ascertain that a given prediction can be considered reliable.47–50 The applicability domain limit value was defined equal to 2.153 based on the equation provided in Materials and methods section. All compounds in the test set had values in the range of 0.019–1.06 except for one which slightly falls outside with a value of 2.29. The predictions for all compounds that fell inside the domain of applicability of the model can be considered reliable.
Our proposed model requires only structural information from the small organic molecules involved and was proven accurate and reliable for given applicability limits. Thus our model could be used as a useful aid to the costly and time consuming experiments for determining cellular uptake of NPs and could further be used to screen existing databases or virtual chemical structures to identify NPs with desired properties. In this effort, the applicability domain will play an important role as it will filter out chemical structures that could not be tolerated by the model.
2.2 Building Enalos InSilicoNano platform
An important aspect that, in the vast majority of examples presented in literature, remains forgotten is the dissemination of the results to the wider community. It is very crucial that the developed model does not remain within the developers' group but is widely disseminated to the community so that it could immediately serve as an important source of information as it was initially designed to be. Moreover as recently highlighted by many initiatives it is crucial that this is done with open source tools that could be easily expanded and adjusted to the special needs of each project.To enable its role and make the model predictions available to the interested users, our proposed model was made publicly available online through Enalos InSilicoNano platform.51 Enalos InSilicoNano platform is a webservice that can host several validated and predictive models that can be utilized in the NPs design process. Our validated model was made publicly available through this platform and can thus be of help to the wider community of end users interested in NP's design. The web service needs no special computational skills and can be easily used by different groups of scientists like chemists, biologists etc. or even non experts involved or interested in the NPs biological evaluation.
Enalos InSilicoNano platform has a user friendly interface with minimum steps required and no authentication and authorization procedure. To initiate a prediction the user must first select the model of interest from the drop down menu provided. When the model “QNAR_PaCa2” is selected the prediction can be initiated when a structure or a batch of structures is uploaded. For that the web service provides three different options described as follows: (i) the user draws a chemical structure of interest using the drawing tool. The user can easily select from the different panels the atoms, bonds or substructures of interest and construct the molecule. What is important is that the user can also open, save and convert files with a variety of chemical formats (i.e. SMILES, IUPAC chemical Identifier, MDL MOL file) using the drop down menu of the online sketcher; (ii) the user enters the SMILES notation of a structure or several structures separated by newlines. Even if the SMILES notation is not initially known it is important that the chemical sketcher included gives the users the opportunity to design the chemical structure and then copy the structure as SMILES from the Edit drop down menu. This is very significant as it facilitates the generation of several structures since the user can make several modifications using the sketcher and copy all structures as SMILES so that a prediction for the whole set of produced structures is generated. The user can thus visualize the modifications and make multiple predictions at once; (iii) the user can select and import an SDF file (.sdf) with several structures.
When structures are uploaded in either way a prediction can be generated by clicking the submit button. The output is then presented in a different html page. The results include the predicted value for each structure entered and an indication of whether the prediction could be considered reliable based on the domain of applicability of the model. A screen shot of the web service and the results page are presented in the following schemes.
