
Isohexide hydroxy esters: synthesis and application of a new class of biobased AB-type building blocks†
Shanmugam Thiyagarajanab,
Jing Wuab,
Rutger J. I. Knoopa,
Jacco van Haverenab,
Martin Lutzc and
Daan S. van Es*ab
aFood & Bio-based Research, Wageningen University and Research Centre, P.O. Box 17, 6700 AA Wageningen, The Netherlands. E-mail: daan.vanes@wur.nl; shanmugam.thiyagarajan@wur.nl; Fax: +31 317 483011; Tel: +31 317 481160
bDutch Polymer Institute, P.O. Box 902, 5600 AX, Eindhoven, The Netherlands
cBijvoet Center for Biomolecular Research, Crystal and Structural Chemistry, Utrecht University, Padualaan 8, 3584, Utrecht, The Netherlands
First published on 19th September 2014
Abstract
Here we present the synthesis of a new family of sugar derived 1,4:3,6-dianhydrohexitol based AB-type monomers, containing one methyl ester group and a secondary hydroxyl group in all four possible stereo isomers (RR, RS, SR, SS). Structural characterization of the monomers (5a–d) was established by 1D and 2D NMR analysis, which was further confirmed by single-crystal X-ray structure determination. The application of these monomers in step-growth polymerization afforded fully isohexide based stereo-regular polyesters. Homo polyesters based on the RR and RS monomers were obtained with reasonable molecular weights by melt polymerization (Mn 2400 and 2500 resp.). These materials showed unexpectedly low glass-transition temperatures of 20 °C and 15 °C respectively. In contrast, the monomers with SR and SS configuration yielded only low molecular weight oligomers. Surprisingly, copolymerization of the RR and SR monomers gave a polyester with higher molecular weight (Mn 4100) and a high Tg of 80 °C. These preliminary results show that isohexide hydroxyesters are an intriguing new class of biobased building blocks with many potential applications.
Introduction
Reducing our dependency on finite fossil resources for the production of durable synthetic materials is one of the main drivers for the development of chemical building blocks from biomass. Starting from renewable, readily available biobased feed stocks such as polysaccharides, sugars and vegetable oils, many different types of building blocks and polymeric materials based upon them have been developed over the years.1 However, the majority of these compounds have a high intrinsic flexibility, which often makes them unsuitable for application in e.g. engineering plastics. Hence there remains a strong industrial interest in biobased rigid building blocks for high Tg (glass transition temperature) polymers. Sugar derived 1,4:3,6-dianhydrohexitols (or isohexides) are one of the few classes of bifunctional biobased monomers that are known to increase the Tg of step-growth polymers due to their intrinsic rigidity.2 Isohexides are V-shaped bicyclic molecules consisting of two cis connected tetrahydrofuran rings with secondary hydroxyl groups in the 2- and 5-positions.3 The orientation of the hydroxyl groups defines the isomeric form, which is related to the parent hexitol (reduced C6-sugar); i.e. endo–endo (RR, isomannide, from D-mannitol), exo–endo (2R5S, isosorbide, from D-sorbitol), and exo–exo (SS, isoidide, from L-iditol) (Fig. 1).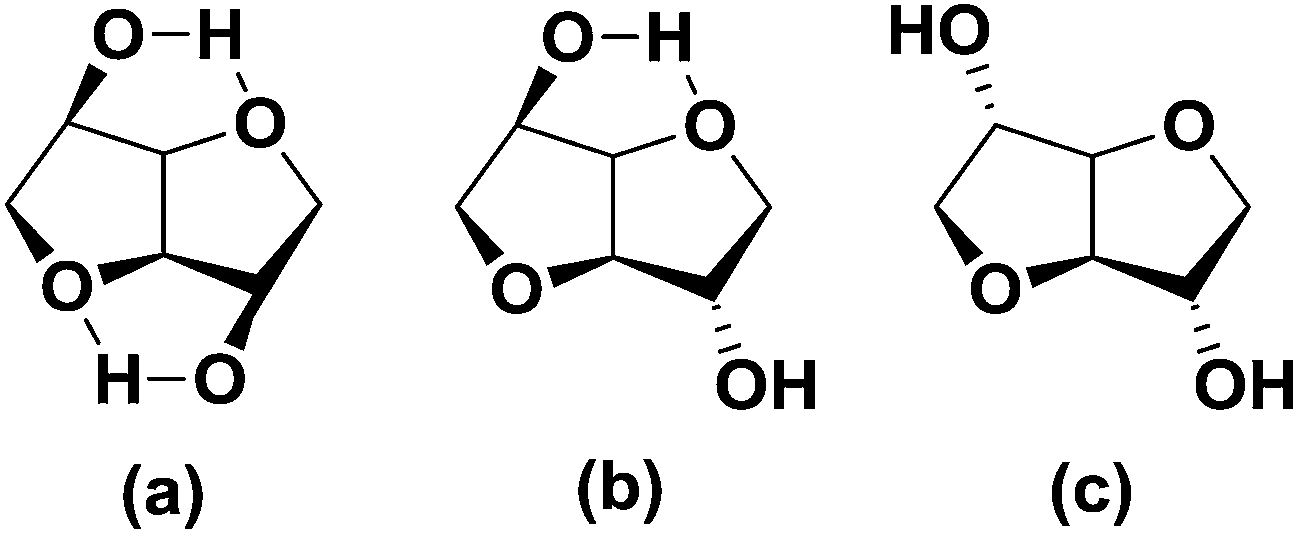 | ||
| Fig. 1 Structure of (a) isomannide, (b) isosorbide and (c) isoidide. | ||
Of these three isomers only isosorbide is produced on commercial scale, since it can be relatively efficiently produced from abundantly available glucose in two steps.3 The orientation of the secondary hydroxyl groups has a strong influence on polymer chemistry and physics. Endo-hydroxyl groups (isomannide/isosorbide) (Fig. 1) are sterically shielded, making it difficult to obtain sufficient molecular weights under mild polymerization reaction conditions. Application of more harsh conditions often results in unwanted side reactions, such as dehydration and/or ring opening, causing discoloration and suppressing molecular weight build-up. Exo-hydroxyl groups (isoidide/isosorbide) on the other hand are sterically more accessible, and therefore more reactive (Fig. 1).2a Whereas isosorbide, due to lack of symmetry, gives stereo irregular polymers, resulting in amorphous materials at high levels of incorporation, the symmetrical isoidide is reported to yield semi-crystalline polymers.4 Unfortunately, since L-idose (the precursor to isoidide) is not produced in any significant levels in plants, historically the availability of isoidide as building block for polymers has been negligible, as are hence examples of isoidide containing polymers. However, recently we have reported a novel efficient method for obtaining isoidide by means of catalytic epimerization of isosorbide, which could stimulate its development as rigid building block.5
Many of the difficulties associated with using isohexides as building blocks in step-growth polymers (low reactivity, low molecular weights, amorphous polymers, discoloration) are associated with the inherent low reactivity of the secondary hydroxyl groups. Several strategies have been pursued to increase the reactivity of isohexides by e.g. functional group substitution or chain extension. Whereas the former approach works well in the case of isohexide diamines or diisocyanates,6 such building blocks cannot be used for polyesters. The success of the latter approach, i.e. chain extension, is strongly depending on the distance between the new reactive group and the isohexide skeleton. We have recently reported on the development of a new family of 1-carbon extended isohexide building blocks (Scheme 1).7 Furthermore we have shown that 1-carbon extension results in increased reactivity, with retention of rigidity, and concomitant increase in Tg.4,8
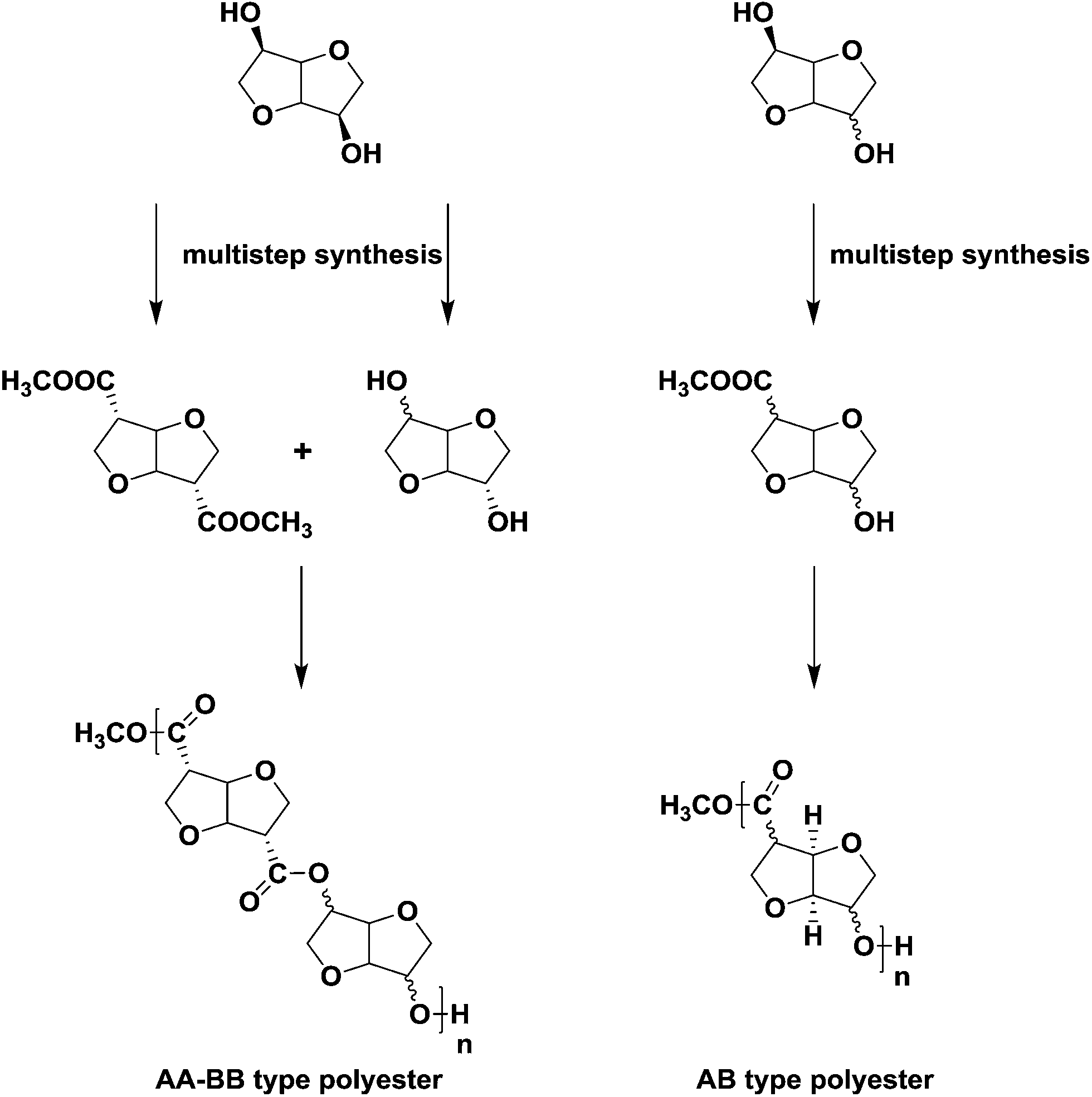 | ||
| Scheme 1 Symmetrical 1-carbon extended AA-BB type building blocks and polyesters derived from them (left side). Stereo-isomers based on AB type building blocks and polyesters derived from them (right side). | ||
Starting from isomannide (Fig. 1a), the AA-type monomer isoidide dimethylcarboxylate (IIDMC) (Scheme 1, left side) is synthesized in 3-steps which involves modification of hydroxyl functionalities into efficient leaving groups (by Tf2O), followed by nucleophilic substitution using cyanide and methanolysis in the last step.7 The similar isoidide (exo–exo) configuration of the AB-type monomer isoidide hydroxy ester (Scheme 1, right side) in the current work is synthesized in 4-steps starting from isosorbide (Fig. 1b). The first-step is selective mono-acetylation of the exo hydroxyl group followed by triflation of the endo hydroxyl functionality in the second-step. In the third-step, inversion of the configuration from sorbide (endo–exo) to idide (exo–exo) is affected by nucleophilic substitution of the triflate group by cyanide. The nitrile and acetate are subsequently solvolyzed to afford the desired monomer. The polyesters were synthesized by a two-step melt-polycondensation procedure as previously reported for AA-BB-type isohexide polyesters.
