
Selective approach to thioesters and thioethers via sp3 C–H activation of methylarenes†
J. Feng,
G.-P. Lu and
C. Cai*
Chemical Engineering College, Nanjing University of Science & Technology, 200 Xiaolingwei, Nanjing 210094, P. R. China. E-mail: c.cai@njust.edu.cn; Fax: +86-25-84315030
First published on 8th October 2014
Abstract
Novel C–S cross-dehydrogenative coupling (CDC) approaches for the selective synthesis of thioesters and thioethers have been developed via sp3 C–H activation of methylarenes and subsequent functionalization. The reaction of methylarenes with thiols resulted in thioesters in the presence of a FeBr2/TBHP system, while treatment of methylarenes with thiols in the Pd(OAc)2/O2/TBHP system led to the formation of thioethers. Both the green protocols demonstrate good functional group tolerance and satisfactory yields.
Introduction
The direct C–H activation path is now being extensively investigated as a short step and greener alternative to the traditional transition-metal-catalyzed couplings.1 One such strategy, cross-dehydrogenative coupling (CDC), is of immense importance because it does not require substrate prefunctionalisation and enjoys the advantage of atom economy.2 Until now the CDC based protocols for the construction of a C–C,3 C–O,4 C–N5 bonds have been easily achieved using transition metal catalysts in combination with various oxidants. However there is still a dearth of information on C–S bond formation.6 In this regard, C–S bond formation and eventual synthesis of thioesters and thioethers, involving C–H bond activation of methylarenes remains the cherished target.Sulfur-containing compounds, especially thioesters and thioethers, represent a class of important synthetic intermediates due to their ease of transformation.7 Moreover, they are excellent building blocks for chemical biology, and hence are frequently found in a number of biologically active and medicinal agents.8 Traditionally, thioethers were formed by coupling reactions between halides and thiols (or sulfur surrogates),9 the SNAr reactions,10 substitution reactions,11 whereas thioesters were commonly prepared via acylation with thiols (or sulfur surrogates) and carboxylic acids,12 carboxylic acid halides,13 carboxylic acid anhydrides14 or aldehydes.15 Although many elegant protocols for the synthesis of thioethers and thioesters have emerged, in accordance with their importance, the synthesis of thioethers or thioesters by unconventional approaches was always appreciable, particularly through the functionalization of inert C–H bonds.
Methylarenes, derived from the oil industry, constitute the most inexpensive and readily available feed stocks for chemistry. Of late, the sp3 C–H activation and subsequent oxidative functionalization of methylarenes has drawn scientist's attention.2e,16 Through this strategy, methylarenes, especially methylbenzenes, are found to be useful precursors and have been used as ArCOO–,17 ArCO–,3h,5b,18 ArCH2O–19 and ArCH2– surrogates.20 Considering the availability and economy of methylarenes and the few reported C–S formation examples via CDC protocol,6a,21 herein we wish to report two novel methods for the synthesis of thioesters and thioethers via sp3 C–H activation of methylarenes. Thus, the use of more traditional coupling partners that require prefunctionalisation, including aryl halide, acyl halide, aryl acid, and aryl aldehyde, are avoided.
Results and discussion
Encouraged by recent Fe-catalyzed synthesis of thioesters from thiols and aldehydes,15k we initiated our work using toluene and 4-chlorobenzenethiol under similar conditions (Fe, TBHP system). The aimed thioesters were observed in low yield by GC-MS along with the formation of benzaldehyde and disulfide22 (Table 1, entry 7). In the process to optimize the reaction conditions, it was found that small amount of thioether was formed when Pd(OAc)2 was used (Table 1, entry 6). Promising to selectively obtain thioesters and thioethers, we optimized both the reactions.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Change from the “standard conditions” | Yieldb | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Standard conditions: toluene 1.5 mmol, 4-chlorobenzenethiol 0.5 mmol, FeBr2 0.01 mmol, TBHP (70% in water) 1.5 mmol, 2 wt% SDS–H2O 1 mL, 100 °C, 24 h.b The yield was determined by GC.c Thiol was added in a dropwise fashion.d The yield of thioether.e The yield of disulfide. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | None | 65(80c) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | No FeBr2 | <1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | FeCl2 instead of FeBr2 | 62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Fe(acac)3 instead of FeBr2 | 43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Cu(OAc)2 instead of FeBr2, no surfactant | 8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Pd(OAc)2 instead of FeBr2, no surfactant | <1(7d) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | No surfactant | 12(80e) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 2 wt% Prij L23 instead of 2 wt% SDS | 10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 2 wt% Trion X100 instead of 2 wt% SDS | 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 0.1 eq. TBAI instead of 2 wt% SDS | 25(69e) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | BPO instead of TBHP | 0(95e) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | DTBP instead of TBHP | 0(10e) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

As the reaction was conducted in water, surfactants may accelerate the reaction process. Screening nonionic and ionic surfactants revealed that nonionic surfactants such as Trion-X 100, and Prij were ineffective, whereas in the presence of TBAI, the yield increased to 25%. The addition of 2 wt% SDS–water boosted the yield to 65% (Table 1, entries 7–10). Next, we examined the metal catalysts in a quest to improve the yield (Table 1 entries 3–6). Among all the catalysts tested, FeBr2 was still found to be the best choice, and oxidants were also checked. In the presence of BPO, 4-chlorobenzenethiol converted to the corresponding disulfide quickly without the formation of desired thioester (Table 1, entry 11), while in the case of DTBP, most starting materials were not converted (Table 1, entry 12). Among all the conditions, the formation of disulfide seemed inevitable. When the thiol was added in a dropwise fashion, the yield of thioester could increase to 80% (Table 1, entry 1).