Our developed KNIME workflow integrated with Enalos InSilicoNano web service made the online prediction of the biological effects of NPs feasible. In the web service presented in Scheme 2 and 3, the user can design or enter a chemical structure and get the prediction. The workflow behind the interface calculates the descriptors and generates the output. It is important that the output will appear on screen within seconds. The user can experiment with different scaffolds and substituents and study the structural characteristics that are responsible to induce a certain effect. The user can take advantage of the proposed QNAR model and immediately scan the structures of interest for a preliminary in silico testing. In this way we overcome a main point of controversy for QSAR models in general, that their results are not available for sharing and implementation. As recently highlighted52 the advantages of making models available for use as software tools will increase in the future and this will enable the re-use of knowledge and will boost further developments. Enalos InSilicoNano platform uses a pipeline tool, KNIME, to address exactly this need of using and testing the models directly available on the web. With this platform we aim to address the need to reduce the amount of time spent by scientists in referencing disparate sources of data to aid decision making related to NPs design and bioactivity profile. Enalos InSilicoNano is launched as an efficient port where models can be developed and published directly on the web using a user friendly interface.
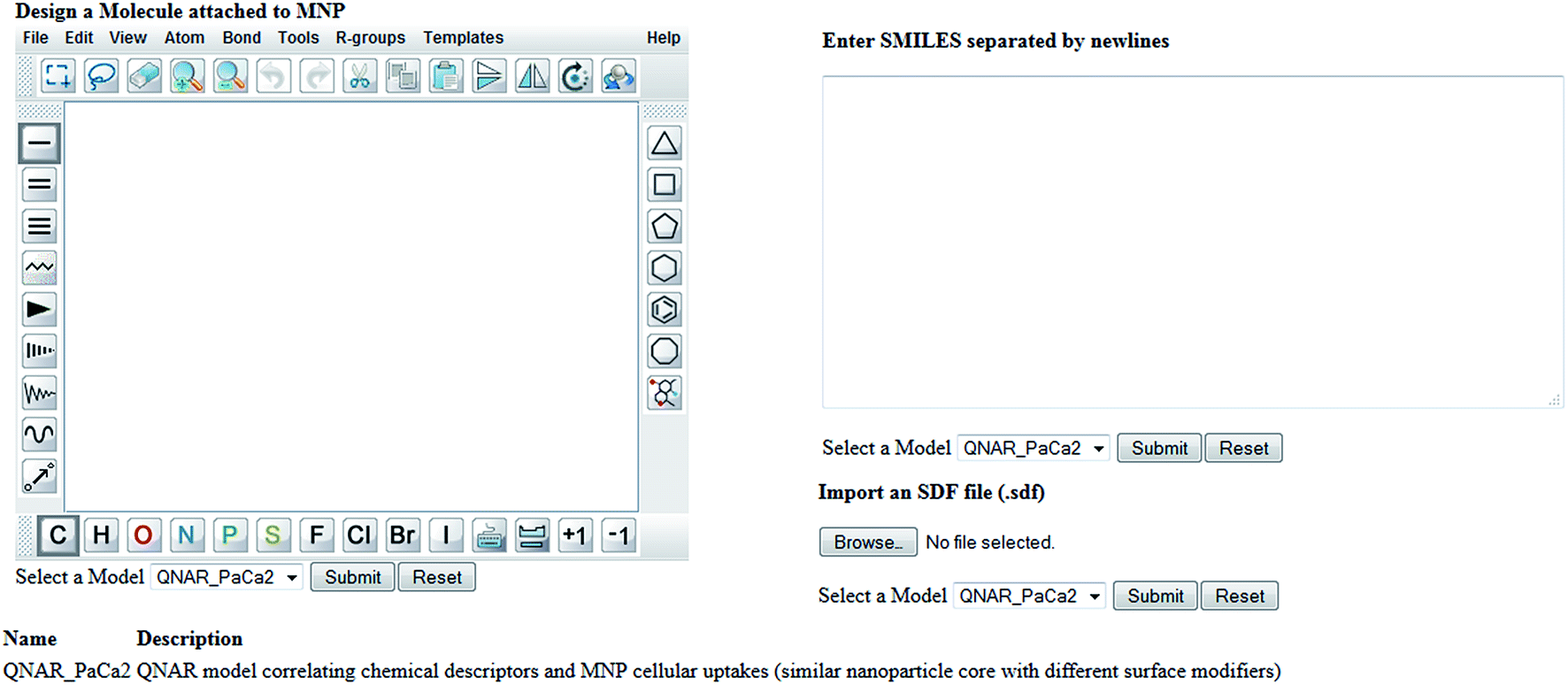 | ||
| Scheme 2 Screen shot of Enalos InSilicoNano platform input page. | ||
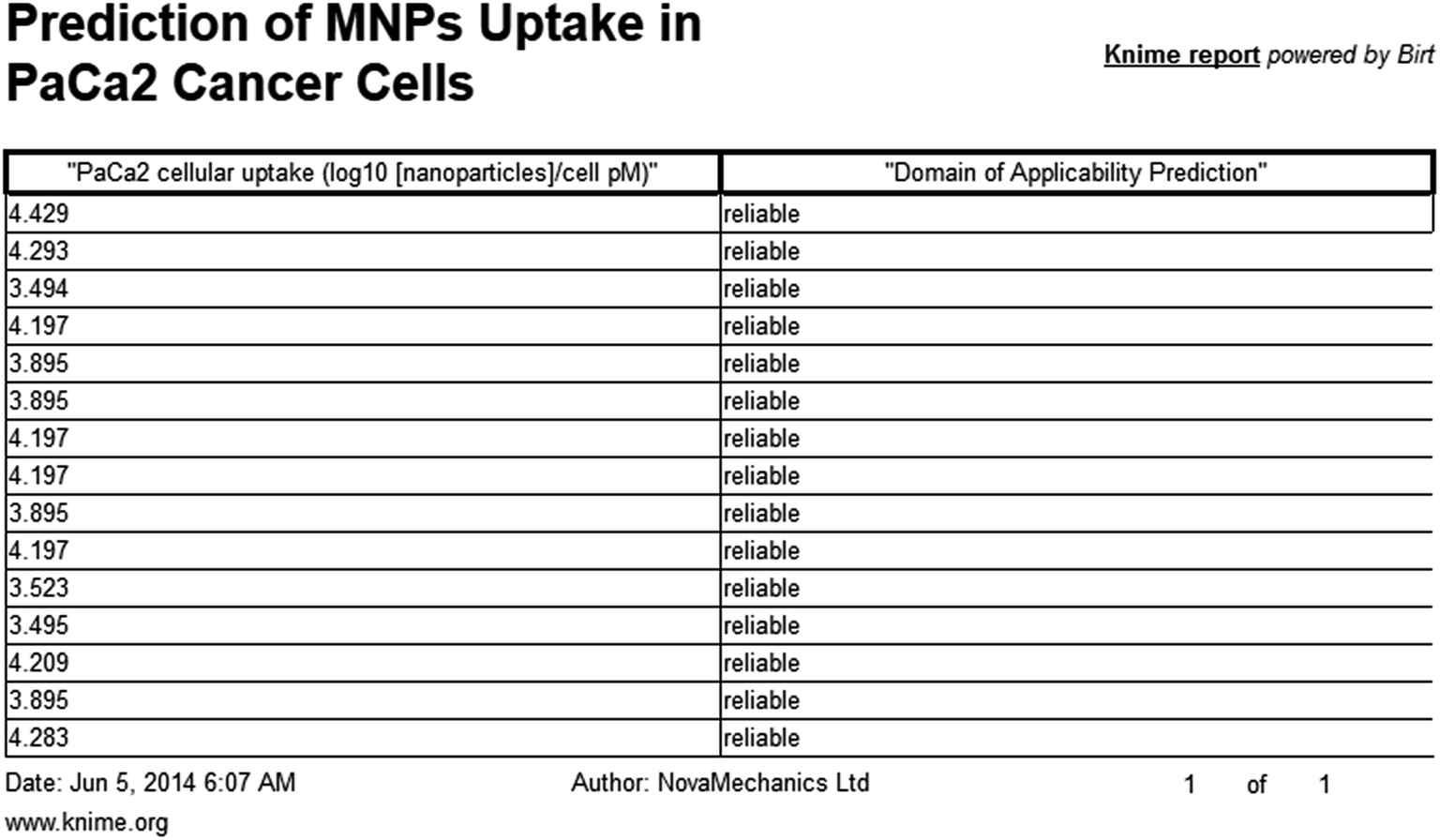 | ||
| Scheme 3 Screen shot of Enalos InSilicoNano platform results. | ||
2.3 Virtual screening
The presented model and web service can be used in a virtual screening framework for the prioritization of novel potent compounds.53,54 To demonstrate the usefulness of the Enalos InSilicoNano platform we have identified novel potent structures within PubChem database using similarity measurements based on Molecular Quantum Numbers (MQNs) as described in Materials and methods section.55 For this purpose we have selected among millions of compounds included in the PubChem56 database the most similar to the most active compound included in our initial dataset, that is compound 36 (isatoic anhydride). The above virtual screening procedure was used for the identification of the first 1000 neighbours of this most active compound included in our dataset in terms of chemical similarity. These first 1000 compounds within the PubChem that were identified as the most similar in the chemical space were evaluated using Enalos InSilicoNano platform for the assessment of their cellular uptake. For this purpose we have uploaded the sdf file containing all the proposed structures to Enalos InSilicoNano platform web service and asked for a prediction. Compounds were then sorted by their increasing potency and the most promising compounds were proposed for screening. The predicted values of the first 20 prioritized compounds are shown in Table 2.Within this proposed strategy Enalos InSilicoNano platform emerges as a key component for evaluating novel nano-structures that have not been experimentally evaluated or even synthesized. It is also important to highlight that our proposed methodology and tools can also be expanded and applied to polymer–nanoparticle composites that are now gaining increasing attention.
We have succeeded to generate a novel computational activity assessment platform for nanoparticles by integrating two open science platforms: KNIME that combines a rich graphical workflow environment for integration of diverse analytics and Enalos InSilicoNano a platform for hosting and publishing models directly on the web allowing the researchers to do virtual screening and/or design of novel nanoparticles. Two milestones have been reached within this work, the first is the development of a validated QNAR model and the second is the development of a web service that will immediately give the opportunity of exploiting the model's results. To demonstrate the usefulness of the model we have also proposed a virtual screening framework that could be used to identify novel potent structures.