Combination of isoidide diacid with rigid diols like isosorbide and isoidide yielded fully isohexide based (AA/BB-type) amorphous aliphatic polyesters with high Tg's (73–85 °C), albeit with relatively low molecular weights (Mn 2500). These results prompted the question whether it would be: (a) possible to use the same synthetic methodology to prepare AB-type isohexide hydroxyacids (or hydroxyesters), and (b) whether AB-type polyesters based upon them would be semi-crystalline (opening up possibilities for solid state post condensation in order to increase molecular weight), and (c) what the influence would be of the stereochemistry of the reactive groups.
To the best of our knowledge there are only two reports of AB-type isohexide based monomers and corresponding polymers in the literature. In 1998, Bachmann et al. obtained semicrystalline polyurethanes prepared either by polyaddition (or) polycondensation of the isocyanate-alcohol and amine-carbamic chloride monomers respectively.9 More recently, Besset et al. synthesized α-azide-ω-alkyne dianhydrohexitol stereoisomers from isohexides. Copper catalyzed azide–alkyne cycloaddition (CuAAC) of these monomers, in DMSO and in melt-polyaddition afforded polytriazoles.10
Here we report the synthesis of a novel class of rigid isohexide based AB-type hydroxy-ester monomers in all four possible stereo isomers (RR, RS, SR, SS) and preliminary results on their application in polyester synthesis. It is the objective of this work to investigate the properties of these new building blocks and the polyesters derived from them. The development of industrially viable, and sustainable routes to these monomers is outside of the scope of the present study.
Results and discussion
Monomer synthesis
Building upon the previously developed synthetic methodology for obtaining AA-type 1-carbon extended isohexides, the isohexide hydroxy methylesters (5a–d) were synthesized in 4-steps starting from isosorbide (1a) or isomannide (1b) (Scheme 2). The first step in the reaction sequence was the selective mono-acetylation of isosorbide (1a) and isomannide (1b) using a stoichiometric amount of acetic acid in the presence of a base 4-dimethylaminopyridine (DMAP) and coupling agent, dicyclohexylcarbodiimide (DCC) at 0 °C. Good isolated yields (65–70%) were obtained after purification by column chromatography (silica gel, EtOAc/petr.ether). The desired products isosorbide 2-acetate (2a) and isomannide 2-acetate (2b) were obtained as white crystalline solids in high purity (>95%, from NMR and GC analyss).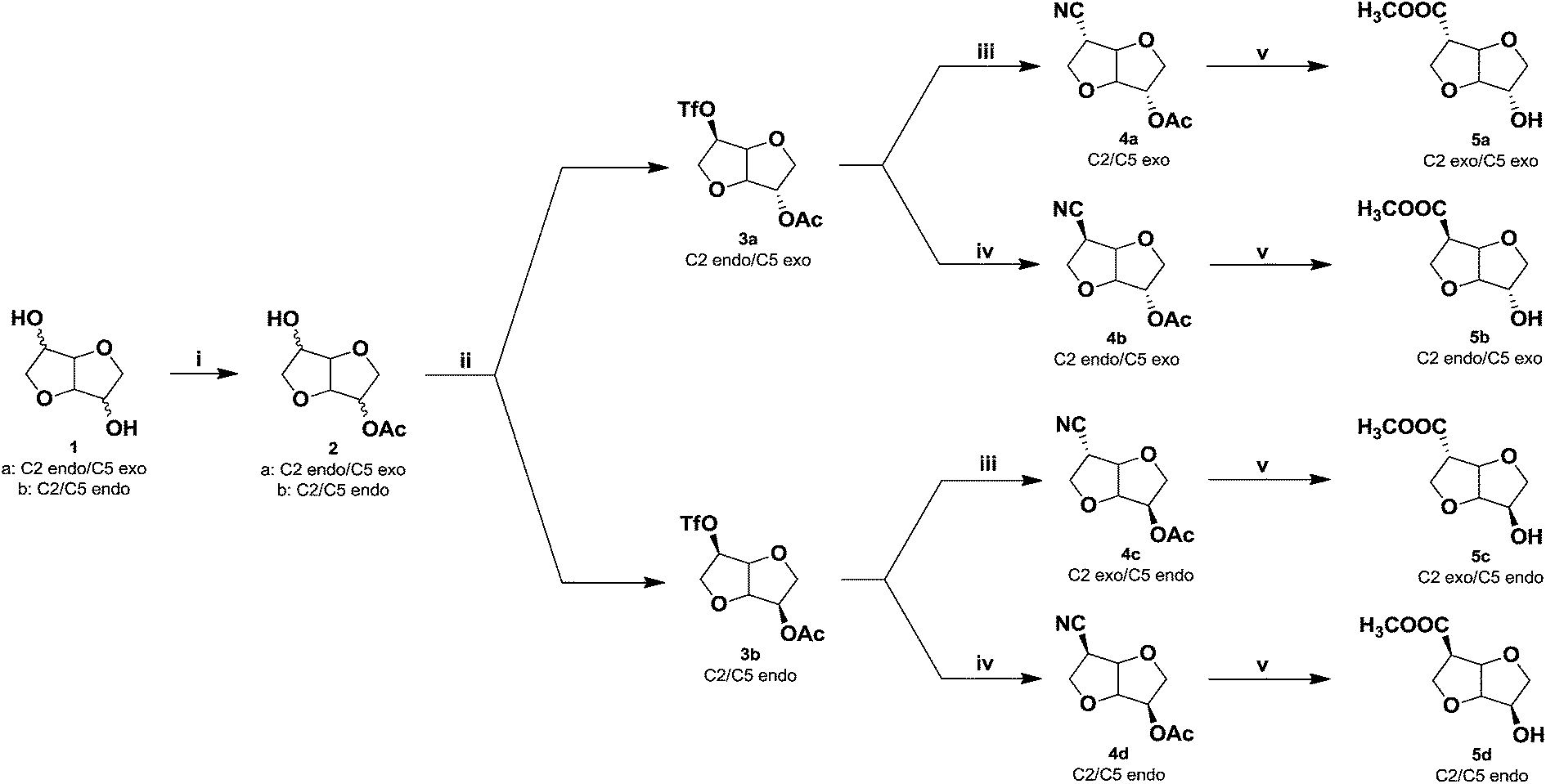 | ||
Scheme 2 Synthesis of the monomers (5a–d). (i) CH3COOH, DMAP/DCC, CH2Cl2, 0 °C, 3 h, 65–70%; (ii) Tf2O, pyridine, CH2Cl2, −10–0 °C, 3 h, 91%; (iii) KCN, 18-Cr-6, dry THF, −5–0 °C, 3 h, 85–92%; (iv) KCN, 18-Cr-6, dry THF, −5–0 °C, 3 h, 40 °C, 1 h, 65%; (v) MeOH–HCl (c) = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture, reflux, 3 h, 85%. :1 mixture, reflux, 3 h, 85%. | ||
Subsequently, the triflate group was introduced in the second step by reacting the isohexide monoacetates (2a & 2b) with trifluorosulfonic anhydride and pyridine in dichloromethane (DCM) at −10 °C. The desired products were obtained in near quantitative yields (>95%). After purification by column chromatography (silica gel, EtOAc/petr.ether), isosorbide 2-acetate-5-triflate (3a) and isomannide 2-acetate-5- triflate (3b) were obtained as a pale yellow crystalline solid and liquid respectively in high isolated yields (90%) and purity (>95%, from NMR and GC analyses). The third step is crucial; i.e. the nucleophilic substitution of the triflate group by cyanide using potassium cyanide and 18-crown-6 in tetrahydrofuran (THF). Strict temperature control (−5° to 0 °C) is required in order to achieve inversion of configuration at C5 affording isoidide nitrile acetate (4a) and isosorbide 5-nitrile-2-acetate (4c), respectively in near quantitative yield (Scheme 2). As we have previously reported, only endo-oriented triflate groups will undergo efficient substitution by cyanide.7
After short path distillation of the crude products using a kugel-rohr oven, the nitrile-acetates (4a & 4c) were obtained in up to 92% isolated yield with high purity (>95%, as determined from NMR and GC analyses). In order to obtain the remaining two stereoisomers, during the third step the reaction temperature was gradually increased to 40 °C for 1 h.
At this temperature, the nitrile group undergoes base induced epimerization yielding isosorbide nitrile acetate (4b) from isoidide nitrile acetate (4a) and isomannide nitrile acetate (4d) from isosorbide nitrile acetate (4c) in 65% yield. Products (4b & 4d) were obtained in high purity by short path distillation using a kugel-rohr oven (>95%, from NMR and GC analyses). In the fourth and final step, the nitrile and acetate groups in the product (4a–d) were solvolyzed in a methanol–conc. HCl (1:1) mixture at reflux to afford the respective hydroxyl methylesters. Thus, the stereo-isomers isoidide hydroxy methylester (5a)-RR, isosorbide hydroxy methylester (5b)-RS, isosorbide hydroxy methylester (5c)-SR and isomannide hydroxy methylester (5d)-SS were obtained in high yields (85%). Short-path distillation of the crude monomers (kugel-rohr oven) afforded the products (5a–d) in high purity (>95%, from NMR and GC analyses) (Fig. 2). Thus, all the monomers were obtained in 4-steps in excellent average isolated overall yields of 33%. Full structural characterization of the novel monomers (5a–d), the known precursors (2a & 2b), and the novel intermediates (3a & 3b) and (4a–d) was established by FTIR, 1H NMR, 13C NMR, 2D-NMR as well as HRMS spectroscopy (see ESI†). Since the orientation of the functional groups on the isohexide rings was expected to have considerable influence on both polymer chemistry as well as polymer properties, special attention was given to the structural characterisation of (5a–d). Due to the rigidity, 1H NMR spectroscopy is an excellent technique for determining the orientation of substituents in isohexides.11 The 1H NMR spectra of the pure monomers (5a–d) are shown in Fig. 2.
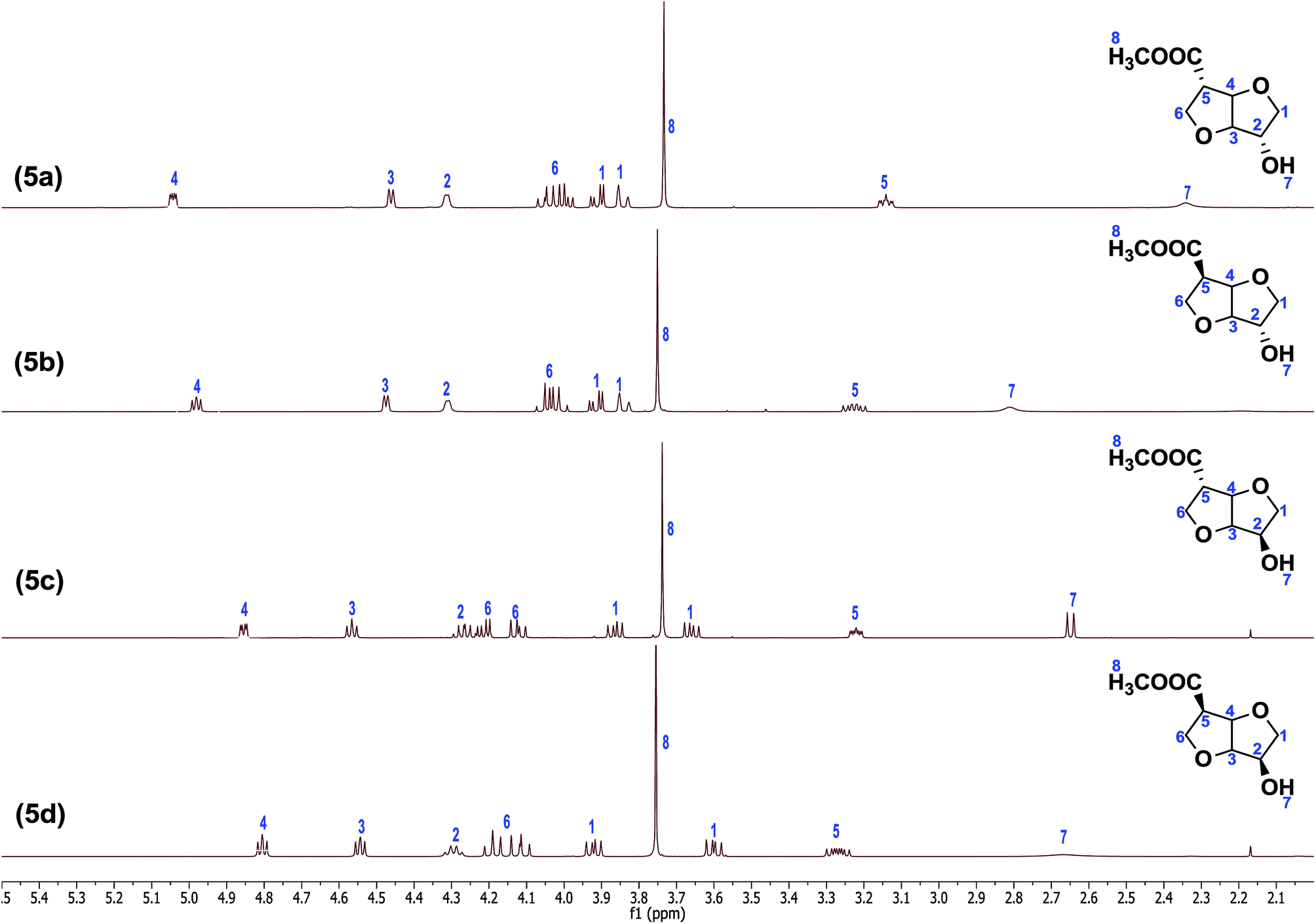 | ||
| Fig. 2 1H NMR spectra of the monomers (5a–d) in CDCl3. | ||
Despite the lack of symmetry in (5a–d), structural assignments are relatively straightforward, building on previous NMR characterization of the parent isohexides. In the exo–exo (RR) isomer (5a), the bridgehead protons at C3 and C4 have no clear dihedral coupling with the respective protons at C2 and C5, resulting in a quasi-doublet multiplicity due to chemical in-equivalence of H3 and H4 (as can also be observed from the 2D NMR spectra, see ESI†). Whereas the proton at C2 has only a small dihedral coupling with one of the protons of the C1 AB-system, the proton at C5 appears to have comparable coupling constants with the AB-system at C6, and therefore comparable dihedral angles. When going from (5a) to (5b) the stereochemistry at C5 is inverted, which mainly affects the protons at C4, C5 and C6.