Having established the optimized reaction conditions, the present oxidative esterification reactions were then implemented on the reaction between toluene and a series of substituted thiophenols and thiols. As shown in Table 2, thiophenols containing electron-donating or electron-withdrawing groups reacted smoothly with toluene, giving thioesters in moderate to good yields (Table 2, 3a–e). There was a decrease of yield when thiophenols bear methyl group at different positions (Table 2, 3b and 3c). It was found that the methyl group can also be activated with the small amount of mercaptobenzaldehyde and mercapto thioesters that were observed. Alkyl thiols can also successfully couple with toluene (Table 2, 3f–k, 3m and 3o). In general, the product yields for the alkyl thiols are lower than those of thiophenols. Especially, sterically demanding substrates yield the product in a lower level (Table 2, 3j). It is worthy to note that thiols bearing fluorous tail can also undergo this coupling, which may be useful in fluorine chemistry23 (Table 2, 3p). We further investigated heterocyclic thiophenols, and to our delight, the reaction between toluene and benzo[d]thiazole-2-thiol afforded the aimed product (Table 2, 3n) in moderate yield. Unfortunately, the reaction failed to proceed in the case of 1H-benzo[d]imidazole-2-thiol, with a collection of byproducts, including the open ring product and the C–N coupling product being obtained instead of the aimed thioester (Table 2, 3q).
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: toluene 1.5 mmol, thiol 0.5 mmol, FeBr2 0.01 mmol, TBHP (70% in water) 1.5 mmol, 2 wt% SDS–H2O 1 mL, 100 °C, 24 h.b Isolated yield.c Reaction conditions: toluene 1.5 mmol, thiol 0.25 mmol, FeBr2 0.02 mmol, TBHP (70% in water) 3 mmol, 2 wt% SDS–H2O 1 mL, 100 °C, 36 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |  |
R = 4-Cl | 3a | 82 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | R = 2-CH3 | 3b | 45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | R = 4-CH3 | 3c | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | R = 4-F | 3d | 66 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | R = 4OCH3 | 3e | 84 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |  |
R = 4-F | 3f | 64 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | R = H | 3g | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | R = 4-CH3 | 3h | 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 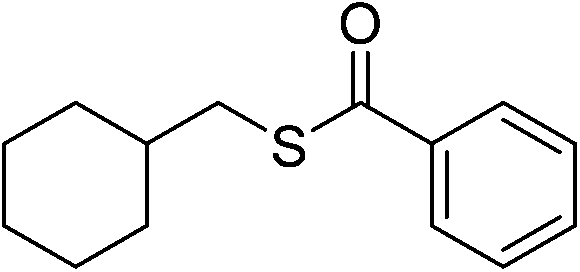 |
3i | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |  |
3j | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 |  |
3k | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 |  |
3l | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 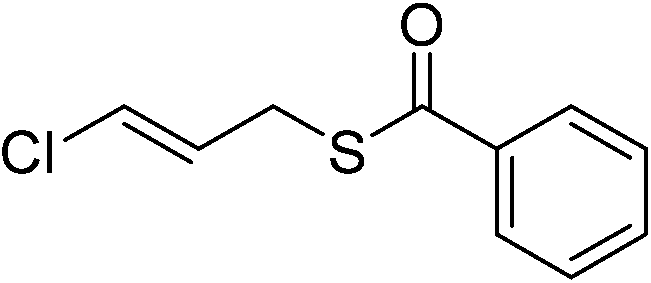 |
3m | 52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 |  |
3n | 46 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 |  |
3o | 42c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 |  |
3p | 45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 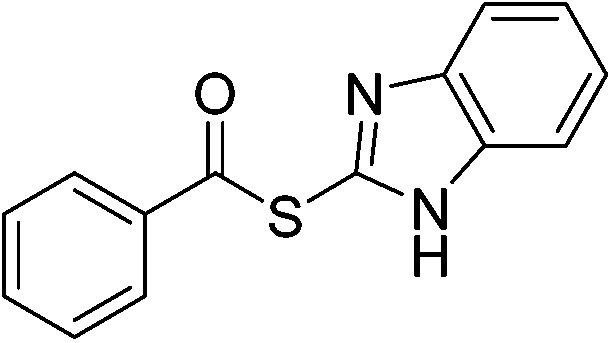 |
3q | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