3. Conclusions
In summary our goal in this work was dual, firstly to build a robust validated QNAR model for NPs and secondly to give immediate access to our model and results using an open access web service. Enalos InSilicoNano platform aspires to be a useful tool for design of novel NPs with desired properties.57,58 To this end we have successfully built and validated a QNAR model that can reliably predict the cellular uptake of a dataset of 109 NPs. The model was made publicly available through Enalos InSilicoNano platform and can be used for the predictions of cellular uptake of new structures that are designed or imported to the server. This online tool was successfully used for the virtual screening of a set of structures within PubChem database that were selected based on MQN descriptors to identify compounds similar to a known active structure. Besides the significance of the validated proposed model, to the best of our knowledge this is the first attempt to develop an online tool for the wider scientific community to use in the computer aided nanoparticles design with desired properties. Our model can now be directly used and easily applied to facilitate the special requirements of the user.4. Materials and methods
4.1 KNIME workflow development
To address the needs for our project we have used the powerful KNIME (Konstanz Information Miner) which is an open source tool for data analysis that allows data integration, processing, analysis, and exploration and enables the user to visually create data flows selectively execute some or all analysis steps, and later investigate the results through interactive views on data and models.35 In this work we have used KNIME platform in order to integrate all the various components of our workflow as described below in details. Within our workflow we have also incorporated our newly developed Enalos KNIME Nodes that were used to facilitate our model development and validation.36 Enalos KNIME nodes include among others: (1) Enalos Mold2 node for the calculation of Mold2 molecular descriptors, (2) Enalos Model Acceptability Criteria node that can be used to validate the Quality of Fit and Predictive Ability of a continuous QSAR Model and (3) Enalos Domain – Similarity node that can be used to define applicability domain (APD) based on the Euclidean distances. The Enalos KNIME Nodes are freely available via the KNIME Community and the company's website.37| ID | Smiles | Observed PaCa2 cellular uptake (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 [NP]/cell) 10 [NP]/cell) |
Predicted PaCa2 cellular uptake (log10 [NP]/cell) |
|---|---|---|---|
| a Test Set. | |||
| 1 | FC(F)(F)C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OC(O)C(F)(F)F O)OC(O)C(F)(F)F |
4.17 | 4.17 |
| 2 | FC(F)(Cl)C(O)OC(O)C(F)(F)Cl |
3.95 | 3.95 |
| 3 | FC(F)(F)C(F)(F)C(O)OC(O)C(F)(F)C(F)(F)F |
4.08 | 4.08 |
| 4 | CC1(C)CC(O)OC1O |
4.11 | 3.80 |
| 5 | OC1OC(O)CC1 |
3.98 | 4.11 |
| 6a | CC1CC(O)OC1O |
3.58 | 3.65 |
| 7 | CC1C(C)C(O)OC1O |
3.48 | 3.80 |
| 8 | CCCCCC(O)OC(O)CCCCC |
3.65 | 3.65 |
| 9 | CC1CC(O)OC1O |
3.64 | 3.65 |
| 10 | OC1OC(O)c2cc(ccc12)C(O)c1ccc2C(O)OC(O)c2c1 |
3.51 | 3.53 |
| 11 | OC1OC(O)c2cc(ccc12)N(O)O |
3.27 | 3.29 |
| 12a | Brc1ccc2C(O)OC(O)c3cccc1c23 |
3.63 | 3.52 |
| 13 | OC1OC(O)c2ccc3C(O)OC(O)c4ccc1c2c34 |
3.67 | 3.68 |
| 14 | Fc1c(F)c(F)c2C(O)OC(O)c2c1F |
3.83 | 3.84 |
| 15 | OC1OC(O)c2cc(cc3cccc1c23)N(O)O |
4.11 | 4.09 |
| 16 | Oc1cccc2C(O)OC(O)c12 |