Inversion results in a change of dihedral angles, and thus multiplicities, and to a lesser extent in chemical shifts. The bridge proton at C4 shows a small downfield shift, while the multiplicity changes to a quasi-triplet structure due to the coupling with the proton at C5, which also displays a radical change in multiplicity, combined with a small downfield shift. Inversion at C2, giving (5c), results in more dramatic changes in chemical shift compared to (5a). Complete inversion at both C2 and C5, giving (5d), results in a spectrum that is relatively comparable to that of (5c). While the bridge proton at C4 has shifted even more upfield, the reverse is true for the proton at C2. Remarkable is also the increase in chemical shift difference between the protons of the AB-system at C1.
All four hydroxy esters (5a–d) were obtained as colorless transparent oily liquids. In order to confirm the structural characterization by NMR spectroscopy, a single crystal X-ray structure determination was performed on the hydroxyl-acid derivative (6c)-SR, obtained by the hydrolysis of hydroxy ester (5c) in 3 N HCl. The absolute configuration of (6c) is shown in Fig. 3.
 | ||
| Fig. 3 Molecular structure of (6c) in the crystal. Displacement ellipsoids are drawn at the 50% probability level. | ||
Synthesis and characterization of polyesters
Fully isohexide-based AB-type polyesters were synthesized by a two-step melt-polycondensation procedure (Scheme 3) as previously reported for AA-BB-type isohexide polyesters.4,8a During the first stage, the reaction is carried out under nitrogen gas to form oligomers. The reaction mixture is heated to 115 °C, and after 15 min the catalyst dibutyltin oxide (DBTO, 10 mol%) is added to the flask under a continuous flow of nitrogen gas. The temperature is then increased to 160 °C under stirring for 12 h. At this stage the reaction mixture became opaque and solidified. Finally the temperature was raised to 180 °C for 2 h to complete the pre-polymerisation reaction. In the second stage, vacuum was applied gradually down to 0.02 mbar at 180 °C for 2 h. After completion, the reaction mixture was cooled down to room temperature under nitrogen atmosphere. The polymers were purified by dissolution in 5 mL of chloroform/TFA (6:1) followed by precipitation in 75 mL of methanol yielding white to yellow coloured amorphous powder-like materials. Typical yields were in the range of 70–80%. The structures of the obtained polyesters were confirmed by 1H, 13C, and 2-D NMR spectroscopy.
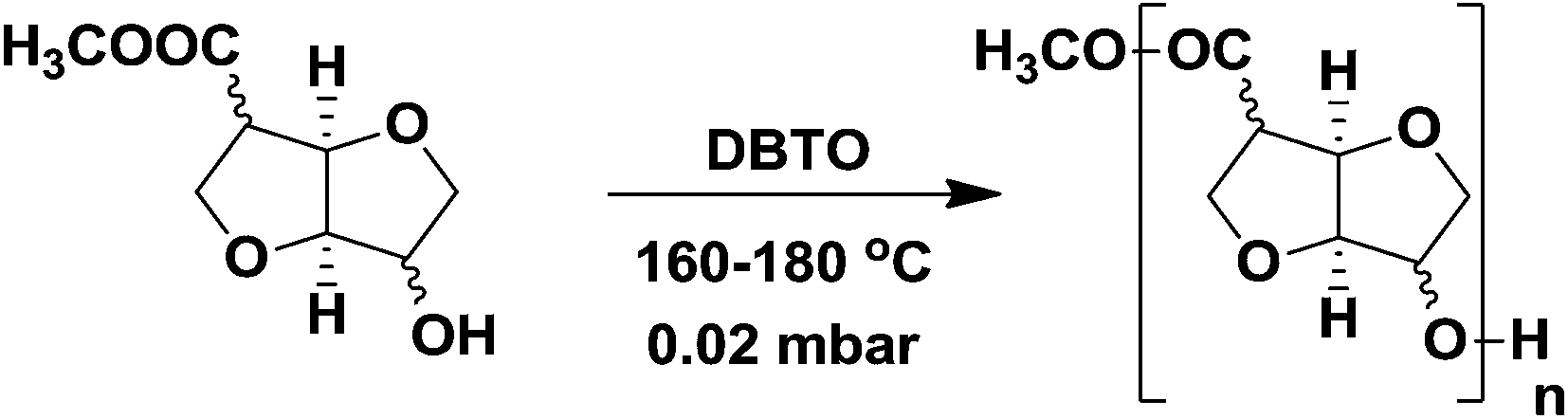 | ||
| Scheme 3 Polymerization of stereo-isomeric monomers yielding aliphatic polyesters. [a] (5a): C2/C5 exo; [b] (5b): C2 endo/C5 exo; [c] (5c): C2 exo/C5 endo; [d] (5d): C2/C5 endo. | ||
The homo-polymerization of the exo-OH monomers (5a)-RR and (5b)-RS afforded the fully isohexide based polyester PE-1 and PE-2 respectively in good yield and with acceptable molecular weights (Table 1). Striking is the very low PDI of these polymers, suggesting low conversion. These results are highly comparable to those obtained with the AA-BB-type polyester based on isosorbide/isoidide and isoidide dicarboxylic acid.4
| Entry | Monomer | GPCa | 1H NMR | Yielde | Colourf | Appearencei | ||
|---|---|---|---|---|---|---|---|---|
| Mn (g mol−1)b | Mw (g mol−1)c | PDId | Mn (g mol−1) | |||||
| a Gel Permeation Chromatography performed using HFIP as solvent.b Mn number-average molecular weight.c Mw weight-average molecular weight.d PDI polydispersity index.e Isolated yield after precipitation in % relative to theoretical maximum.f Colour of the polyester after precipitation.g Pale yellow.h Deep yellow.i Appearence of the crude polyester in the reactor.j Opaque.k Transparent.l Translucent.m Mn could not be calculated due to overlapping signals. | ||||||||
| PE-1 | 5a (RR) | 2400 | 2500 | 1.04 | 2500 | 83 | p.y.g | Op.j |
| PE-2 | 5b (RS) | 2500 | 2700 | 1.08 | 2300 | 76 | p.y. | Op. |
| PE-3 | 5c (SR) | 770 (oligomers) | 1000 | 1.29 | — | 40 | Brown | Op. |
| PE-4 | 5d (SS) | 790 (oligomers) | 980 | 1.24 | — | 35 | Brown | Op. |
| PE-5 | 5a:5c (RR:SR) | 4100 | 10500 |
2.55 | n.c.m | 89 | d.y.h | Tp.k |
| PE-6 | 5a:5d (RR:SS) | 2700 | 11100 |
4.10 | n.c. | 78 | Brown | Tl.l |
In contrast the endo-OH isomers (5c)-SR and (5d)-SS yielded only low MW oligomeric products (PE-3 and PE-4) under the typical polymerization conditions. Again, a very low PDI was obtained. The low reactivity of these monomers is also apparent from the low isolated yields after precipitation (Table 1). Increasing the temperature in order to increase reactivity and molecular weight build-up proved to be detrimental, resulting in severe discoloration and gel formation, comparable to previously reported results with the AA-BB system.4
Clearly the orientation of the hydroxyl group has a dramatic influence on the reactivity of the monomers. Whereas (5a) and (5b) with exo-OH functionalities show comparably moderate reactivity, monomers (5c) and (5d) with endo-OH groups are much more unreactive. In contrast, the orientation of the methylester substituent does not appear to have a significant influence on reactivity.
The reactivity of the mixture of stereo-isomers was also investigated by co-polymerisation of the more reactive (5a)-RR isomer with the poorly reactive isomers (5c)-SR and (5d)-SS. The combination of monomers (5a) and (5c) in a 1:1 ratio surprisingly resulted in an increase in molecular weight as well as in PDI. The high isolated yield of 89% of co-polyester PE-5 after precipitation proves that a significant amount of unreactive monomer (5c) is incorporated into the polymer. In contrast, combination of monomers (5a) and (5d) (1:1 ratio) resulted in a dramatic increase in the PDI, indicating branching had occurred (mind however that these data are from the precipitated polymer, so the high PDI material was still soluble in CHCl3/TFA). Representative 1H NMR spectra of PE-1 and PE-2 are given in Fig. 4 (GPC profiles are included in the ESI†).