After that we turned our attention to the functional group compatibility of methylarenes. Methylarenes containing a methyl-, chloro-, bromo- or methoxy-substituent were all tolerated under the reaction conditions (Table 3, 4a–d, 4f and 4g). However, methylarene bearing a –N(CH3)2 substituent failed to couple with the thiophenol. The demethylation of the N–CH3 was observed under the standard conditions. The steric hindrance had little influence with similar yields obtained with 4b, 4g. When there are more than one methyl groups in the benzene ring (for example 4k), the “dithioester” could be afforded when more TBHP was added and the reaction time extended. Accompanied with 4k, S-(p-tolyl)-3-formylbenzothioate 4k′ was also observed. Remarkably, naphthalene-containing methylarenes could also serve as coupling partners with thiols (Table 3, 4j). However, unfortunately, only trace amount of the aimed product was observed when pyridine-containing substrate was used.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: toluene 1.5 mmol, thiol 0.5 mmol, FeBr2 0.01 mmol, TBHP (70% in water) 1.5 mmol, 2 wt% SDS–H2O 1 mL, 100 °C, 24 h.b Isolated yield.c Reaction conditions: toluene 1.5 mmol, thiol 1 mmol, FeBr2 0.02 mmol, TBHP (70% in water) 3 mmol, 2 wt% SDS–H2O 1 mL, 100 °C, 36 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |  |
R = 4-CH3 | 4a | 69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | R = 2,4-Cl | 4b | 79 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | R = 3-Br | 4c | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | R = 4-OCH3 | 4d | 83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | R = 4-N(CH3)2 | 4e | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | R = 4-NO2 | 4f | 52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | R = 4-Cl | 4g | 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | R = 4-CF3 | 4h | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |  |
4i | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |  |
4j | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11c |  |
4k | 45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
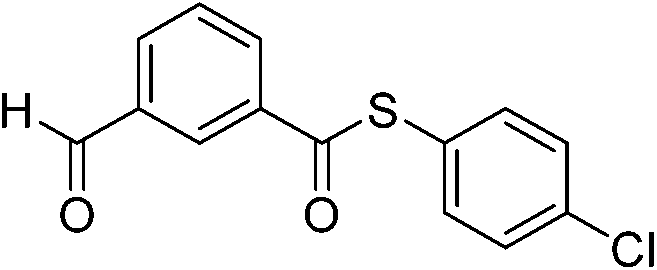 |
4k′ | 15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As mentioned above, thioether instead of thioester was obtained when FeBr2 was replaced by Pd(OAc)2. Aiming for the selective preparation of thioethers, we further explored the palladium-catalyzed CDC thiolation. Details of optimizations are shown in Table 4. The yield of the desired thioether slightly increased when the reaction was moved from the argon atmosphere to the open air, albeit in a low yield (Table 4, entry 9). This indicated that oxidants may play a key role in the CDC process, thus we placed the reaction in a pure oxygen atmosphere (Table 4, entry 1) and increased the equivalent of TBHP (Table 4, entry 11). The addition of excess TBHP had no positive effect to the present reaction but produced more benzaldehyde and sulfur oxidation products in contrast. When the reaction was conducted under oxygen atmosphere, the yield increased to 86%. In view of this result, TBHP may not be necessary; however, the yield dropped sharply in the absence of TBHP (Table 4, entry 7). It is known that thioether may be oxidised by TBHP under heated conditions. However, only trace amount of sulfur oxidation products was observed with the present conditions. Similar to the above reaction, surfactant was also used to accelerate the reaction, but it did not show any improvement (Table 4, entry 6). Finally, some commercially available palladium catalysts and copper catalysts were examined. Still, Pd(OAc)2 was the best choice (Table 4, entries 3–5), and a decrease in catalyst loading (5 mol%) had an adverse effect on product yield (Table 4, entry 12).