3.97 | 3.97 |
| 17 | OC1OC(O)C2C3CCC(CC3)C12 |
3.9 | 3.87 |
| 18 | Clc1ccc2NC(O)OC(O)c2c1 |
4.18 | 4.17 |
| 19 | OC1OS(O)(O)c2ccccc12 |
3.88 | 3.93 |
| 20 | ClC1C(Cl)C(O)OC1O |
3.84 | 3.87 |
| 21a | CC(O)SC1CC(O)OC1O |
3.59 | 3.85 |
| 22 | Clc1cc2C(O)OC(O)c2cc1Cl |
4.12 | 4.07 |
| 23 | OC1OC(O)C2C3OC(CC3)C12 |
3.82 | 3.80 |
| 24 | OC1OC(O)C2C3CCC(C12)C1C3C(O)OC1O |
3.63 | 3.65 |
| 25 | OC1OC(O)C2CCCCC12 |
3.89 | 3.86 |
| 26 | OC1OC(O)c2ccccc2-c2ccccc12 |
3.77 | 3.77 |
| 27 | OC1OC(O)c2ccc(c3cccc1c23)N(O)O |
3.93 | 3.92 |
| 28 | OC1OC(O)C2C1C1C2C(O)OC1O |
3.77 | 3.86 |
| 29 | CCCCCCCCCCCC(O)OC(O)CCCCCCCCCCC |
3.82 | 3.82 |
| 30a | OC(O)c1ccc2C(O)OC(O)c2c1 |
3.55 | 3.62 |
| 31 | Cc1ccc2C(O)OC(O)c2c1 |
3.98 | 3.97 |
| 32 | OC1OC(O)c2c1cccc2N(O)O |
3.5 | 3.54 |
| 33 | OC1Cc2ccccc2C(O)O1 |
3.78 | 3.81 |
| 34 | OC1CCCC(O)O1 |
4.07 | 4.06 |
| 35a | OC1CN(CCN2CC(O)OC(O)C2)CC(O)O1 |
3.93 | 3.76 |
| 36 | OC1Nc2ccccc2C(O)O1 |
4.44 | 4.43 |
| 37 | CN1C(O)OC(O)c2ccccc12 |
3.36 | 3.38 |
| 38a | CC1CC(O)OC(O)C1 |
3.91 | 3.68 |
| 39 | OC1OC(O)C2C1CCCC2 |
3.73 | 3.74 |
| 40 | CC(O)OC1C(OC(C)O)C(O)OC1O |
3.91 | 3.91 |
| 41a | Brc1c(Br)c(Br)c2C(O)OC(O)c2c1Br |
3.8 | 3.66 |
| 42a | OC1OC(O)C2CCCCC12 |
3.93 | 3.88 |
| 43 | OC1OC(O)C2C1CCC2 |
3.69 | 3.71 |
| 44 | ICC(O)OC(O)CI |
3.42 | 3.42 |
| 45 | ClCC(O)OC(O)CCl |
3.63 | 3.62 |
| 46 | ClC1C(Cl)C2(Cl)C3C(C(O)OC3O)C1(Cl)C2(Cl)Cl |
3.47 | 3.49 |
| 47a | CCCCCCCCCCCCCCCC(O)OC(O)CCCCCCCCCCCCCCC |
3.55 | 3.78 |
| 48 | Nc1ccc2C(O)OC(O)c3cccc1c23 |
3.64 | 3.63 |
| 49a | CCCCCCCCCC(O)OC(O)CCCCCCCCC |
4.03 | 3.78 |
| 50a | OC1CC2(CCCC2)CC(O)O1 |
4.06 | 3.88 |
| 51 | OC1OC(O)C2C3CCC(C3)C12 |
3.94 | 3.91 |
| 52 | OC1OC(O)c2cccc3cccc1c23 |
3.96 | 3.95 |
| 53 | OC1CCC(C(O)O1)c1ccccc1 |
4.02 | 4.00 |
| 54a | Clc1c(Cl)c(Cl)c2C(O)OC(O)c2c1Cl |
3.83 | 3.66 |
| 55 | Clc1ccc(Cl)c2C(O)OC(O)c12 |
3.9 | 3.88 |
| 56a | CC1(C)CCC(O)OC1O |
3.94 | 3.80 |
| 57 | CCCCCN | 3.78 | 3.78 |
| 58 | CC(C)CC(C)N | 3.85 | 3.85 |
| 59 | NC1C(O)CC(CO)C(O)C1O | 3.36 | 3.36 |
| 60a | CCCCCCN | 3.75 | 3.77 |
| 61 | CC(C)(C)N | 3.86 | 3.87 |
| 62 | CC(C)CN | 3.72 | 3.74 |
| 63 | CC(C)(C)CN | 3.75 | 3.91 |
| 64 | CC(C)CCN | 3.83 | 3.82 |
| 65 | CCC(N)CC | 3.81 | 3.82 |
| 66 | CCC(C)(C)N | 4.07 | 3.91 |
| 67 | NCCN | 3.46 | 3.46 |
| 68 | CCCCCCCCCCCCCCCN | 4.06 | 4.03 |
| 69 | NCCCN | 3.49 | 3.49 |
| 70 | NCCCCN | 3.48 | 3.48 |
| 71 | NCCCCCCN | 3.62 | 3.62 |
| 72 | CCCCC(CC)CN | 3.95 | 3.94 |
| 73 | CCCCCCCCCCCCCCCCN | 3.97 | 4.00 |
| 74 | CCCCCC(C)N | 3.63 | 3.64 |
| 75a | CCCCCCCCCCCCCCN | 4.27 | 4.02 |
| 76 | NCCNCCN | 3.77 | 3.77 |
| 77 | NCC12CC3CC(CC(C3)C1)C2 | 2.84 | 2.87 |
| 78 | NCCc1ccc(O)c(O)c1 | 2.53 | 2.53 |
| 79 | NCCc1ccc(O)cc1 | 2.77 | 2.77 |
| 80a | NCCCNCCCCNCCN | 2.41 | 2.37 |
| 81 | NCCNCCCNCCN | 2.23 | 2.24 |
| 82 | NCCNCCNCCNCCNCCN | 2.54 | 2.54 |
| 83 | NC12CC3CC(CC1C3)C2 | 3.12 | 3.14 |
| 84 | NC1C2CC3CC(C2)CC1C3 | 3.18 | 3.15 |
| 85 | NCC(O)O |
2.57 | 2.58 |
| 86 | COC(O)C(N)Cc1ccccc1 |
3.39 | 3.39 |
| 87 | NC(CO)C(O)O |
3.36 | 3.35 |
| 88 | CC(O)C(N)C(O)O |
3.21 | 3.21 |
| 89 | NC(Cc1c[nH]c2ccccc12)C(O)O |
3.19 | 3.19 |
| 90 | NC(Cc1ccc(O)cc1)C(O)O |
3.07 | 3.07 |
| 91a | CC(C)C(N)C(O)O |
3.27 | 3.04 |
| 92 | NCCCCC(N)C(O)O |
3.25 | 3.25 |
| 93 | NC(C(O)O)c1ccc(Cl)cc1 |
3.06 | 3.07 |
| 94 | CC(N)C(O)O |
2.9 | 2.90 |
| 95a | NC(CCCNC(N)N)C(O)O |
3.15 | 3.28 |
| 96a | NC(CC(O)O)C(O)O |
3.29 | 3.35 |
| 97 | NC(CCC(N)O)C(O)O |
3.32 | 3.32 |
| 98 | NC(CCC(O)O)C(O)O |
3.4 | 3.40 |