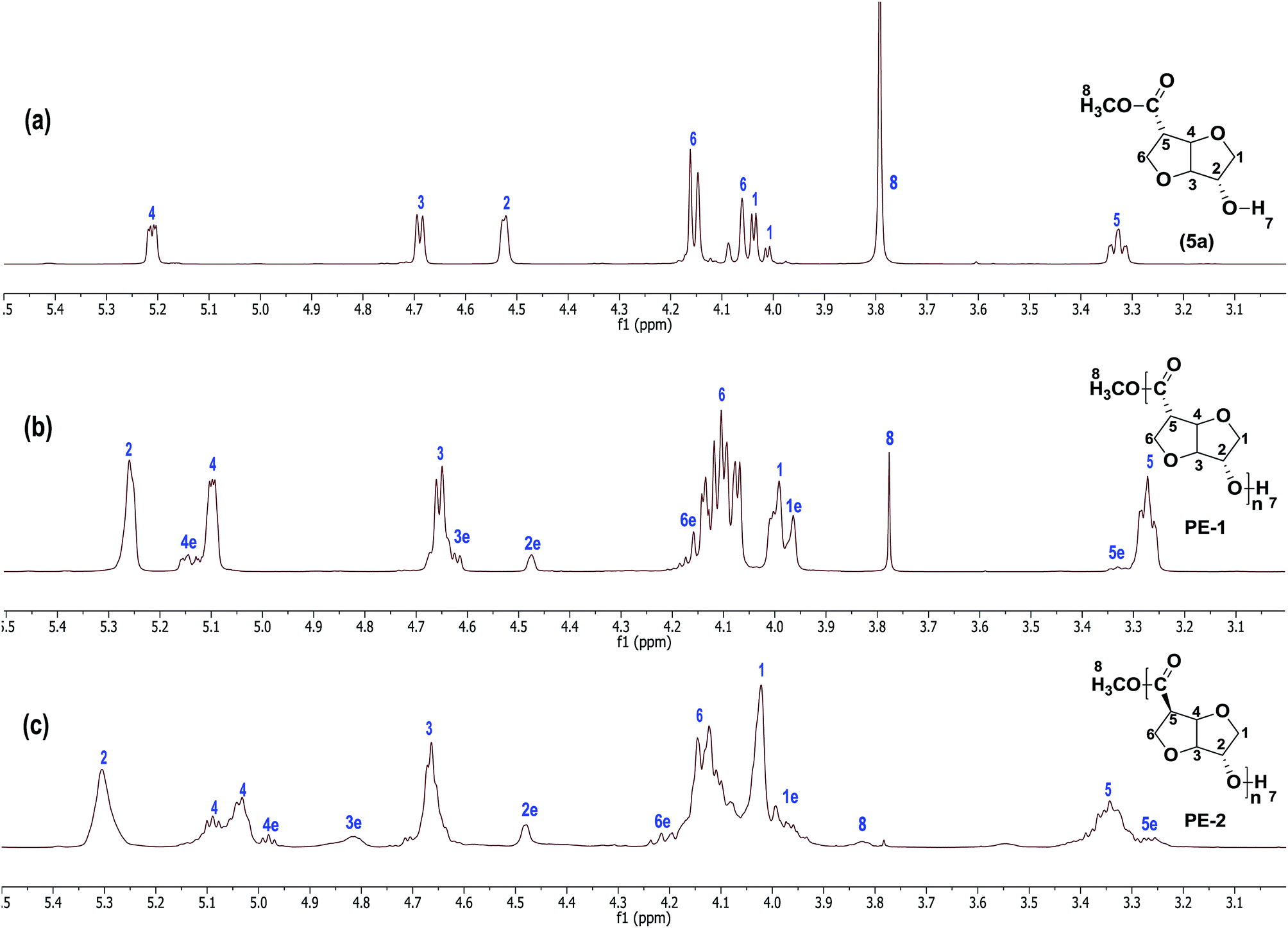 | ||
| Fig. 4 1H NMR spectra of (a) monomer (5a); (b) PE-1 and (c) PE-2, recorded in CDCl3/TFA (trifluoroacetic acid-d) (6:1) mixture. The numbers 1–7 in the 1H NMR spectra denote the corresponding protons (as indicated in the structure) and numbers 1e–7e refer to the end groups present in the polyester. | ||
1H NMR spectroscopy is a powerful tool to characterize the structure of stereo isomers. Since the 1H NMR spectra of homopolymers PE-1 and PE-2 show clear and distinctive signals, characterisation is relatively simple. For instance in the case of PE-1, the proton at C1 has a negligible dihedral coupling with the bridge proton at C2, indicative of an isoidide configuration. Similarly, the proton at C4 has no significant dihedral coupling with the bridge proton at C3, confirming retention of the isoidide configuration at C4. Hence, the stereochemistry of the monomer (5a) is retained in the polymer. The low intensity signals designated with an “e” belong to the end-group. Remarkable is the upfield shift of over 1 ppm of the proton at C4 in the end-groups compared the same proton in the repeating units in the polymer. Due to severe signal overlap, the 1H NMR spectra of copolyesters PE-5 and PE-6 are too complicated to allow for quantification of the incorporation ratio of the two co-monomers in the copolyesters (see ESI†).
The new polyesters were also characterized by MALDI-ToF-MS. Representative mass spectra of homopolymers PE-1 and PE-2, as well as copolymer PE-5 are shown in Fig. 5. The mass spectra of PE-1, PE-2 and PE-5 show that the linear polymer (hydroxy and ester terminated) is the predominant species (n*repeating unit of 156 D + MeOH). Furthermore, in the case of PE-1 and PE-5 macrolactones (n*repeating unit) can be observed (designated “(b)” in Fig. 5). In contrast, the mass spectrum of PE-2, does not show any macrolactones. This can be rationalized by the more linear character of monomer (5b) compared to (5a), which would more easily allow for the formation of cyclics for the latter. The mass spectrum of PE-1 also shows repeating signals for a species corresponding to the linear polyester (a) + 28 a.u. This would correspond to the tentative structure (c), a formate terminated linear polyester, as shown in Fig. 5.
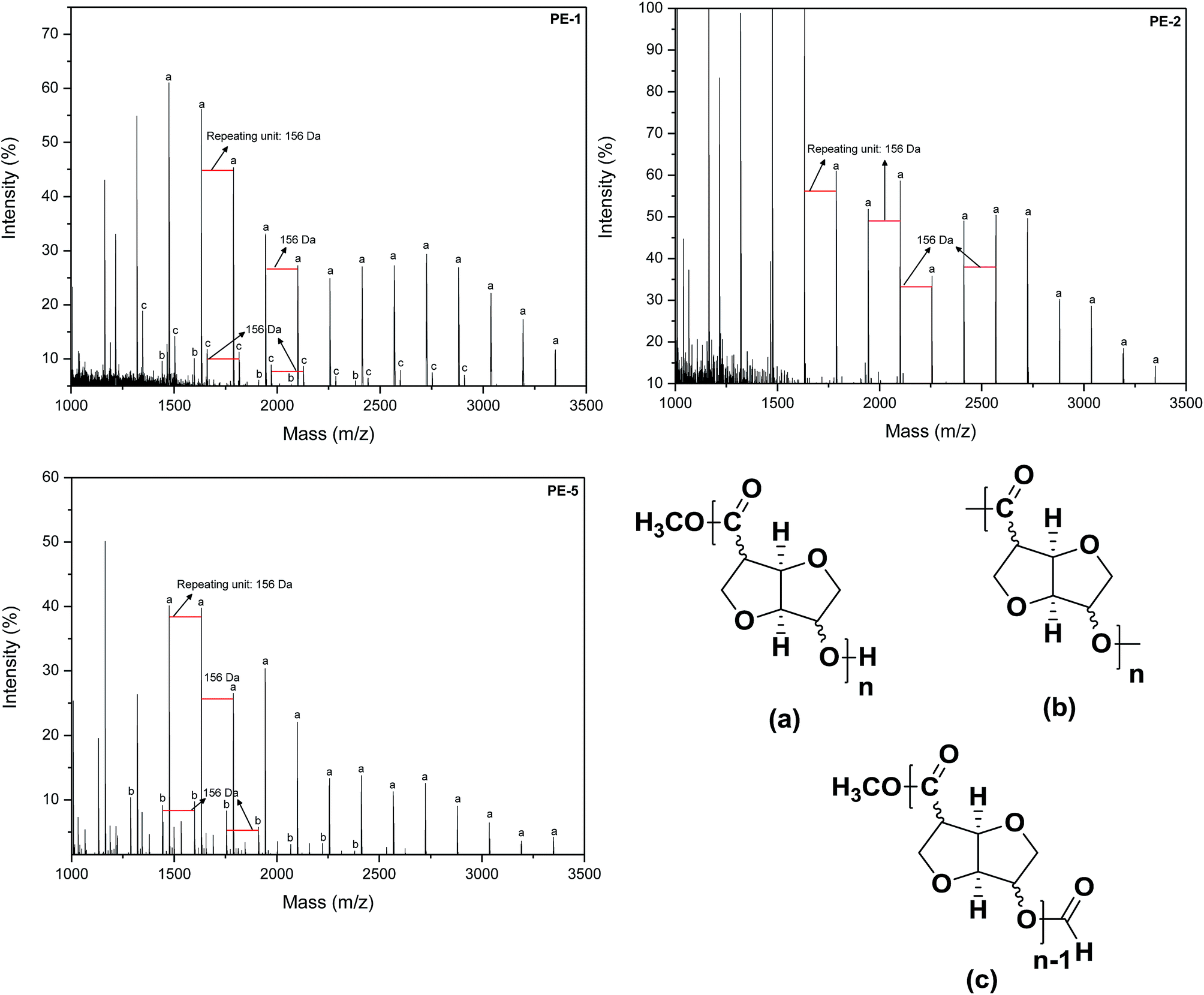 | ||
| Fig. 5 Sections of the MALDI-ToF-MS spectra of (i) homopolymer PE-1 (RR); (ii) homopolymer PE-2 (RS) and (iii) copolymer PE-5 (RR-SR), signals cationised with K+: (a) linear polyester with methyl ester and hydroxyl end-groups; (b) macrolactone; (c) tentative structure for signal observed in PE-1 spectrum. | ||
Thermal properties of polyesters
Thermal properties of the new homo- and co-polyesters were determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The poor reactivity of the endo-hydroxyl groups in monomers (5c) and (5d) resulted in the oligomeric products only; hence the thermal properties of PE-3 and PE-4 will not be discussed here. TGA measurements were performed from 30 °C to 600 °C under nitrogen atmosphere (Fig. 6). The temperature at 5% weight loss and the maximum decomposition rates are shown in Table 2.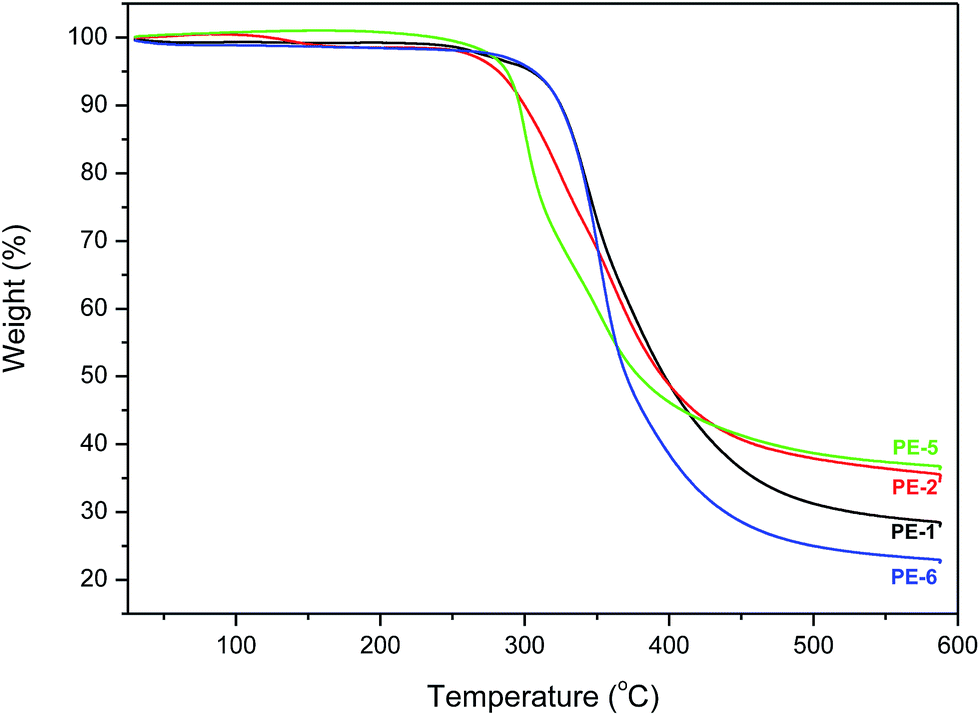 | ||
| Fig. 6 TGA traces of homopolyesters PE-1 and PE-2, and co-polyesters PE-5 and PE-6 are recorded from 30 to 600 °C at 10 °C min−1 under N2 atmosphere. | ||
| Polymer | TGAa | DSCd | Dh (%) | ΔHmi (J g−1) | ΔH0m (J g−1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| First heating | Second heating | |||||||||
| T5% (°C)b | Tmax (°C)c | Tg (°C)e | Tm (°C)f | ΔHm (J g−1) | Tg (°C)e | Tm (°C)f | ||||
| a TGA was performed from 30 °C to 600 °C under nitrogen atmosphere.b Temperature at 5% weight loss.c Temperature at maximum rate of decomposition.d DSC was performed from −60 °C to 200 °C with two heating and cooling runs at 10 °C min−1.e Glass-transition temperature.f Melting temperature.g No melting endotherm was observed up to the maximum temperature (200 °C), however STA analyses showed a melting endotherm at 286 °C.h Degree of crystallinity from WAXD.i From DSC analysis.j Fully amorphous. | ||||||||||
| PE-1 | 275 | 460 | 24 | — | — | 20 | —g | 47 | — | — |
| PE-2 | 253 | 419 | 16 | 172 | 29.5 | 15 | — | 22 | 29.5 | 134 |
| PE-5 | 277 | 428 | 93 | 177 | 3.2 | 80 | — | —j | 3.2 | — |
| PE-6 | 259 | 401 | 30 | — | — | 24 | — | |||
| P-δ-VL12 | — | — | −66 | 58 | — | — | — | — | — | 182 |
| PIsI (AA-BB)4 | 274 | 310/382 | 75 | 125–168 | 4.3 | 73 | — | — | — | — |
| PIiI (AA-BB)4 | 286 | 308/384 | — | 109–143 | 15.4 | 85 | — | 32 | 15.4 | 48 |
| PLLA | — | — | — | — | — | — | — | — | — | 90 |
TGA analysis shows that despite their relatively low molecular weights the homopolymers PE-1 and PE-2 are still quite thermally stable. The difference between the TGA curves of PE-1 and PE-2 is striking, given the fact that both polymers are very similar with regard to molecular weight and PDI. PE-2 has a significantly lower T5% and Tmax compared to the isomeric PE-1. Compared to their AA-BB type analogues reported by Wu et al. PE-1 and PE-2 appear to be have a higher Tmax, as well as higher residual masses. Furthermore, while the AA-BB type polyesters display distinct 2-step decomposition mechanisms, the AB analogues show a single stage decomposition.