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Change from the “standard conditions” | Yieldb | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Standard conditions: toluene 1.5 mmol, 4-chlorobenzenethiol 0.5 mmol, Pd(OAc)2 0.05 mmol, TBHP (70% in water) 0.5 mmol, 1 atom O2, 115 °C, 24 h.b The yield was determined by GC.c The yield of benzaldehyde.d The yield of thioester. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | None | 86 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | No Pd(OAc)2 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Cu(OAc)2 instead of Pd(OAc)2 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Pd(PPh3)2Cl2 instead of Pd(OAc)2 | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Pd(dppf)Cl2 instead of Pd(OAc)2 | 37 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | The addition of SDS (0.02 eq.) as a surfactant | 84 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | No TBHP, 48 h | 16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Under Ar atmosphere | 8(12c) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | In the air | 30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | The addition of FeBr2 | 26(35d) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 3 eq. TBHP | 21(55c, 9d) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | Pd(OAc)2 loading decreased to 5% | 62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Then, these optimized conditions were applied to the reaction of structurally different methylarenes and thiophenols or thiols. A series of unsymmetrical aryl alkyl thioethers could be successfully prepared from substituted methylarenes whereby electronic effects showed no significant association with the yield (Table 4, 5a–g). Unlike the thioesterification described above, the steric hindrance affected the product yield significantly (Table 4, 5b and 5g). Like the above esterification reaction, the “di-thioether” could be obtained under harsher reaction conditions (Table 5, 5q). We have also investigated the applicability of our method using different thiols or thiophenols. As expected, thiophenols could easily convert to the aimed products for all the electron-donating and the electron-withdrawing substituents (Table 5, 5i–l). Alkyl thiols, including benzyl thiol and cyclohexanethiol were also applied for the coupling. Symmetric thioether was obtained in good yield with benzyl thiol (Table 5, 5n), while cyclohexanethiol reacted sluggishly and a 55% yield was obtained by prolonging the reaction time (Table 5, 5m). At last, heterocyclic thiols were tested. Similarly, benzo[d]thiazole-2-thiol underwent the present reaction smoothly while benzo[d]imidazole-2-thiol failed this reaction (Table 5, 5o and 5p).
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: methylarenes 1.5 mmol, thiol or thiophenol 0.5 mmol, Pd(OAc)2 0.05 mmol, TBHP (70% in water) 0.5 mmol, 1 atm O2, 115 °C, 24 h.b Isolated yield.c The yield of 1,4-bis(((4-chlorophenyl)thio)methyl)benzene.d Reaction time 48 h.e Reaction conditions: m-xylene 1.5 mmol, thiophenol 1 mmol, Pd(OAc)2 0.1 mmol, TBHP (70% in water) 1 mmol, 1 atm O2, 120 °C, 36 h. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |  |
R = –H | 5a | 84 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | R = 4-F | 5b | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | R = 4-CH3 | 5c | 62(4c) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | R = 4-NO2 | 5d | 58 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | R = 4-CN | 5e | 62 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | R = 4-Br | 5f | 83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | R = 2-F | 5g | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |  |
5h | 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 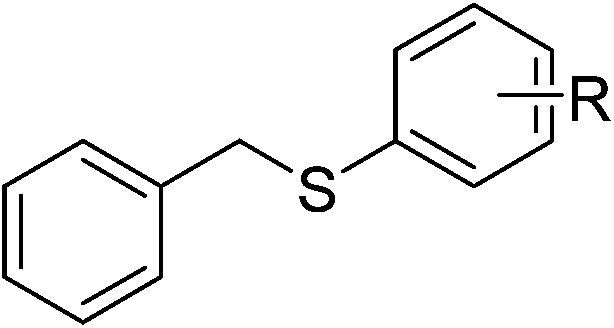 |
R = 2-CH3 | 5i | 72 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | R = 4-CH3 | 5j | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | R = 4-OCH3 | 5k | 70 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | R = 4-F | 5l | 46 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 |  |
5m | 55d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 |  |
5n | 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 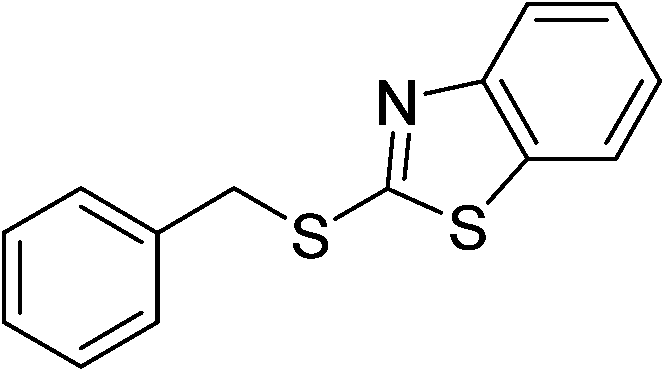 |
5o | 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 |  |
5p | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 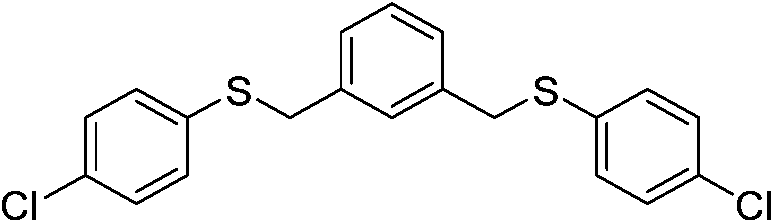 |
5q | 28e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

To gain insight into the catalytic pathway of the two reactions, we conducted some mechanistic experiments (Scheme 1). In the presence of TBHP, thiophenol could easily convert to the disulfide.