| 99 | NC(Cc1c[nH]cn1)C(O)O |
3.38 | 3.38 |
| 100 | CSCCC(N)C(O)O |
3.23 | 3.23 |
| 101 | NC(Cc1ccccc1)C(O)O |
3.29 | 3.29 |
| 102 | OC1CCC(O)O1 |
4.24 | 4.11 |
| 103a | CC(O)OC(C)O |
4.05 | 3.80 |
| 104 | CC1CC(O)OC1O |
4.04 | 4.06 |
| 105 | OC1COCC(O)O1 |
3.99 | 3.96 |
| 106 | OC1OC(O)c2ccccc12 |
3.9 | 3.92 |
| 107 | OC(O)CC1CC(O)OC1O |
4.03 | 4.03 |
| 108 | Fc1ccc(F)c2C(O)OC(O)c12 |
3.91 | 3.87 |
| 109 | OC(O)CN(CCN1CC(O)OC(O)C1)CCN1CC(O)OC(O)C1 |
4.1 | 4.09 |
| ID | Compound | Predicted value PaCa2 cellular uptake (log10 [NP]/cell) |
Domain of applicability (limit: 2.153) |
|---|---|---|---|
| 679 |  |
4.41 | 0.03 |
| 604 |  |
4.41 | 0.01 |
| 958 |  |
4.41 | 0.06 |
| 676 | 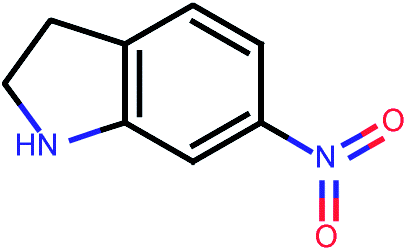 |
4.40 | 0.01 |
| 678 |  |
4.40 | 0.05 |
| 677 | 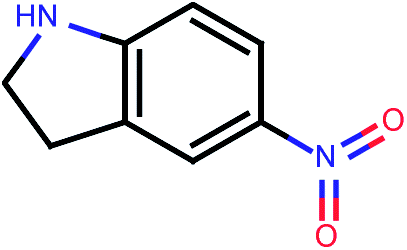 |
4.39 | 0.02 |
| 107 | 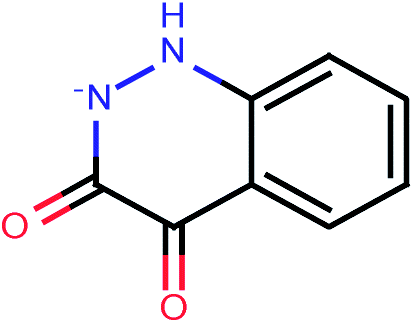 |
4.39 | 0.02 |
| 368 | 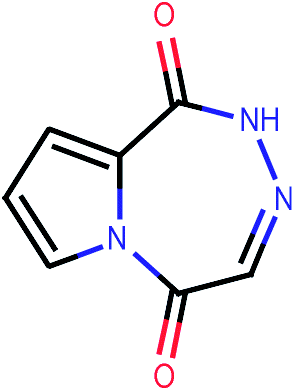 |
4.38 | 0.10 |
| 293 |  |
4.37 | 0.09 |
| 493 | 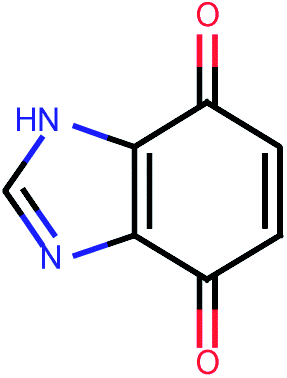 |
4.37 | 0.10 |
| 494 | 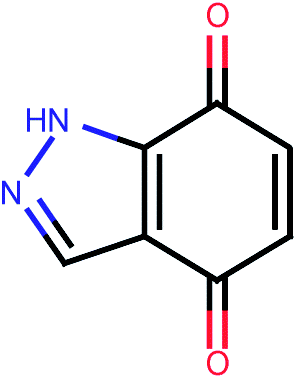 |
4.37 | 0.10 |
| 550 | 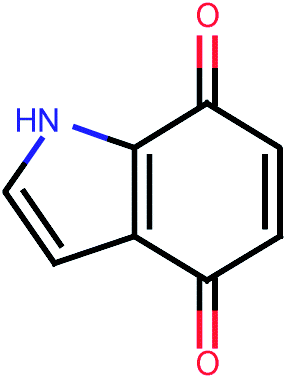 |
4.36 | 0.11 |
| 196 |  |
4.35 | 0.06 |
| 200 | 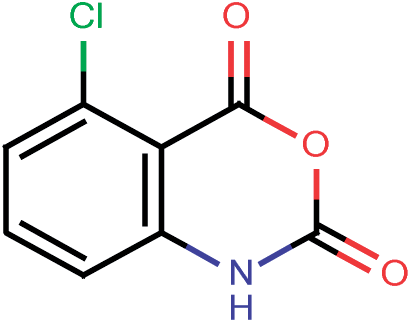 |
4.35 | 0.06 |
| 626 | 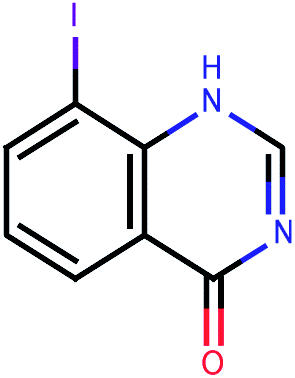 |
4.35 | 0.06 |
| 602 | 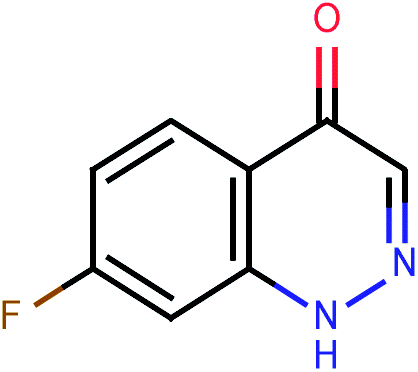 |
4.35 | 0.06 |
| 981 | 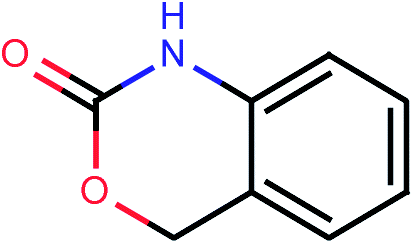 |
4.34 | 0.10 |
| 925 |  |
4.34 | 0.10 |
| 192 | 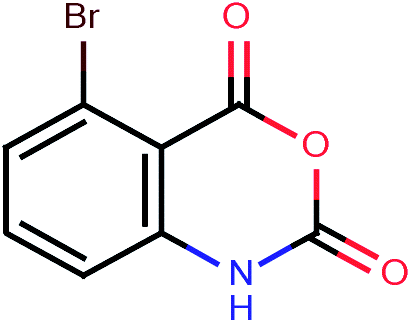 |
4.34 | 0.06 |
| 65 | 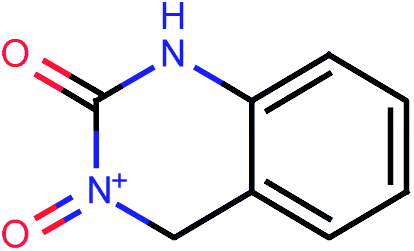 |
4.34 | 0.05 |
Within our KNIME workflow we have included Enalos Mold2 KNIME node36 that is able to calculate a number of 777 descriptors that account for the topological, geometric and structural characteristics of the small molecules. From this original pool of descriptors a number was removed as some of the descriptors do not have any discrimination power (no variation) and for this a node called ‘Low Variance Filter’ was applied.39
Subsequently a machine learning method that could best model the available dataset was applied. We have thus incorporated in our KNIME workflow k-nearest neighbors (kNN) methodology.43 kNN methodology belongs to instance-based (or lazy) learning that classifies objects based on the closest training examples in the feature space. An object is classified by a majority vote of its neighbors, with the object being assigned to the class most common amongst its k nearest neighbors (a positive integer, typically small). For our dataset we have used an optimal k value and Euclidean distance with all descriptors and contributions of neighbors weighted by the inverse of distance.
For external validation the partitioning KNIME node was applied and the dataset was separated into training and validation set leaving a number of 20 compounds for the external validation of the model. All compounds included in the test set were not involved by any means in the training procedure.
To evaluate the models performance the following statistical criteria were used: the coefficient of determination between experimental values and model predictions (R2), validation through an external test set, leave-many-out cross validation procedure and Quality of Fit and Predictive Ability of a continuous QSAR Model according to Tropsha's tests.45,46 The latter was made feasible by including Enalos Model Acceptability Criteria node in our workflow.