Comparison of the thermograms of copolyesters PE-5 and PE-6 shows interesting differences. PE-5 shows 2-stage decomposition behavior versus single stage for PE-6. Furthermore, while the initial thermal stability of PE-6 appears higher (which could be related to the higher MW) PE-5 yields almost 20% more residual weight at 600 °C, indicating significantly different decomposition mechanisms. Comparison of the TGA results of the homo- and copolyesters shows that unfortunately there is no clear relationship between TGA data and isomeric composition of the polymers.
Differential scanning calorimetry (DSC) of the polyesters gave highly surprising results (Table 2). Previously Wu et al. reported a Tg of 85 °C for the semi-crystalline AA-BB-type polyester of isoidide and isoidide dicarboxylic acid (PIiI, Table 2, Mn 2500, PDI 1.4). Unexpectedly, the AB-type analogue PE-1 showed a Tg of only 20 °C (see also Fig. 7). Furthermore, no melting endotherm was observed for PE-1, neither in the first nor in the second heating DSC run measured upto 200 °C. However, STA analyses (Simultaneous Thermal Analyzer - combined analyses of TGA and DSC to a single sample at same time) revealed an endotherm at 286 °C (onset temperature), which is rapidly followed by the degradation of the polymer (see ESI†). Unfortunately, increasing the polymerization temperature above 180 °C, in order to accommodate for the high melting point, resulted in severe discoloration of the polymers. The visual observation of PE-1 during melt polymerization indicates semi-crystallinity, since the material becomes opaque during reaction.
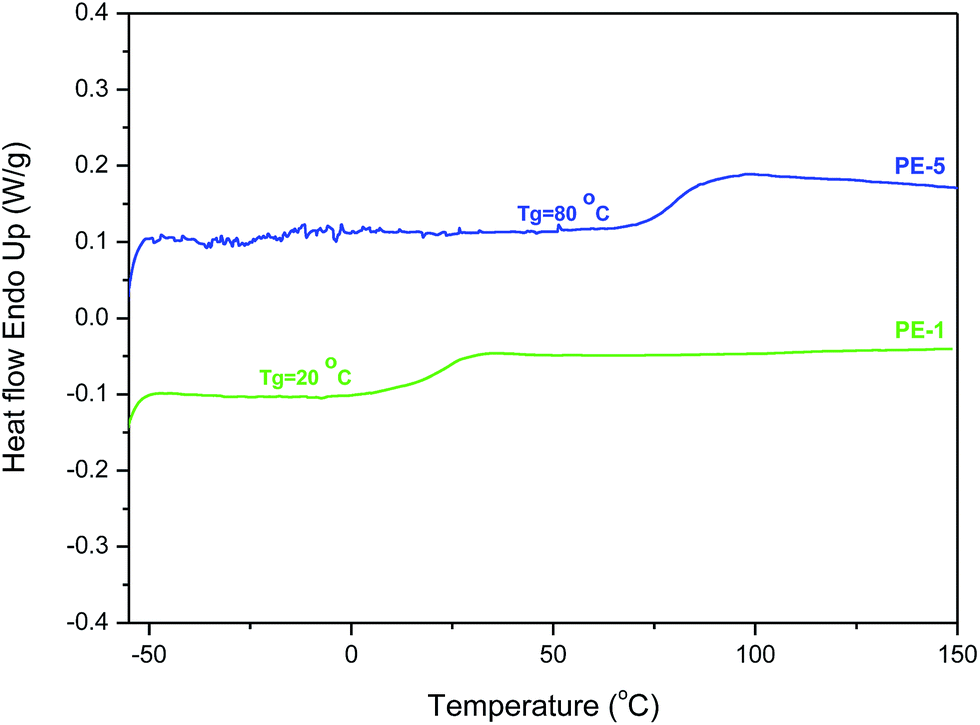 | ||
| Fig. 7 Second heating DSC curves of homopolymer PE-1 and copolymer PE-5. | ||
In contrast, the isomeric PE-2 did show melting behavior in the first run, but yet also an unexpectedly low Tg value of 15 °C. The melting point at 172 °C is somewhat comparable to the value of 125–168 °C reported by Wu et al. for the polyester of isosorbide and isoidide dicarboxylic acid (PIsI, Table 2). Despite a significant melting enthalpy of approx. 30 J g−1, the absence of a melting endotherm in the second heating run (as well as (cold) crystallization) indicates a low rate of crystallization for this polymer. Nevertheless, the Tg enhancing effect of the bicyclic isohexide skeleton is evident by comparison with the conformationally unrestricted analogue poly-δ-valerolactone, which results in an increase of Tg of 81–86 °C.12
Even more surprising than the low Tgs of PE-1 and PE-2 is the Tg value of 80 °C recorded for co-polyester PE-5, which is fully in line with the results of Wu et al. Also the observation of a melting endotherm (first run only, very low melting enthalpy) is unexpected since a high degree of incorporation of monomer (5c) in PE-5 is expected to result in a stereoirregular, and hence amorphous polymer. The observed low Tg for PE-6 is in line with that of homopolyester PE-1. In contrast to PE-1, co-polyester PE-6 does not have semi-crystalline character, which would be expected from the high PDI value.
Wide-angle X-ray diffraction
In order to address some of the intriguing questions that arose from the DSC analysis, preliminary Wide-Angle X-ray Diffraction (WAXD) measurements were performed on homopolyesters PE-1 and PE-2, as well as on co-polyester PE-5 (Fig. 8).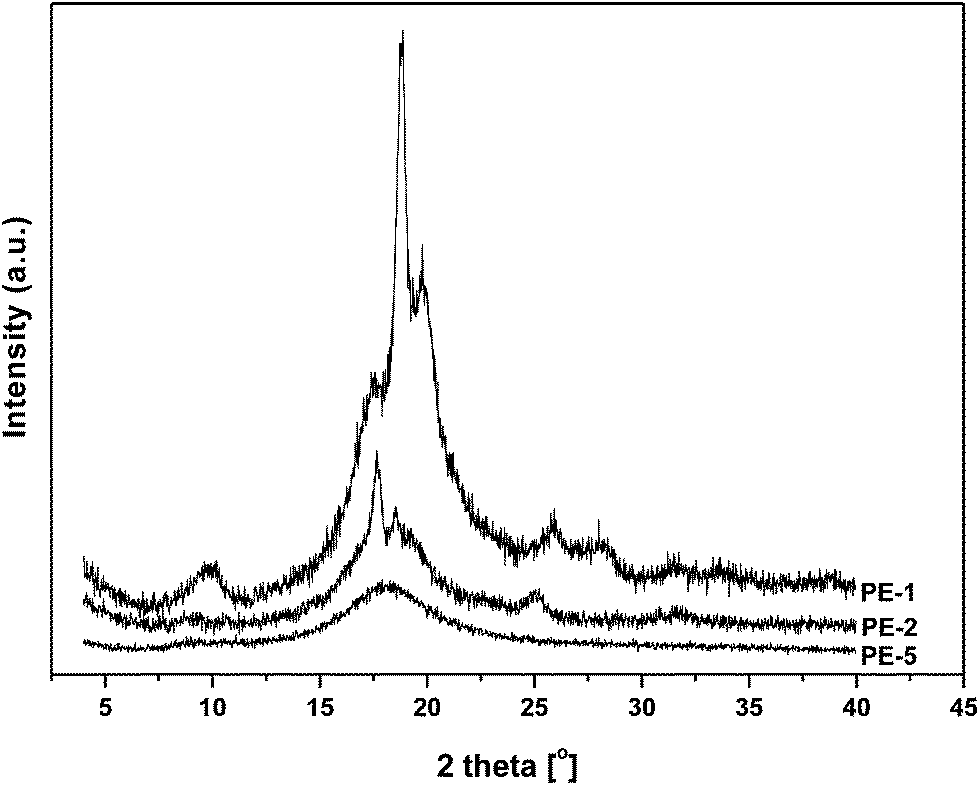 | ||
| Fig. 8 Wide angle X-ray powder diffraction profiles of PE-1, PE-2 and PE-5. | ||
The XRD diffractogram of PE-1 clearly shows a high degree of crystallinity, which is in agreement with the visual observation of high opacity of the crude material obtained from the melt polymerization. According to DSC analysis no melting occurred up to 200 °C, while TGA-DSC analysis showed a melting endotherm at 286 °C. Hence, the highly crystalline PE-1 apparently has a melting point very close to the decomposition temperature. The diffractogram of PE-2 also indicates crystallinity, which is in agreement with the observed melting endotherm (and significant melting enthalpy) in the DSC analysis. In contrast, WAXD analysis of copolyester PE-5 merely shows an amorphous halo, which is consistent with the DSC data, showing only a very low melting enthalpy of 3.2 J g−1.
From the WAXD diffractograms the degree of crystallinity of polyesters PE-1 and PE-2 were derived (Table 2). PE-1 displayed a high degree of crystallinity, which is in agreement with the observation that the material is opaque. Since according to DSC and TGA-DSC analysis the melting point is close to the decomposition temperature no heat of fusion can be calculated. In contrast, since PE-2 did show clear melting behavior and a relatively low degree of crystallinity, the estimated heat of fusion is 134 J g−1 which is significantly higher than the calculated value for the AA-BB analogue reported by Wu et al. When compared to other AB-polyesters it is clear that the heat of fusion of PE-2 is similar to that of its straight-chain analogue P-δ-VL, yet significantly higher compared to e.g. poly-L-lactic acid (PLLA).
Combining the available data (GPC, DSC and WAXD) we propose that the anomalously low glass-transition temperatures observed for PE-1 and PE-2 find their origin in the relatively low Mn and PDI (degree of polymerisation = 15–16), combined with a high degree of crystallinity. As a result, the amorphous phase will have a high concentration of end-groups (and hence free-volume), as well as insufficient possibilities for chain entanglement. Wu et al. reported Tg values of 85 °C and 73 °C for the AA-BB analogues of PE-1 and PE-2 respectively. Although the Mn and PDI values of those materials are comparable to those of PE-1 and PE-2, the observed melting enthalpies are significantly lower, while WAXD analysis also indicated a lower degree of crystallinity (32% for the isoidide polyester, amorphous for isosorbide). Increasing the molecular weight of the homopolyesters PE-1 and PE-2 by either adapted melt polymerization techniques or SSPC is therefore expected to have significant effects on the Tg.