 | ||
| Scheme 1 Controlled experiments for preliminary mechanistic studies. | ||
Thus, disulfide was used for the developed two reactions to replace the thiophenol. Under the standard condition, both the thioesterification and thioetherification could proceed, but with lower yields. This means thiophenol is more appropriate for the two reactions, although the formation of disulfide is inevitable. During the reaction of toluene with FeBr2/TBHP, both aldehyde and benzoic acid were observed while in the Pd(OAc)2/O2 conditions, and only trace amount of aldehyde was observed. Thus, we further placed benzaldehyde and 4-chlorobenzoic acid simultaneously into the thioesterification reaction. The crossover experiment showed that benzaldehyde reacted with 4-chlorobenzenethiol under the present experimental conditions, which ruled out the usual esterification of acid and thiol. Moreover, the thioesterification reaction was treated with a radical inhibitor. When excess TEMPO (5 equivalents) was added, no thioester could be observed, which indicated that the iron-catalyzed thioesterification may follow a radical mechanism.
Based on the above results and in combination with the already known mechanism in the literature,15j a plausible mechanism was proposed as shown in Scheme 2. In the iron-catalyzed reaction, toluene was first oxidized to form benzaldehyde, which further reacted with t-BuOO˙ to give an acyl radical. On the other hand, iron salts promoted TBHP to form t-BuOO˙ and the FeBr2 converted to intermediate A, which further reacts with the thiol or disulfide to give an iron(III) thiolate complex B and release HBr. After that the complex B reacted with acyl radical which provide the aimed thioester and complex C, and C reacted with HBr to regenerate FeBr2 to complete the circle.
 | ||
| Scheme 2 Proposed mechanism for the Fe-catalyzed thioesterification and Pd-catalyzed thioetherification via sp3 C–H activation of methylarenes. | ||
As for the Pd-catalyzed cycle, the catalytic cycle starts with PdII-mediated C–H cleavage to form benzyl Pd complex A′. The benzylation products may result from nucleophilic attack on the benzylic carbon by thiophenol (Path A). Alternatively, they can undergo ligand exchange to afford benzyl Pd(II)carboxylate B′, and subsequent reductive elimination yields thioether products (Path B). The Pd0 species generated in Path A or B is oxidised to PdII by O2 to continue the catalytic cycle. It should be noted that the existing TBHP may accelerate the C–H cleavage and the palladium oxidation process (Pd0–PdII).
Experimental section
Melting points are uncorrected. All commercial materials were used without further purification. Thiols were synthesized according to the literature;24 TBHP used was 70% TBHP in water. GC-MS analyses were performed on an Agilent 7890A-5975C instrument (Column: DB-5 MS). NMR was recorded on Bruker DRX 500 and tetramethylsilane (TMS) was used as a reference. Elemental analysis was performed on a Yanagimoto MT3CHN recorder. Preparative high performance liquid chromatography was performed on an Agilent Technologies 1200 Column: XDB-C18 9.6 × 250 mm.General procedure for synthesis of thioesters
A 5 mL vial with condenser was charged with toluene (1.5 mmol), FeBr2 (0.01 mmol), TBHP (1.5 mmol), 2 wt% SDS–H2O (2 mL). Thiol (or thiophenol, 0.5 mmol) was then added dropwise (in portion for solids) at 100 °C. The reaction mixture was stirred at 100 °C for 24 h. Upon completion, the reaction mixture was then cooled, extracted with ethyl acetate, and dried over anhydrous Na2SO4. After filtration, the organic solutions were concentrated and the residue was purified by column chromatography on silica gel to give the pure product (hexane–ethyl acetate = 20/1).General procedure for synthesis of thioethers
A mixture of toluene (1.5 mmol), Pd(OAc)2 (0.05 mmol), TBHP (1.5 mmol), and thiol (thiophenol) (0.5 mmol) was introduced in a pressure reactor. The suspension was stirred at 115 °C for 24 h under 1 atm of oxygen. After this, the pressure was released and the mixture was extracted with ethyl acetate, and dried over anhydrous Na2SO4. After filtration, the organic solutions were concentrated and the residue was purified by column chromatography on silica gel to give the pure product (hexane–ethyl acetate = 50/1).Conclusions
In summary, we developed two facile and efficient protocols, by which thioesters and thioethers can be selectively prepared via the Fe-catalyzed C–H functionalization thioesterification and Pd-catalyzed C–H functionalization thioetherification, respectively. The major advantages of these methods include the use of toluene as the benzylation or benzoylation reagent and oxygen or TBHP as the green oxidant, which has high atom economy, avoids the prefunctionalization of substrates and reduces the production of chemical wastes. In addition, the two rarely reported protocols demonstrate good functional group tolerance and moderate to good yields.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of Jiangsu Province (BK 20131346) for financial support.Notes and references
- For selected reviews or books on transition-metal-catalyzed C–H functionalization, see: (a) G. Dyker, Handbook of C-H Transformations: Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) J.-Q. Yu and Z.-J. Shi, C-H Activation, Springer, Berlin, Germany, 2010 Search PubMed; (c) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (d) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- (a) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS PubMed; (c) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS PubMed; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (f) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC.