In particular the formulas for calculating Tropsha's tests45 are given below:
 | (1) |
 | (2) |
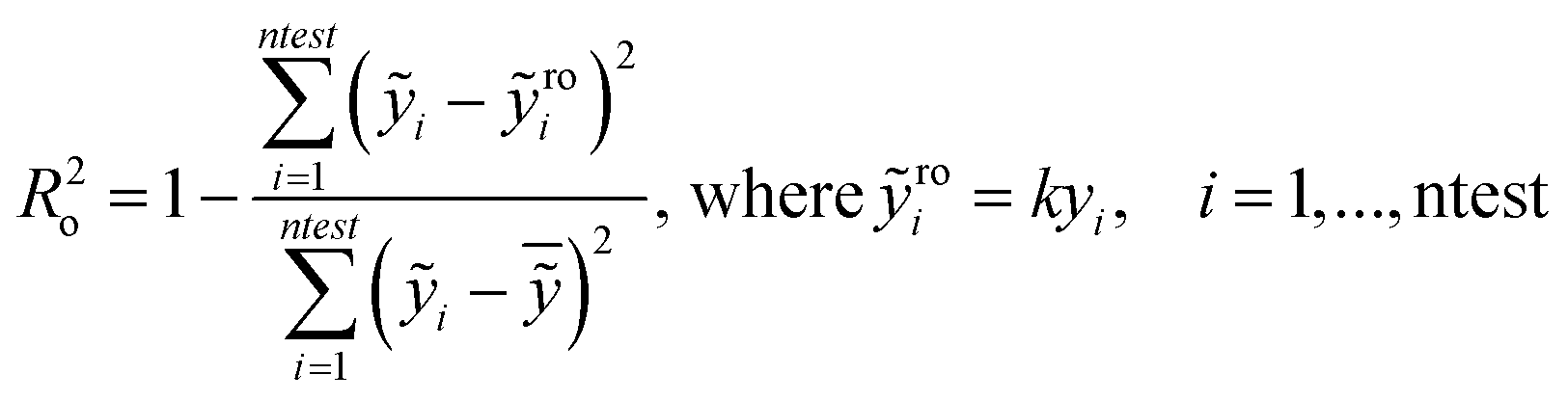 | (3) |
In the above equation ntest is the number of compounds that constitute the validation data set, ȳtr is the averaged value for the dependent variable for the training set, yi, ỹ, i = 1, …, ntest are the measured values and the QSAR model predictions of the dependent variable over the available validation set and 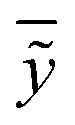 is the average over all ỹ, i = 1, …, ntest.
is the average over all ỹ, i = 1, …, ntest.
Tropsha et al.45 considered a QSAR model predictive, if the following conditions are satisfied:
| R2cvext > 0.5 | (4) |
| R2pred > 0.6 | (5) |
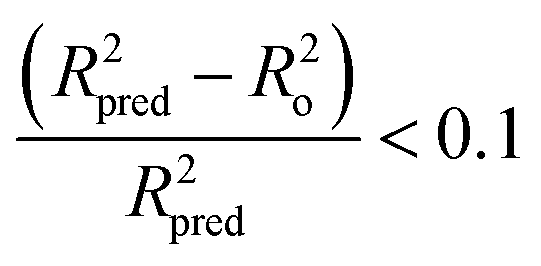 | (6) |
| 0.85 ≤ k ≤ 1.15 | (7) |
| APD = 〈d〉 +Zσ | (8) |
Calculation of 〈d〉 and σ was performed as follows: first, the average of Euclidean distances between all pairs of training compounds was calculated. Next, the set of distances that were lower than the average was formulated. 〈d〉 and σ were finally calculated as the average and standard deviation of all distances included in this set. Z was an empirical cutoff value and for this work, it was chosen equal to 0.5.47–50 Enalos Domain – Similarity node that executes the aforementioned procedure is included in our workflow and was used to assess domain of applicability of the proposed model.47–50
4.2 Enalos InSilicoTox platform
Novamechanics Ltd has recently launched Enalos InSilico platform, a new toxicity and drug discovery platform freely available online.51 Enalos InSilico platform aims to address the need to reduce the amount of time spent by scientists in referencing disparate sources of data to aid decision making related to NPs design and bioactivity profile and it offers an efficient and cost-effective response to the EU REACH legislation and the desire to reduce animal testing. The available workflows are built based on diverse and reliable data sources and integrate advanced in silico tools to provide accurate predictions. The web service is solely based on open source and freely available software including the powerful KNIME (Konstanz Information Miner)35 which is a user friendly and comprehensive open-source platform for data analysis including also all analysis modules of the well-known Weka data mining.41 In this work we have used KNIME platform in order to simultaneously run and compare different modeling methodologies and explore which of the available methods (or combination) best suites our data. As previously mentioned, to address our needs for robust and accurate model development targeting structural optimization and design we have developed the Enalos family nodes that were made publicly available for all KNIME users.Through Enalos InSilico platform, toxicity, biological activity and property predictions can be obtained for chemical structure provided by the user. Structures can be designed, entered as SMILES or imported in SDF format. The QNAR model described in this work can be selected from the pull down menu of the available workflows already developed and provided by the Enalos InSilicoNano platform.
4.3 Virtual screening
In an effort to identify novel potent compounds, a virtual screening study was initiated for compounds included in PubChem database.55 PubChem is a publicly available database that archives the molecular structures and bioassay data within the National Institute of Health (NIH) Roadmap for Medical Research Initiative. PubChem is currently the largest publicly available molecular database with millions of entries.PubChem database was used to retrieve potent compounds in the described virtual screening framework using the most active compound in our original database, compound 36, which has a PaCa2 cellular uptake value equal to 4.44 expressed as decadic logarithm of the concentration (pM) of NP per cell (log10[NP]/cell). All compounds included in the PubChem database were compared to compound 36 in a similarity context on the basis of 42 integer value descriptors of molecular structure, called Molecular Quantum Numbers (MQNs). MQNs count elementary features that matter most for the properties of organic molecules: atoms, bonds, polar groups, and topological features.56 The MQN-space organises molecules by their global structural features, but also by their similarity in biological activity. Distances in MQN-space can be used to search for analogues of known drugs. The MQNs form a scalar fingerprint which can be used to measure the similarity between pairs of molecules and enable ligand-based virtual screening.
Acknowledgements
This project has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no 310451 (Project NanoMILE).References
- A. Gajewicz, B. Rasulev, T. C. Dinadayalane, P. Urbaszek, T. Puzyn, D. Leszczynska and J. Leszczynsk, Advancing risk assessment of engineered nanomaterials: Application of computational approaches, Adv. Drug Delivery Rev., 2012, 64, 1663–1693 CrossRef CAS PubMed.
- Y. Cohen, R. Rallo, R. Liu and H. H. Liu, In silico analysis of nanomaterials hazard and risk, Acc. Chem. Res., 2013, 46, 802–812 CrossRef CAS PubMed.
- A. E. Nel, Implementation of alternative test strategies for the safety assessment of engineered nanomaterials, J. Intern. Med., 2013, 274, 561–577 CrossRef CAS PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. Lin, Z. Ji and H. Zhang, Nanomaterial toxicity testing in the 21st century: Use of a predictive toxicological approach and high-throughput screening, Acc. Chem. Res., 2013, 46, 607–621 CrossRef CAS PubMed.
- L. Lubinski, P. Urbaszek, A. Gajewicz, M. T. D. Cronin, S. J. Enoch, J. C. Madden, D. Leszczynska, J. Leszczynski and T. Puzyn, Evaluation criteria for the quality of published experimental data on nanomaterials and their usefulness for QSAR modeling, SAR QSAR Environ. Res., 2013, 24, 995–1008 CrossRef CAS PubMed.
- D. A. Winkler, E. Mombelli, A. Pietroiusti, L. Tran, A. Worth, B. Fadeel and M. J. McCall, Applying quantitative structure-activity relationship approaches to nanotoxicology: Current status and future potential, Toxicology, 2013, 313, 15–23 CrossRef CAS PubMed.