Conclusions
We have successfully synthesized and characterized a new type of 1-carbon extended isohexide based hydroxy esters. Careful control of the reactions conditions allowed us to obtain all four possible isomers in four steps in satisfactory yields and high purity. The influence of the orientation of the substituents on the isohexide skeleton on polymerization of these monomers is dramatic. Whereas the orientation of the carboxyl group has only minor influence on reactivity, the orientation of the hydroxyl group has a profound effect. Comparable to the parent isohexides, endo-hydroxyl groups are detrimental to reactivity and thermal stability. The molecular weights obtained of the AB-type homo polyesters of the exo-OH monomers (5a) and (5b) are highly comparable to those reported for the analogous AA-BB type polyester. In contrast, the thermal stabilities of the AB-type homopolyesters are higher than those of the AA-BB analogues. Surprisingly, the homopolymers obtained from monomers (5a) and (5b) showed unexpectedly low Tgs, especially when compared to their AA-BB analogues. Co-polymerization of AB-monomers (5a) and (5c) resulted in a significant increase in MW of the ensuing polyester compared to the homopolymer. The significantly increased Tg of the copolyester is comparable to that of the AA-BB analogue. (TGA) DSC and WAXD analysis of the homopolymers PE-1 and PE-2 shows that these materials are highly crystalline with high melting enthalpies, and significantly higher melting points than the AA-BB analogues. Our preliminary polymerization results show that these new rigid biobased hydroxyesters have intriguing properties that warrant further research.Experimental section
Materials
Isomannide (Sigma-Aldrich), isosorbide (ex-Roquette), acetic acid (Merck, p.a.), 4-(dimethyl amino) pyridine (DMAP, reagent plus 99% Sigma-Aldrich), N,N′-dicyclohexylcarbodiimide (DCC, 99% Sigma-Aldrich), trifluoromethanesulfonic anhydride (TFA, ≥99%, Aldrich), dichloromethane (DCM, Merck, p.a.), pyridine (Merck, p.a.), hydrochloric acid (reagent grade, 37%, Sigma-Aldrich), tetrahydrofuran (THF, anhydrous, ≥99.9%, Sigma-Aldrich), potassium cyanide (extra pure, Merck), 18-Crown-6 (≥99%, Fluka), chloroform (Merck, p.a.), methanol (Merck, p.a.), petroleum ether (PE, Acros Organics, 40–60 °C), ethyl acetate (Acros Organics, 99+%), silica gel (Alfa Aesar, 230–400 mesh), chloroform-D (99.8 atom% D Aldrich), activated carbon (Norit, CN1), magnesium sulfate (Acros Organics, 99% extra pure, dried, contains 3 to 4 moles of water), Celite® 545 coarse (Fluka). All chemicals were used as received, unless denoted otherwise.Methods
Fourier transform infrared (FT-IR) spectra were obtained on a Varian Scimitar 1000 FT-IR spectrometer equipped with a Pike MIRacle ATR Diamond/ZnSe single reflection plate and a DTSG-detector. The measurement resolution was set at 4 cm−1, and the spectra were collected in the range 4000–650 cm−1 with 32 co-added scans. NMR spectra were recorded on a Bruker Avance III spectrometer operating at 400.17 MHz (1H) and 100.62 MHz (13C). Gas chromatography was performed on an Interscience Focus GC equipped with an AS 3000 series auto sampler. Injection volume 1 μL. Injector temperature 275 °C. Split ratio 1:33. Column flow (at 275 °C) 50 mL min−1 helium. GC column: Restek Rxi-5ms, 30 m × 0.25 mm × 0.25 μm. GC program (2,5-FDA.mth); hold 2 min at 70 °C, ramp 10 °C min−1, final temperature 300 °C, hold 2 min. Total run time 27 min. Detector; FID at 300 °C. GC-MS analysis was performed on a Interscience TraceGC Ultra GC with AS3000 II autosampler (He carrier gas, flow 1 ml min−1, split flow 20 ml min−1; Restek GC column Rxi-5ms 30m × 0.25 mm × 0.25 μm; GC program hold 3 min at 50 °C, ramp 7.5 °C min−1, final temperature 330 °C) connected to a Interscience TraceDSQ II XL quadrupole mass selective detector (EI, mass range 35–500 Dalton, 150 ms sample speed). Electrospray Ionisation (ESI) mass spectrometry was carried out using a Bruker micrOTOF-Q instrument in positive ion mode (capillary potential of 4500 V). Melting points were measured on a Thermal Fisher Scientific IA 9000 Series digital melting point apparatus. Differential Scanning Calorimetry (DSC) measurements were conducted on a Perkin Elmer Diamond series DSC. The temperature range used was 20 °C up to 200 °C at a heating rate of 10 °C min−1. Thermogravimetric analysis (TGA) was measured with an STA 6000 (Simultaneous Thermal Analyser) from Perkin Elmer Instrument. The samples were heated from 30 to 600 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 40 mL min−1. Short path distillations were carried out using a Buchi Glass Oven B-585 (Kugel-rohr). Gel Permeation Chromatography (GPC) was performed on a Viscotek HP-SEC system, VE-2001 GPCmax (pump and auto sampler) equipped with TDA305 Triple Detector Array (Right Angle Light Scattering (RALS) + Low Angle Light Scattering (LALS), Refractive Index (RI) Detector and Viscometer. 2x GPC column PSS PFG analytical linear M and guard column, molecular range ∼250–2.5 × 106 D (PMMA in HFIP). Data were calculated with OmniSECTM, Version 4.6 software. Hexafluoroisopropanol (HFIP) containing 0.02 M potassium trifluoroacetate was used as eluent with a flow-rate of 0.7 ml min−1. PMMA samples (60 K and 95 K, viscotek Polycal TDS) were used to calibrate the SEC setup. Control measurements were performed with Easyvial PMMA standards from Agilent. Wide angle X-ray scattering (WAXS) powder diffractograms were recorded on a Philips PC-APD diffractometer in the reflection geometry in the angular range 4–40° (2θ), with a step size of 0.02° (2θ) and an acquisition time of 1.0 s per step. The Cu Ka1 radiation from the anode, generated at 40 kV and 30 mA, was monochromatized using a 15μm Ni foil (λ) 0.1542 nm). The diffractometer was equipped with a 1° divergence slit, a 0.2 mm receiving slit, and a 1° scatter slit. Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry analysis was performed on a Bruker UltraFlextreme (Bruker Daltonics, Germany) in reflective mode and negative ions were examined. The instrument was calibrated with a mixture of peptide standards with known molecular masses from Bruker Daltonics. Samples were ten times diluted in the matrix solution containing 10 mg mL−1 2,5-dihydroxybenzoic acid in 50% (v/v) acetonitrile. For analysis, 2 μL of the mixture was transferred to the target plate and dried under a stream of dry air. The lowest laser intensity needed to obtain a good quality spectrum was applied and 9 times 50 laser shots, randomly obtained from the sample, were accumulated.
X-ray crystal structure determination of (6c)
C7H10O5, Fw = 174.15, colourless block, 0.30 × 0.28 × 0.12 mm3, triclinic, P1 (no. 1), a = 5.0392(2), b = 5.4022(2), c = 7.5864(3) Å, α = 99.295(3), β = 102.547(2), γ = 102.956(3)°, V = 191.598(15) Å3, Z = 1, Dx = 1.509 g cm−3, μ = 0.13 mm−1. 5668 reflections were measured on a Bruker Kappa ApexII diffractometer with sealed tube and Triumph monochromator (λ = 0.71073 Å) at a temperature of 100(2) K up to a resolution of (sinθ/λ)max = 0.65 Å−1. Intensity data were integrated with the Eval15 software.13 Absorption correction and scaling was performed with SADABS (correction range 0.70–0.75).14 1706 Reflections were unique (Rint = 0.014), of which 1691 were observed [I > 2σ(I)]. The structure was solved with the program SHELXS-97.15 Least-squares refinement was performed with SHELXL-2013(ref. 15) against F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were located in difference Fourier maps. O–H hydrogen atoms were refined freely with isotropic displacement parameters, C–H hydrogen atoms were refined with a riding model. 117 Parameters were refined with 3 restraints (floating origin). R1/wR2 [I > 2σ(I)]: 0.0247/0.0642. R1/wR2 [all refl.]: 0.0250/0.0646. S = 1.070. The absolute structure could not be determined reliably from anomalous differences. Residual electron density between 0.15 and 0.30 e/Å3. Geometry calculations and checking for higher symmetry was performed with the PLATON program.16 .
(3S,6R)-6-hydroxyhexahydrofuro[3,2 b]furan-3-yl acetate (2a)
A 500 mL two-necked round-bottom flask, equipped with a magnetic stirrer, and a reflux condenser with a nitrogen inlet, was charged with isosorbide (1a, 25.0 g, 0.171 mol), acetic acid (10.7 mL, 0.188 mol) and DCM (300 mL). The reaction mixture was stirred at room temperature under nitrogen atmosphere for 10 min. The solution was then cooled down to 0 °C and DCC (38.8 g, 0.188 mol) followed by DMAP (0.20 g, 1.710 mmol) was added portion-wise. A white crystalline precipitate of N,N′-dicyclohexylurea was formed immediately. The reaction progress was monitored by TLC analysis (ethylacetate/PE, 2:3). After 3 h, when the reaction was completed, the mixture was filtered off to remove N,N′-dicyclohexylurea and washed with DCM (2 × 100 mL). The combined filtrates were washed with water (2 × 500 mL), dried over magnesium sulfate and evaporated under reduced pressure to give the crude mixture of isosorbide 2-acetate, isosorbide 5-acetate and isosorbide 2,5-diacetate. The desired product, isosorbide 2-acetate (2a) was separated by column chromatography on silica gel (ethylacetate/PE, 1:4) to obtain (2a) as a white crystalline solid.
Yield: 20.6 g, 64%; m.p. 79–81 °C (lit.17 m.p. 78 °C); 1H NMR (400 MHz, CDCl3): δ = 5.27–5.17 (m, 1H), 4.63 (t, J = 4.9 Hz, 1H), 4.48 (d, J = 4.3 Hz, 1H), 4.31 (dd, J = 7.1, 5.6 Hz, 1H), 4.02 (t, J = 2.5 Hz, 2H), 3.89 (dd, J = 9.5, 6.0 Hz, 1H), 3.57 (dd, J = 9.5, 6.0 Hz, 1H), 2.65 (d, J = 7.3 Hz, 1H), 2.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 167.77, 83.45, 79.79, 76.23, 71.43, 71.35, 70.12, 18.71 ppm; IR (neat): ν = 3404, 2925, 2884, 1733, 1372, 1254, 1047, 1010, 989 cm−1; HRMS (ESI): MH+, found 189.0757. C8H13O5 requires 189.0763.
(3R,6R)-6-hydroxyhexahydrofuro[3,2 b]furan-3-yl acetate (2b)
A 500 mL two-necked round-bottom flask, equipped with a magnetic stirrer, and a reflux condenser with a nitrogen inlet, was charged with isomannide (1b, 25.0 g, 0.171 mol), acetic acid (10.7 mL, 0.188 mol) and DCM (300 mL). The reaction mixture was stirred at room temperature under nitrogen atmosphere for 10 min. The solution was then cooled down to 0 °C and DCC (38.8 g, 0.188 mol) followed by DMAP (0.20 g, 1.710 mmol) was added portion-wise. A white crystalline precipitate of N,N′-dicyclohexylurea was formed immediately. The reaction progress was followed by TLC analysis (ethylacetate/PE, 3:2). After 3 h, when the reaction was completed, the mixture was filtered off to remove N,N′-dicyclohexylurea and washed with DCM (2 × 100 mL). The combined filtrates were washed with water (2 × 500 mL), dried over magnesium sulfate and evaporated under reduced pressure to give the crude mixture of isomannide 2-acetate and isomannide 2,5-diacetate. The desired product, isomannide 2-acetate (2b) was separated by column chromatography on silica gel (ethylacetate/PE, 2:3). to obtain (2b) as a white crystalline semi-solid.
Yield: 22.5 g, 70%; 1H NMR (400 MHz, CDCl3): δ = 5.14 (td, J = 6.5, 5.4 Hz, 1H), 4.69 (t, J = 5.1 Hz, 1H), 4.48 (t, J = 5.2 Hz, 1H), 4.36–4.24 (m, 1H), 4.11 (dd, J = 9.4, 6.5 Hz, 1H), 3.97 (dd, J = 9.1, 6.3 Hz, 1H), 3.84 (dd, J = 9.4, 6.6 Hz, 1H), 3.58 (dd, J = 9.1, 7.1 Hz, 1H), 2.81 (d, J = 8.2 Hz, 1H), 2.13 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 170.11, 81.35, 80.19, 73.89, 73.55, 72.05, 70.39, 20.36 ppm; IR (neat): ν = 3518, 2956, 2878, 1726, 1386, 1228, 1099, 1059, 1021, 931 cm−1; HRMS (ESI):MH+, found 189.0757. C8H13O5 requires 189.0763.
(3S,6R)-6-(((trifluoromethyl)sulfonyl)oxy)hexahydrofuro[3,2 b]furan-3-yl acetate (3a)
A 500 mL 3-necked round-bottom flask, equipped with a magnetic stirrer and a dropping funnel, was charged with (2a) (15.0 g, 0.079 mol), pyridine (10 mL, 0.119 mol) and DCM (350 mL). The colorless solution was cooled down to −10 °C under nitrogen atmosphere. Trifluoromethanesulfonic anhydride (20 mL, 0.119 mol) was added drop-wise over 1 h. The progress of the reaction was followed by GC-MS analysis, which indicated that the reaction had gone to completion after 3 h. The reaction mixture was then poured onto ice-water (500 mL) and stirred for 15 min. The organic layer was separated, and the water layer was extracted with DCM (2 × 150 mL). The combined organic layers were subsequently washed with aqueous HCl (1.0 M, 3 × 300 mL), water (2 × 300 mL), dried over MgSO4, and decolorized with activated carbon. After filtration through a G-3 glass filter funnel containing Celite, the resulting clear solution was evaporated under reduced pressure using a rotary evaporator. Thus obtained crude product was further purified by column chromatography on silica gel (ethylacetate/PE, 1:4)., to obtain (3a) as a pale yellow colored crystalline solid.