- For selected recent references on CDC C–C formation see: (a) Y. Wu, B. Li, F. Mao, X. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed; (b) F. Péron, C. Fossey, T. Cailly and F. Fabis, Org. Lett., 2012, 14, 1827 CrossRef PubMed; (c) R. K. Chinnagolla and M. Jeganmohan, Org. Lett., 2012, 14, 5246 CrossRef CAS PubMed; (d) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef CAS PubMed; (e) M. Brasse, J. Campora, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2013, 135, 6427 CrossRef CAS PubMed; (f) L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 2906 CrossRef CAS PubMed; (g) X. Cong, J. You, G. Gao and J. Lan, Chem. Commun., 2013, 49, 662 RSC; (h) S. Guin, S. K. Rout, A. Banerjee, S. Nandi and B. K. Patel, Org. Lett., 2012, 14, 5294 CrossRef CAS PubMed.
- For selected recent references on CDC C–O formation see: (a) W. Zhou, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7094 CrossRef CAS PubMed; (b) S. K. Rout, S. Guin, K. K. Ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982 CrossRef CAS PubMed; (c) X. F. Cheng, Y. Li, Y. M. Su, F. Yin, J. Y. Wang, J. Sheng, H. U. Vora, X. S. Wang and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (d) D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448 CrossRef CAS PubMed; (e) P. Novak, A. Correa, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef CAS PubMed; (f) K. Takenaka, M. Akita, Y. Tanigaki, S. Takizawa and H. Sasai, Org. Lett., 2011, 13, 3506 CrossRef CAS PubMed; (g) Y. Wei and N. Yoshikai, Org. Lett., 2011, 13, 5504 CrossRef CAS PubMed; (h) E. Shi, Y. Shao, S. Chen, H. Hu, Z. Liu, J. Zhang and X. Wan, Org. Lett., 2012, 14, 3384 CrossRef CAS PubMed; (i) W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484 CrossRef CAS PubMed; (j) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef CAS PubMed; (k) T. S. Jiang and G. W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed; (l) W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362–8366 CrossRef CAS PubMed.
- For selected recent references on CDC C–N formation see: (a) K. Kamata, T. Yamaura and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7250 CrossRef PubMed; (b) R. Vanjari, T. Guntreddi and K. N. Singh, Org. Lett., 2013, 15, 4908 CrossRef CAS PubMed; (c) M. L. Louillat and F. W. Patureau, Org. Lett., 2013, 15, 164 CrossRef CAS PubMed; (d) J. Hu, S. Chen, Y. Sun, J. Yang and Y. Rao, Org. Lett., 2012, 14, 5030 CrossRef CAS PubMed; (e) J. L. Liang, S. X. Yuan, J. S. Huang, W. Y. Yu and C. M. Che, Angew. Chem., Int. Ed., 2002, 41, 3465 CrossRef CAS; (f) M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638 CrossRef CAS PubMed; (g) R. J. Tang, C. P. Luo, L. Yang and C. J. Li, Adv. Synth. Catal., 2013, 355, 869 CrossRef CAS; (h) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed; (i) J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953 RSC; (j) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2012, 14, 656 CrossRef CAS PubMed; (k) K. H. Ng, Z. Zhou and W. Y. Yu, Org. Lett., 2012, 14, 272 CrossRef CAS PubMed.