- D. Fourches, D. Pu and A. Tropsha, Exploring Quantitative Nanostructure-Activity Relationships (QNAR) Modeling as a Tool for Predicting Biological Effects of Manufactured Nanoparticles, Comb. Chem. High Throughput Screening, 2011, 14, 217–225 CrossRef CAS.
- C. P. Roca, R. Rallo, A. Fernández and F. Giralt, Nanoinformatics for safe-by-design engineered nanomaterials, in Towards Efficient Designing of Safe Nanomaterials: Innovative Merge of Computational Approaches and Experimental Techniques, ed. J. Leszczynski and T. Puzyn, RSC Nanoscience and Nanotechnology, Cambridge UK, 2012, pp. 89–107 Search PubMed.
- R. Liu, R. Rallo, S. George, Z. Ji, S. Nair, A. E. Nel and Y. Cohen, Classification NanoSAR development for cytotoxicity of metal oxide nanoparticle, Small, 2011, 7, 1118–1126 CrossRef CAS PubMed.
- B. Rasulev, A. Gajewicz, T. Puzyn, D. Leszczynska and J. Leszczynski, Nano-QSAR: Advances and challenges, in Towards Efficient Designing of Safe Nanomaterials: Innovative Merge of Computational Approaches and Experimental Techniques, ed. J. Leszczynski and T. Puzyn, RSC Nanoscience and Nanotechnology, Cambridge UK, 2012, pp. 220–256 Search PubMed.
- R. Liu, B. France, S. George, R. Rallo, H. Zhang, T. Xia, A. E. Nel, K. Bradley and Y. Cohen, Association rule mining of cellular responses induced by metal and metal oxide nanoparticles, Analyst, 2014, 139, 943–953 RSC.
- S. L. Harper, J. E. Hutchison, N. Baker, M. Ostraat, S. Tinkle, J. Steevens, M. D. Hoover, J. Adamick, K. Rajan, S. Gaheen, Y. Cohen, A. Nel, R. E. Cachau and M. Tuominen, Nanoinformatics workshop report: Current resources, community needs and the proposal of a collaborative framework for data sharing and information integration, Comput. Sci. Discovery, 2013, 6, 1–16 Search PubMed , (art. no. 014008).
- R. Liu, H. Y. Zhang, Z. X. Ji, R. Rallo, T. Xia, C. H. Chang, A. Nel and Y. Cohen, Development of structure-activity relationship for metal oxide nanoparticles, Nanoscale, 2013, 5, 5644–5653 RSC.
- T. Patel, D. Telesca, R. Rallo, S. George, T. Xia and A. E. Nel, Hierarchical Rank Aggregation with Applications to Nanotoxicology, J. Agr. Biol. Environ. Stat., 2013, 18, 159–177 CrossRef PubMed.
- R. Rallo, B. France, R. Liu, S. Nair, S. George, R. Damoiseaux, F. Giralt, A. Nel, K. Bradley and Y. Cohen, Self-organizing map analysis of toxicity-related cell signaling pathways for metal and metal oxide nanoparticles, Environ. Sci. Technol., 2011, 45, 1695–1702 CrossRef CAS PubMed.
- J. Ehret, M. Vijver and W. Peijnenburg, The Application of QSAR Approaches to Nanoparticles, ATLA, Altern. Lab. Anim., 2014, 42, 43–50 CAS.
- C.-Y. Shao, S.-Z. Chen, B.-H. Su, Y. J. Tseng, E. X. Esposito and A. J. Hopfinger, Dependence of QSAR models on the selection of trial descriptor sets: A demonstration using nanotoxicity endpoints of decorated nanotubes, J. Chem. Inf. Model., 2013, 53, 142–158 CrossRef CAS PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Using nano-QSAR to predict the cytotoxicity of metal oxide nanoparticles, Nat. Nanotechnol., 2011, 6, 175–178 CrossRef CAS PubMed.
- S. George, T. Xia, R. Rallo, Y. Zhao, Z. Ji, S. Lin, X. Wang, H. Zhang, B. France, D. Schoenfeld, R. Damoiseaux, R. Liu, S. Lin, K. A. Bradley, Y. Cohen and A. E. Nel, Use of a high-throughput screening approach coupled with in vivo zebrafish embryo screening to develop hazard ranking for engineered nanomaterials, ACS Nano, 2011, 5, 1805–1817 CrossRef CAS PubMed.
- A. P. Toropova, A. A. Toropov, E. Benfenati and R. Korenstein, QSAR model for cytotoxicity of SiO2 nanoparticles on human lung fibroblasts, J. Nanopart. Res., 2014, 16, 2282 CrossRef.
- A. P. Toropova and A. A. Toropov, Optimal descriptor as a translator of eclectic information into the prediction of membrane damage by means of various TiO2 nanoparticles, Chemosphere, 2013, 93, 2650–2655 CrossRef CAS PubMed.
- R. Liu, H. Y. Zhang, Z. X. Ji, R. Rallo, T. Xia, C. H. Chang, A. Nel and Y. Cohen, Development of structure-activity relationship for metal oxide nanoparticles, Nanoscale, 2013, 5, 5644–5653 RSC.
- R. Liu, R. Rallo, R. Weissleder, C. Tassa, S. Shaw and Y. Cohen, Nano-SAR development for bioactivity of nanoparticles with considerations of decision boundaries, Small, 2013, 9, 1842–1852 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, Use of metal oxide nanoparticle band gap to develop a predictive paradigm for oxidative stress and acute pulmonary inflammation, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- R. Weissleder, K. Kelly, E. Y. Sun, T. Shtatland and L. Josephson, Cell-specific targeting of nanoparticles by multivalent attachment of small molecules, Nat. Biotechnol., 2005, 23, 1418–1423 CrossRef CAS PubMed.
- D. Fourches, D. Q. Y. Pu, C. Tassa, R. Weissleder, S. Y. Shaw, R. J. Mumper and A. Tropsha, Quantitative Nanostructure Activity Relationship Modeling, ACS Nano, 2010, 4, 5703–5712 CrossRef CAS PubMed.
- V. C. Epa, F. R. Burden, C. Tassa, R. Weissleder, S. Shaw and D. A. Winkler, Modeling Biological Activities of Nanoparticles, Nano Lett., 2012, 12, 5808–5812 CrossRef CAS PubMed.
- D. A. Winkler, F. R. Burden, B. Yan, R. Weissleder, C. Tassa, S. Shaw and V. C. Epa, Modelling and predicting the biological effects of nanomaterials, SAR QSAR Environ. Res., 2014, 25, 161–172 CrossRef CAS PubMed.
- Y. T. Chau and C. W. Yap, Quantitative Nanostructure–Activity Relationship modelling of nanoparticles, RSC Adv., 2012, 2, 8489–8496 RSC.
- A. A. Toropov, A. P. Toropova, T. Puzyn, E. Benfenati, G. Gini, D. Leszczynska and J. Leszczynski, QSAR as a random event: Modeling of nanoparticles uptake in PaCa2 cancer cells, Chemosphere, 2013, 92, 31–37 CrossRef CAS PubMed.