Yield: 23.0 g, 90.1%; m.p. 32–34 °C; 1H NMR (400 MHz, CDCl3): δ = 5.23 (t, J = 4.1 Hz, 2H), 4.90 (t, J = 5.3 Hz, 1H), 4.49 (d, J = 5.0 Hz, 1H), 4.16–4.01 (m, 3H), 3.90 (dd, J = 11.3, 4.8 Hz, 1H), 2.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.64, 123.00, 119.83, 116.65, 113.48, 86.05, 85.29, 80.57, 73.47, 70.49, 20.53 ppm; IR (neat): ν = 3517, 2949, 2877, 1727, 1410, 1234, 1198, 1143, 1097, 966 cm−1; HRMS (ESI):MH+, found 321.0237. C9H12F3O7S requires 321.0256.
(3R,6R)-6-(((trifluoromethyl)sulfonyl)oxy)hexahydrofuro[3,2 b]furan-3-yl acetate (3b)
A 500 mL 3-necked round-bottom flask, equipped with a magnetic stirrer and a dropping funnel, was charged with (2b) (15.0 g, 0.079 mol), pyridine (10 mL, 0.119 mol) and DCM (350 mL). The colorless solution was cooled down to −10 °C under nitrogen atmosphere. Trifluoromethanesulfonic anhydride (20 mL, 0.119 mol) was added drop-wise over 1 h. The progress of the reaction was followed by GC-MS analysis, which indicated that the reaction had gone to completion after 3 h. The reaction mixture was then poured onto ice-water (500 mL) and stirred for 15 min. The organic layer was separated, and the water layer was extracted with dichloromethane (2 × 150 mL). The combined organic layers were subsequently washed with aqueous HCl (1.0 M, 3 × 300 mL), water (2 × 300 mL), dried over MgSO4, and decolorized with activated carbon. After filtration through a G-3 glass filter funnel containing Celite, the resulting clear solution was evaporated under reduced pressure using a rotary evaporator. Thus obtained crude product was further purified by column chromatography on silica gel (ethylacetate/PE, 1:4)., to obtain (3b) as a pale yellow liquid.
Yield: 23.2 g, 91%; 1H NMR (400 MHz, CDCl3): δ = 5.20 (d, J = 5.1 Hz, 1H), 5.13–5.03 (m, 1H), 4.73 (dd, J = 13.5, 5.4 Hz, 2H), 4.15 (dd, J = 9.4, 6.6 Hz, 1H), 4.12–3.97 (m, 2H), 3.88 (dd, J = 9.4, 7.6 Hz, 1H), 2.13 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 170.23, 123.27, 120.09, 116.92, 113.74, 84.81, 80.37, 72.88, 70.80, 70.45, 20.47 ppm; IR (neat): ν = 3511, 2953, 2888, 1740, 1410, 1201, 1143, 1077, 970 cm−1; HRMS (ESI):MH+, found 321.0233. C9H12F3O7S requires 321.0256.
General procedure for cyanation (4a & 4c)
A 500 mL three-necked round-bottom flask, equipped with a magnetic stirrer, internal thermo couple, pressure equalizing dropping funnel and a reflux condenser, was charged with potassium cyanide (4.0 g, 0.062 mol), 18-crown-6 (16.4 g, 0.062 mol) and dry THF (250 mL). The suspension was cooled down to between −5 °C and 0 °C, and a solution of (3a or 3b) (10.0 g, 0.031 mol) in THF (100 mL) was added drop-wise over 1 h with stirring under a continuous flow of nitrogen. After addition was complete, the slightly exothermic reaction was strictly maintained at the specified temperature to avoid epimerization. The progress of the reaction was followed by GC-MS analysis, which indicated that the reaction had gone to completion after 3 h. The reaction mixture was then poured onto cold water (350 mL) and extracted with chloroform (3 × 250 mL). The combined organic layers were dried over MgSO4 and decolorized with activated carbon. After filtration through a G-3 glass filter funnel containing Celite, the resulting colorless solution was evaporated under reduced pressure using a rotary evaporator to give the crude product. Finally, pure mono nitrile acetates were obtained by distillation using a kugel-rohr glass oven to afford (4a) as a colorless liquid and (4c) as a white crystalline solid.General procedure for cyanation followed by epimerization (4b & 4d)
A 500 mL three-necked round-bottom flask, equipped with a magnetic stirrer, internal thermo couple, pressure equalizing dropping funnel and a reflux condenser, was charged with potassium cyanide (4.0 g, 0.062 mol), 18-crown-6 (16.4 g, 0.062 mol) and dry THF (250 mL). The suspension was cooled down to between –5 °C and 0 °C, and a solution of (3a or 3b) (10.0 g, 0.031 mol) in THF (100 mL) was added drop-wise over 1 h with stirring under a continuous flow of nitrogen. After addition was complete, the reaction was stirred at room temperature for 1.5 h and gradually the temperature was raised to 40 °C which allows epimerization to take place. The progress of the reaction was followed by GC-MS analysis, which indicated that the epimerization reaction had proceeded to 65–70% completion after 2 h. The reaction mixture was then poured onto cold water (350 mL) and extracted with chloroform (3 × 250 mL). The combined organic layers were dried over MgSO4 and decolorized with activated carbon. After filtration through a G-3 glass filter funnel containing Celite, the resulting pale yellow colored solution was evaporated under reduced pressure using a rotary evaporator to give the crude product. Finally, pure mono nitrile acetates were obtained by distillation using a kugel-rohr glass oven to afford (4b) as a white solid and (4d) as a white crystalline solid.(3S,6S)-6-cyanohexahydrofuro[3,2 b]furan-3-yl acetate (4a)
Distillation conditions: 120 °C at 0.02 mbar for 1.5 h. Yield: 5.2 g, 85%; 1H NMR (400 MHz, CDCl3): δ = 5.24–5.13 (m, 1H), 5.00 (dd, J = 4.2, 1.8 Hz, 1H), 4.66 (d, J = 4.1 Hz, 1H), 4.16–3.86 (m, 4H), 3.19 (ddd, J = 6.2, 5.0, 1.8 Hz, 1H), 2.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 171.69, 119.76, 88.39, 87.39, 78.87, 74.90, 72.08, 39.07, 22.68 ppm; IR (neat): ν = 2977, 2886, 2246, 1741, 1370, 1229, 1080, 1061, 914 cm−1; HRMS (ESI):MH+, found 198.0759. C9H12NO4 requires 198.0766.(3S,6R)-6-cyanohexahydrofuro[3,2 b]furan-3-yl acetate (4b)
Distillation conditions: 125 °C at 0.02 for 1.5 h. Yield: 3.7 g, 60%; m.p. 70–72 °C; 1H NMR (400 MHz, CDCl3): δ = 5.21 (dt, J = 4.0, 1.3 Hz, 1H), 4.90 (t, J = 4.5 Hz, 1H), 4.58 (dt, J = 4.1, 1.0 Hz, 1H), 4.25–4.08 (m, 2H), 4.05–3.86 (m, 2H), 3.20 (ddd, J = 9.3, 7.9, 4.9 Hz, 1H), 2.09 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 167.81, 113.77, 85.06, 79.77, 75.57, 71.78, 67.47, 34.50, 18.82 ppm; IR (neat): ν = 2945, 2886, 2247, 1725, 1374, 1232, 1109, 1055, 1011, 952 cm−1; HRMS (ESI):MH+, found 198.0754. C9H12NO4 requires 198.0766.(3R,6S)-6-cyanohexahydrofuro[3,2 b]furan-3-yl acetate (4c)
Distillation conditions: 130 °C at 0.02 mbar for 1.5 h. Yield: 5.6 g, 92%; m.p. 69–71 °C; 1H NMR (400 MHz, CDCl3): δ = 5.16 (q, J = 5.5 Hz, 1H), 4.88 (t, J = 5.2 Hz, 1H), 4.83 (dd, J = 4.9, 1.4 Hz, 1H), 4.16 (dd, J = 9.1, 3.1 Hz, 1H), 4.05 (dd, J = 9.1, 5.9 Hz, 1H), 3.96 (dd, J = 10.0, 5.9 Hz, 1H), 3.86 (dd, J = 10.0, 5.1 Hz, 1H), 3.21 (ddd, J = 5.9, 3.1, 1.4 Hz, 1H), 2.12 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 170.04, 118.01, 85.62, 81.40, 73.02, 71.06, 70.90, 37.51, 20.54 ppm; IR (neat): ν = 2957, 2880, 2250, 1735, 1363, 1239, 1110, 1080, 1063, 1015, 902 cm−1; HRMS (ESI):MH+, found 198.0757. C9H12NO4 requires 198.0766.(3R,6R)-6-cyanohexahydrofuro[3,2 b]furan-3-yl acetate (4d)
Distillation conditions: 130 °C at 0.02 mbar for 1.5 h. Yield: 3.5 g, 57%; m.p. 53–55 °C; 1H NMR (400 MHz, CDCl3): δ = 5.17 (q, J = 5.6 Hz, 1H), 4.82 (dd, J = 5.8, 4.8 Hz, 1H), 4.72 (t, J = 5.0 Hz, 1H), 4.23 (t, J = 8.1 Hz, 1H), 4.01 (t, J = 5.4 Hz, 2H), 3.92 (dd, J = 10.3, 8.5 Hz, 1H), 3.13 (ddd, J = 10.3, 7.8, 5.1 Hz, 1H), 2.13 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ = 168.14, 113.20, 79.89, 79.64, 71.30, 69.49, 68.20, 34.16, 18.50 ppm; IR (neat): ν = 2950, 2890, 2249, 1733, 1373, 1250, 1102, 1060, 1019, 973 cm−1; HRMS (ESI):MH+, found 198.0756. C9H12NO4 requires 198.0766.General procedure for solvolysis (5a–d)
A 250 mL one-necked round-bottom flask was equipped with a magnetic stirrer and a reflux condenser. Methanol (100 mL), HCl (37%, 100 mL) and (4a–d) (5.0 g, 0.025 mol) were sequentially charged to the flask. After stirring at reflux temperature for 3 h, the reaction mixture was cooled down to room temperature and extracted with chloroform (3 × 150 mL). The combined organic layers were dried over MgSO4, and the resulting solution was evaporated under reduced pressure using a rotary evaporator to give the crude product. Pure (5a–d) was obtained as a colorless viscous liquid by distillation using a kugel-rohr glass oven.(3S,6S)-methyl 6-hydroxyhexahydrofuro[3,2 b]furan-3-carboxylate (5a)
Distillation conditions: 115 °C at 0.02 mbar for 1.5 h. Yield: 4.1 g, 86%; 1H NMR (400 MHz, CDCl3): δ = 5.04 (dd, J = 4.4, 2.1 Hz, 1H), 4.46 (d, J = 4.4 Hz, 1H), 4.31 (d, J = 3.3 Hz, 1H), 4.12–3.96 (m, 2H), 3.95–3.87 (m, 1H), 3.84 (dt, J = 10.1, 1.1 Hz, 1H), 3.73 (s, 3H), 3.14 (ddd, J = 7.2, 5.1, 2.1 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.92, 86.81, 82.88, 73.87, 72.12, 68.22, 50.25, 49.65 ppm; IR (neat): ν = 3426, 2955, 2881, 1730, 1437, 1202, 1073, 974 cm−1; HRMS (ESI):MH+, found 189.0748. C8H13O5 requires 189.0763.(3R,6S)-methyl 6-hydroxyhexahydrofuro[3,2 b]furan-3-carboxylate (5b)
Distillation conditions: 125 °C at 0.02 mbar for 1.5 h. Yield: 4.0 g, 84%; 1H NMR (400 MHz, CDCl3): δ = 4.98 (dd, J = 5.1, 4.0 Hz, 1H), 4.47 (dt, J = 3.9, 1.0 Hz, 1H), 4.38–4.25 (m, 1H), 4.03 (dd, J = 9.2, 5.6 Hz, 2H), 3.91 (dd, J = 10.2, 3.5 Hz, 1H), 3.84 (d, J = 10.1 Hz, 1H), 3.75 (s, 3H), 3.23 (dd, J = 5.1, 1.2 Hz, 1H), 2.81 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ = 169.54, 89.11, 82.78, 76.27, 75.59, 68.42, 52.05, 50.64 ppm; IR (neat): ν = 3431, 2953, 2885, 1733, 1439, 1206, 1080, 1007 cm−1; HRMS (ESI):MH+, found 189.0752. C8H13O5 requires 189.0763.(3S,6R)-methyl 6-hydroxyhexahydrofuro[3,2 b]furan-3-carboxylate (5c)
Distillation conditions: 120 °C at 0.02 mbar for 1.5 h. Yield: 3.8 g, 80%; 1H NMR (400 MHz, CDCl3): δ = 4.85 (dd, J = 4.9, 1.8 Hz, 1H), 4.57 (t, J = 5.2 Hz, 1H), 4.34–4.18 (m, 2H), 4.12 (dd, J = 9.0, 6.7 Hz, 1H), 3.85 (d, J = 5.7 Hz, 1H), 3.74 (s, 3H), 3.66 (dd, J = 9.6, 5.6 Hz, 1H), 3.22 (ddd, J = 6.3, 4.0, 1.8 Hz, 1H), 2.65 (d, J = 6.8 Hz, 1H). ppm; 13C NMR (100 MHz, CDCl3): δ = 171.64, 85.15, 82.51, 73.61, 71.94, 71.58, 52.34, 52.16 ppm; IR (neat): ν = 3443, 2954, 2888, 1730, 1437, 1199, 1072, 1010 cm−1; HRMS (ESI):MH+, found 189.0754. C8H13O5 requires 189.0763.(3R,6R)-methyl 6-hydroxyhexahydrofuro[3,2 b]furan-3-carboxylate (5d)
Distillation conditions: 115 °C at 0.02 mbar for 1.5 h. Yield: 4.0 g, 84%; 1H NMR (400 MHz, CDCl3): δ = 4.80 (t, J = 4.8 Hz, 1H), 4.54 (dd, J = 5.4, 4.4 Hz, 1H), 4.29 (q, J = 6.1 Hz, 1H), 4.24–4.06 (m, 2H), 3.92 (dd, J = 9.4, 6.2 Hz, 1H), 3.75 (s, 3H), 3.60 (dd, J = 9.4, 6.6 Hz, 1H), 3.27 (ddd, J = 10.6, 8.3, 5.3 Hz, 1H), 2.67 (br, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ = 167.84, 82.02, 81.89, 73.28, 71.34, 68.67, 51.07, 50.16 ppm; IR (neat): ν = 3432, 2960, 2890, 1742, 1439, 1212, 1090, 985 cm−1; HRMS (ESI):MH+, found 189.0752. C8H13O5 requires 189.0763.(3S,6R)-6-hydroxyhexahydrofuro[3,2 b]furan-3-carboxylic acid (6c)
A 100 mL one-necked round-bottom flask equipped with a magnetic stirrer and a reflux condenser, was charged with (5c) (1.0 g, 5.3 mmol) and aqueous HCl (3.0 M, 20 mL). The reaction mixture was stirred under reflux for 24 h and then cooled down to room temperature. After removal of the solvent under reduced pressure using a rotary evaporator, the crude hydroxyl carboxylic acid was subsequently recrystallized from diethyl ether affording pure (6c) as a white solid. Single crystals were obtained from ethanol by slow evaporation. Yield: 0.75 g, 82% (purity: 99%, GC analysis); 1H NMR (400 MHz, Deuterium Oxide): δ = 4.87 (dd, J = 5.2, 2.1 Hz, 1H), 4.54 (t, J = 5.1 Hz, 1H), 4.30 (q, J = 6.1 Hz, 1H), 4.14 (dq, J = 5.4, 3.3 Hz, 1H), 4.05 (dd, J = 9.2, 6.3 Hz, 1H), 3.87 (dd, J = 9.4, 5.8 Hz, 1H), 3.57 (dd, J = 9.3, 6.5 Hz, 1H), 3.22 (dq, J = 8.2, 2.8, 2.2 Hz, 1H); 13C NMR (100 MHz, Deuterium Oxide): δ = 175.10, 85.22, 82.24, 71.41, 71.29, 71.01, 51.99 ppm; IR (neat): ν= 1692 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; HR-MS (ESI):MH+, found 175.0601. C7H11O5 requires 175.0606.