- (a) B. N. Du, B. Jin and P. P. Sun, Org. Lett., 2014, 16, 3032 CrossRef CAS PubMed; (b) S. R. Guo, Y. Q. Yuan and J. N. Xiang, Org. Lett., 2013, 15, 4654 CrossRef CAS PubMed; (c) B. Basu, B. Mandal, S. Das and S. Kundu, Tetrahedron Lett., 2009, 50, 5523 CrossRef CAS; (d) In the process for preparation of the paper, Prof. Bhisma K. Patel reported similar thioesterification using copper catalysts see: W. Ali, S. Guin, S. K. Rout, A. Gogoi and B. K. Patel, Adv. Synth. Catal., 2014, 356, 3099 CrossRef CAS.
- (a) H. Tokuyama, S. Yokoshima, T. Yamashita and T. Fukuyama, Tetrahedron Lett., 1998, 39, 3189 CrossRef CAS; (b) V. P. Mehta, A. Sharma and E. Van der Eycken, Org. Lett., 2008, 10, 1147 CrossRef CAS PubMed; (c) I. A. Os'kina and V. M. Vlasov, Russ. J. Org. Chem., 2009, 4, 523 CrossRef; (d) H. Azuma, K. Okano and H. Tokuyama, Chem. Lett., 2011, 40, 959 CrossRef CAS; (e) M. N. Burhardt, A. Ahlburg and T. Skrydstrup, J. Org. Chem. DOI:10.1021/jo5009965; (f) R. J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons, Chichester, 1996 Search PubMed.
- (a) D. N. Jones, Comprehensive Organic Chemistry, ed. D. H. Barton and D. W. Ollis, Pergamon, NewYork, 1979 Search PubMed; (b) A. Marcincal-Lefebvre, J. C. Gesquiere and C. Lemer, J. Med. Chem., 1981, 24, 889 CrossRef CAS; (c) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (d) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (e) S. Magens and B. Plietker, Chem.–Eur. J., 2011, 17, 8807 CrossRef CAS PubMed; (f) A. R. Katritzky, A. A. Shestopalov and K. Suzuki, Synthesis, 2004, 1806 CrossRef CAS; (g) S. Limura, K. Manabe and S. Kobayashi, Chem. Commun., 2002, 94 Search PubMed; (h) C. C. Silveira, A. L. Braga and E. L. Larghi, Organometallics, 1999, 18, 5183 CrossRef CAS; (i) H. M. Meshram, G. S. Reddy, K. H. Mindu and J. S. Yadav, Synlett, 1998, 877 CrossRef CAS.
- (a) A. V. Bierbeek and M. Gingras, Tetrahedron Lett., 1998, 39, 6283 CrossRef; (b) C. C. Eichman and J. P. Stambuli, J. Org. Chem., 2009, 74, 4005 CrossRef CAS PubMed; (c) T. Dahl, C. W. Tornøe, B. Bang-Andersen, P. Nielsen and M. Jørgensen, Angew. Chem., 2008, 120, 1726 CrossRef PubMed; (d) V. Percec, J. Y. Bae and D. H. Hill, J. Org. Chem., 1995, 60, 6895 CrossRef CAS; (e) Y. C. Wong, T. T. Jayanth and C. H. Cheng, Org. Lett., 2006, 8, 5613 CrossRef CAS PubMed; (f) N. Taniguchi and T. Onami, J. Org. Chem., 2004, 69, 915 CrossRef CAS PubMed; (g) F. Y. Kwong and S. L. Buchwald, Org. Lett., 2002, 4, 3517 CrossRef CAS PubMed; (h) Y. J. Chen and H. H. Chen, Org. Lett., 2006, 8, 5609 CrossRef CAS PubMed; (i) V. P. Reddy, K. Swapna, A. V. Kumar and K. R. Rao, J. Org. Chem., 2009, 74, 3189 CrossRef CAS PubMed; (j) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 1697 CrossRef CAS PubMed; (k) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef CAS PubMed; (l) J. R. Wu, C. H. Lin and C. F. Lee, Chem. Commun., 2009, 4450 RSC.
- J. R. Campbell, J. Org. Chem., 1964, 29, 1830 CrossRef CAS.
- J. Ham, I. Yang and H. Kang, J. Org. Chem., 2004, 69, 3236 CrossRef CAS PubMed.
- (a) B. C. Ranu and R. Jana, Adv. Synth. Catal., 2005, 347, 1811 CrossRef CAS; (b) T. Uno, T. Inokuma and Y. Takemoto, Chem. Commun., 2012, 48, 1901 RSC; (c) C. Simiona, I. Hashimotob, Y. Mitomac, A. M. Simiona and N. Egashirac, Phosphorus, Sulfur, 2010, 185, 2480; (d) S. Narayanaperumal, E. E. Alberto, K. Gul, C. Y. Kawasoko, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Tetrahedron, 2011, 67, 4723 CrossRef CAS; (e) B. Basu, S. Paul and A. K. Nanda, Green Chem., 2010, 12, 767 RSC.