- M. Ghorbanzadeh, M. H. Fatemi and M. Karimpour, Modeling the Cellular Uptake of Magnetofluorescent Nanoparticles in Pancreatic Cancer Cells: A Quantitative Structure Activity Relationship Study, Ind. Eng. Chem. Res., 2012, 51, 10712–10718 CrossRef CAS.
- R. Liu, R. Rallo and Y. Cohen, Quantitative Structure-Activity-Relationships for cellular uptake of nanoparticles, Proceedings of the 13th IEEE Conference on Nanotechnology, 2013, 154–157 Search PubMed , (art. no. 6720861).
- K. P. Singh and S. Gupta, Nano-QSAR modeling for predicting biological activity of diverse nanomaterials, RSC Adv., 2014, 4, 13215–13230 RSC.
- S. Kar, A. Gajewicz, T. Puzyn and K. Roy, Nano-quantitative structure-activity relationship modeling using easily computable and interpretable descriptors for uptake of magnetofluorescent engineered nanoparticles in pancreatic cancer cells, Toxicol. in Vitro, 2014, 28, 600–606 CrossRef CAS PubMed.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kotter, T. Meinl, P. Ohl, C. Sieb, K. Thiel and B. Wiswedel, KNIME: The Konstanz Information Miner, Studies in Classification, Data Analysis, and Knowledge Organization, ed. C. Preisach, H. Burkhardt, L. Schmidt-Thieme and R. Decker, GfKl: Springer, 2007, pp. 319–326 Search PubMed.
- G. Melagraki and A. Afantitis, Enalos KNIME nodes: Exploring corrosion inhibition of steel in acidic medium, Chemom. Intell. Lab. Syst., 2013, 123, 9–14 CrossRef CAS PubMed.
- http://www.novamechanics.com/knime.php.
- H. Hong, Q. Xie, W. Ge, F. Qian, F. Fang, L. Shi, Z. Su, R. Perkins and W. Tong, Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics, J. Chem. Inf. Model., 2008, 48, 1337–1344 CrossRef CAS PubMed.
- P. K. Ojha and K. Roy, Comparative QSARs for antimalarial endochins: Importance of descriptor-thinning and noise reduction prior to feature selection, Chemom. Intell. Lab. Syst., 2011, 109, 146–161 CrossRef CAS PubMed.
- I. H. Witten and E. Frank, Data mining, practical machine learning tools and techniques Microsoft Research, in The Morgan Kaufmann Series in Data Management Systems, ed. J. Gray, Elsevier, 2nd edn, 2005 Search PubMed.
- M. Hall, E. Frank, G. Holmes, B. Pfahringer, P. Reutemann and I. H. Witten, The WEKA data mining software: an update, SIGKDD Explor. Newsl., 2009, 11, 10–18 CrossRef.
- R. Todeschini and V. Consonni, in Molecular Descriptors for Chemoinformatics, ed. R. Mannhold, H. Kubinyi and G. Folkers, Wiley - VCH, Weinheim, 2009 Search PubMed.
- H. Franco-Lopez, A. R. Ek and M. E. Bauer, Estimation and mapping of forest stand density, volume, and cover type using the k-nearest neighbors method, Rem. Sens. Environ., 2001, 77, 251–274 CrossRef.
- OECD Principles for the validation, for regulatory purposes of (Quantitative) Structure Activity Relationship Models (http://www.oecd.org).
- A. Tropsha, Best practices for QSAR model development, validation, and exploitation, Mol. Inf., 2010, 29, 476–488 CrossRef CAS.
- A. Afantitis, G. Melagraki, H. Sarimveis, P. A. Koutentis, J. Markopoulos and O. Igglessi-Markopoulou, Development and evaluation of a QSPR model for the prediction of diamagnetic susceptibility, QSAR Comb. Sci., 2008, 27, 432–436 CAS.
- S. Zhang, A. Golbraikh, S. Oloff, H. Kohn and A. Tropsha, Novel Automated Lazy Learning QSAR (ALL-QSAR) Approach: Method Development, Applications, and Virtual Screening of Chemical Databases Using Validated ALL-QSAR Models, J. Chem. Inf. Model., 2006, 46, 1984–1995 CrossRef CAS PubMed.
- E. Papa, S. Kovarich and P. Gramatica, Development, Validation and Inspection of the Applicability Domain of QSPR Models for Physicochemical Properties of Polybrominated Diphenyl Ethers, QSAR Comb. Sci., 2009, 28, 790–796 CAS.
- H. Liu, X. Yao and P. Gramatica, The applications of machine learning algorithms in the modeling of estrogen-like chemicals, Comb. Chem. High Throughput Screening, 2009, 12, 490–496 CrossRef CAS.
- V. D. Mouchlis, G. Melagraki, T. Mavromoustakos, G. Kollias and A. Afantitis, Molecular modeling on pyrimidine-urea inhibitors of TNF-α production: An integrated approach using a combination of molecular docking, classification techniques, and 3D-QSAR CoMSIA, J. Chem. Inf. Model., 2012, 52, 711–723 CrossRef CAS PubMed.
- http://enalos.insilicotox.com/QNAR_PaCa2/.
- I. V. Tetko, The perspectives of computational chemistry modeling, J. Comput.-Aided Mol. Des., 2012, 26, 135–136 CrossRef CAS PubMed.
- E. Vrontaki, G. Melagraki, T. Mavromoustakos and A. Afantitis, Exploiting ChEMBL database to identify indole analogs as HCV replication inhibitors, Methods, 2014 DOI:10.1016/j.ymeth.2014.03.021.
- G. Melagraki, A. Afantitis, H. Sarimveis, O. Igglessi-Markopoulou, P. A. Koutentis and G. Kollias, In silico exploration for identifying structure-activity relationship of MEK inhibition and oral bioavailability for isothiazole derivatives, Chem. Biol. Drug Des., 2010, 76, 397–406 CAS.
- E. Bolton, Y. Wang, P. A. Thiessen and S. H. Bryant, PubChem: Integrated Platform of Small Molecules and Biological Activities, in Annual Reports in Computational Chemistry, Elsevier, Oxford, UK, 2008, vol. 4, ch. 12, pp. 217–240 Search PubMed.
- M. Awale, R. van Deursen and J.-L. Reymond, The MQN-Mapplet: Visualization of Chemical Space with Interactive Maps of DrugBank, ChEMBL, PubChem, GDB-11 and GDB-13, J. Chem. Inf. Model., 2013, 53, 509–518 CrossRef CAS PubMed.
- A. D. Zdetsis, Designing novel Sn-Bi, Si-C and Ge-C nanostructures, using simple theoretical chemical similarities, Nanoscale Res. Lett., 2011, 6, 1–9 CrossRef PubMed.
- V. Jain and P. V. Bharatam, Pharmacoinformatic approaches to understand complexation of dendrimeric nanoparticles with drugs, Nanoscale, 2014, 6(5), 2476–2501 RSC.
| This journal is © The Royal Society of Chemistry 2014 |