O) cm−1; HR-MS (ESI):MH+, found 175.0601. C7H11O5 requires 175.0606.
General polymerization procedure
Stereo-isomer (5a–d) (0.202 g, 1 mmol) and dibutyltin oxide (DBTO) (10 mol%) was charged into a 10 mL RB flask and inserted in to the kugelrohr oven. The set-up was vented with 3 cycles of nitrogen/vacuum to remove the oxygen. During the first stage, the reaction is carried out under nitrogen gas to form oligomers. The reaction mixture is heated to 115 °C, and after 15 min the catalyst dibutyltin oxide (DBTO, 10 mol%) is added to the flask under a continuous flow of nitrogen gas. The temperature is then increased to 160 °C under stirring for 12 h. At this stage the reaction mixture became opaque and solidified. Finally the temperature was raised to 180 °C for 2 h to complete the pre-polymerisation reaction. In the second stage, vacuum was applied gradually down to 0.02 mbar at 180 °C for 2 h. The resulting polymer was cooled down to room temperature under the continuous flow of nitrogen, dissolved in CHCl3:TFA (6:1) mixture and precipitated into methanol, filtered and dried in vacuo at 40 °C for 15 h.
Poly(isoidide methylcarboxylate) (PE-1)
1H NMR (400 MHz, CDCl3+TFA-d): δ = 5.26 (d, J = 3.7 Hz, 1H), 5.10 (dt, J = 3.9, 1.9 Hz, 1H), 4.64 (dq, J = 8.8, 4.8, 4.2 Hz, 1H), 4.05 (m, 3H), 4.04–3.92 (m, 1H), 3.27 (ddd, J = 6.7, 4.7, 2.0 Hz, 1H).; 13C NMR (100 MHz, CDCl3+TFA-d): δ = 170.80, 86.73, 85.59, 78.73, 72.5, 70.87, 51.83 ppm; IR (neat): ν = 3462, 2960, 2884, 1730, 1183, 1075, 1024 cm−1.Poly(isosorbide methylcarboxylate) (PE-2)
1H NMR (400 MHz, CDCl3+TFA-d): δ = 5.30 (p, J = 4.4, 3.8 Hz, 1H), 5.18–5.00 (m, 1H), 4.66 (h, J = 4.1 Hz, 1H), 4.21–4.00 (m, 4H), 3.48–3.19 (m, 1H); δ = 166.31, 84.80, 81.44, 76.76, 71.52, 66.93, 48.58; IR (neat): ν = 3468, 2959, 2885, 1736, 1177, 1080, 1007 cm−1.Poly(isoidide-isosorbide methylcarboxylate) (PE-5)
1H NMR (400 MHz, CDCl3+TFA-d): δ = 5.33 (s, 1H), 5.31–5.25 (m, 1H), 5.22 (ddt, J = 7.0, 4.4, 2.0 Hz, 1H), 5.11–4.92 (m, 2H), 4.78 (tt, J = 8.5, 3.8 Hz, 1H), 4.41–4.29 (m, 1H), 4.26–3.98 (m, 7H), 3.47–3.32 (m, 2H). δ = ; IR (neat): ν = 3458, 2956, 2886, 1732, 1174, 1077, 1008 cm−1.Poly(isoidide-isomannide methylcarboxylate) (PE-6)
1H NMR (400 MHz, CDCl3+TFA-d): δ = 5.20 (dd, J = 7.4, 3.7 Hz, 1H), 5.15 (t, J = 5.0 Hz, 1H), 5.09–4.93 (m, 1H), 4.85 (dd, J = 14.6, 4.9 Hz, 1H), 4.77 (d, J = 5.6 Hz, 1H), 4.56 (dt, J = 9.2, 4.6 Hz, 1H), 4.35–3.97 (m, 5H), 3.96–3.79 (m, 3H), 3.21 (ddt, J = 16.6, 6.7, 3.0 Hz, 2H); IR (neat): ν = 3462, 2958, 2879, 1752, 1181, 1075, 1002 cm−1.Acknowledgements
Dr A. E. Frissen and L. Gootjes, BSc. are gratefully acknowledged for NMR and XRD analyses, W. Teunissen and H. de Beukelaer, BSc. for GC-MS and DSC analyses, Dr L. A. M. van den Broek for MALDI-ToF measurements and E. Janssen, MSc. for HR-MS measurements. The X-ray diffractometer for the single crystal structure determination was financed by the Netherlands Organization for Scientific Research (NWO). This work is part of the research program of the Dutch Polymer Institute (DPI), # 656 GREENER.References
- (a) P. Stoss, R. Hemmer and H. Derek, in Adv. Carbohydr. Chem. Biochem., Academic Press, 1991, vol. 67, pp. 93–173 Search PubMed; (b) A. C. Cope and T. Y. Shen, J. Am. Chem. Soc., 1956, 78, 3177–3182 CrossRef CAS; (c) H. R. Kricheldorf, J. Macromol. Sci., Part C, 1997, 37, 599–631 CrossRef; (d) F. Xianhong, J. E. Anthony, H. Willis and J. Michael, in Contemporary Science of Polymeric Materials, American Chemical Society, 2010, vol. 1061, pp. 3–27 Search PubMed; (e) C. Lavilla, A. Alla, A. Martínez de Ilarduya and S. Muñoz-Guerra, Biomacromolecules, 2013, 14, 781–793 CrossRef CAS PubMed; (f) C. Lavilla, A. Alla, A. Martínez de Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Biomacromolecules, 2011, 12, 2642–2652 CrossRef CAS PubMed.
- (a) F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS PubMed; (b) R. l. Sablong, R. Duchateau, C. E. Koning, G. d. Wit, D. S. van Es, R. Koelewijn and J. v. Haveren, Biomacromolecules, 2008, 9, 3090–3097 CrossRef CAS PubMed.
- (a) D. S. van Es, J. Renewable Mater., 2013, 1, 61–72 CrossRef CAS; (b) M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- J. Wu, P. Eduard, L. Jasinska-Walc, A. Rozanski, B. A. J. Noordover, D. S. van Es and C. E. Koning, Macromolecules, 2012, 46, 384–394 CrossRef.
- J. Le Nôtre, J. van Haveren and D. S. van Es, ChemSusChem, 2013, 6, 693–700 CrossRef PubMed.
- (a) S. Thiyagarajan, L. Gootjes, W. Vogelzang, J. Van Haveren, M. Lutz and D. S. Van Es, ChemSusChem, 2011, 4, 1823–1829 CrossRef CAS PubMed; (b) S. Thiyagarajan, L. Gootjes, W. Vogelzang, J. Wu, J. Van Haveren and D. S. Van Es, Tetrahedron, 2011, 67, 383–389 CrossRef CAS PubMed; (c) J. Wu, L. Jasinska-Walc, D. Dudenko, A. Rozanski, M. R. Hansen, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 9333–9346 CrossRef CAS; (d) D. Tang, S. Thiyagarajan, B. A. J. Noordover, C. E. Koning, D. S. van Es and J. van Haveren, J. Renewable Mater., 2013, 1, 222–229 CrossRef CAS.
- J. Wu, P. Eduard, S. Thiyagarajan, J. Van Haveren, D. S. Van Es, C. E. Koning, M. Lutz and C. Fonseca Guerra, ChemSusChem, 2011, 4, 599–603 CrossRef CAS PubMed.
- (a) J. Wu, P. Eduard, S. Thiyagarajan, L. Jasinska-Walc, A. Rozanski, C. F. Guerra, B. A. J. Noordover, J. van Haveren, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 5069–5080 CrossRef CAS; (b) J. Wu, PhD thesis, Eindhoven : Technische Universiteit Eindhoven, 2012.
- F. Bachmann, J. Reimer, M. Ruppenstein and J. Thiem, Macromol. Rapid Commun., 1998, 19, 21–26 CrossRef CAS.
- C. l. Besset, S. Binauld, M. Ibert, P. Fuertes, J.-P. Pascault, E. Fleury, J. Bernard and E. Drockenmuller, Macromolecules, 2009, 43, 17–19 CrossRef.
- F. J. Hopton and G. H. S. Thomas, Can. J. Chem., 1969, 47, 2395–2401 CrossRef CAS.
- A. A. Yevstropov, B. V. Lebedev, T. G. Kulagina and N. K. Lebedev, Polym. Sci. U.S.S.R., 1982, 24, 628–636 CrossRef.
- A. M. M. Schreurs, X. Xian and L. M. J. Kroon-Batenburg, J. Appl. Crystallogr., 2010, 43, 70–82 CrossRef CAS.
- G. M. Sheldrick, SADABS: Area-Detector Absorption Correction, Universität Göttingen, Germany, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., 2009, D65, 148–155 CrossRef PubMed.
- J. Lub, W. P. M. Nijssen, R. T. Wegh, I. De Francisco, M. P. Ezquerro and B. Malo, Liq. Cryst., 2005, 32, 1031–1044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 964358. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra07896a |
| This journal is © The Royal Society of Chemistry 2014 |