- (a) S. Iimura, K. Manabe and S. Kobayashi, Chem. Commun., 2002, 94 RSC; (b) N. Iranpoor, H. Firouzabadi, D. Khalili and S. Motevalli, J. Org. Chem., 2008, 73, 4882 CrossRef CAS PubMed.
- (a) A. R. Massah, R. J. Kalbasi, M. Toghiani, B. Hojati, M. Adibnejad and A. R. Massah, J. Chem., 2012, 9, 2501 CAS; (b) S. K. Prajapti, A. Nagarsenkar and B. N. Babu, Tetrahedron Lett., 2014, 55, 1784 CrossRef CAS.
- Selected recent references see: (a) S. Rossi, M. Benaglia, F. Cozzi, A. Genoni and T. Benincori, Adv. Synth. Catal., 2011, 353, 848 CrossRef CAS; (b) D. Crich and I. Sharma, Angew. Chem., Int. Ed., 2009, 48, 2355 CrossRef CAS PubMed; (c) D. Crich and K. Sasaki, Org. Lett., 2009, 11, 3514 CrossRef CAS PubMed; (d) K. Matsuo and M. Sindo, Org. Lett., 2010, 12, 5346 CrossRef CAS PubMed; (e) K. Kunchithapatham, C. C. Eichman and J. P. Stambuli, Chem. Commun., 2011, 47, 12679 RSC; (f) T. Uno, T. Inokuma and Y. Takemoto, Chem. Commun., 2012, 48, 1901 RSC; (g) X. Zhu, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2013, 355, 3558 CrossRef CAS; (h) S. Iwahana, H. Iida and E. Yashima, Chem.–Eur. J., 2011, 17, 8009 CrossRef CAS PubMed; (i) C.-L. Yi, Y.-T. Huang and C.-F. Lee, Green Chem., 2013, 15, 2476 RSC; (j) J.-W. Zeng, Y.-C. Liu, P.-A. Hsieh, Y.-T. Huang, C.-L. Yi, S. S. Badsara and C.-F. Lee, Green Chem., 2014, 16, 2644 RSC; (k) Y.-T. Huang, S.-Y. Lu, C.-L. Yi and C.-F. Lee, J. Org. Chem., 2014, 79, 4561 CrossRef CAS PubMed.
- Selected reviews see: (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253 RSC; (c) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC.
- (a) G. Majji, S. Guin, A. Gogoi, S. K. Rout and B. K. Patel, Chem. Commun., 2013, 49, 3031 RSC; (b) L. Liu, L. Yun, Z. Wang, X. Fu and C.-H. Yan, Tetrahedron Lett., 2013, 54, 5383 CrossRef CAS; (c) S. K. Rout, S. Guin, W. Ali, A. Gogoi and B. K. Patel, Org. Lett., 2014, 16, 3086 CrossRef CAS PubMed.
- (a) Y.-J. Bian, C.-B. Xiang, Z.-M. Chen and Z.-Z. Huang, Synlett, 2011, 16, 2407 Search PubMed; (b) S. K. Rout, S. Guin, A. Banerjee, N. Khatun, A. Gogoi and B. K. Patel, Org. Lett., 2013, 15, 4106 CrossRef CAS PubMed; (c) Z. Yin and P. Sun, J. Org. Chem., 2012, 77, 11339 CrossRef CAS PubMed; (d) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (e) D. L. Priebbenow and C. Bolm, Org. Lett., 2014, 16, 1650 CrossRef CAS PubMed.
- S. K. Rout, S. Guin, K. K. Ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982 CrossRef CAS PubMed.
- (a) H. Liu, G. Shi, S. Pan, Y. Jiang and Y. Zhang, Org. Lett., 2013, 15, 4098 CrossRef CAS PubMed; (b) L. Ju, J. Yao, Z. Wu, Z. Liu and Y. Zhang, J. Org. Chem., 2013, 78, 10821 CrossRef CAS PubMed.
- (a) R. Tang, Y. Xie, Y. Xie, J. Xiang and J. Li, Chem. Commun., 2011, 47, 12867 RSC; (b) Y. P. Zhu, M. Lian, F. C. Jia, M. C. Liu, J. J. Yuan, Q. H. Gao and A. X. Wu, Chem. Commun., 2012, 48, 9086 RSC; (c) Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC; (d) F. Yang and S. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed.
- B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854 RSC.
- T. Darmanin, E. T. de Givenchy, S. Amigoni and F. Guittard, J. Fluorine Chem., 2012, 134, 85 CrossRef CAS.
- C.-C. Han and R. Balakumar, Tetrahedron Lett., 2006, 47, 8255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; spectral data for all reported compounds. See DOI: 10.1039/c4ra09450f |
| This journal is © The Royal Society of Chemistry 2014 |