
A conformationally switchable fluorescent oligophenol foldamer for selective sensing of copper(II) ions†
Jiaqiang Liu‡
a,
Chang Sun‡b,
Wenliang Maa,
Yu-Jing Lua,
Lin Yua,
Kun Zhanga and
Huaqiang Zeng*c
aFaculty of Chemical Engineering and Light Industry, Guang Dong University of Technology, Guang Dong, 510006, China
bCollege of Textiles and Clothing, Jiangnan University, 1800 Lihu Avenue, Wuxi, Jiangsu 214122, China
cInstitute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, 138669, Singapore. E-mail: hqzeng@ibn.a-star.edu.sg; Tel: +65-6824-7115
First published on 17th October 2014
Abstract
A stimuli-responsive hexameric oligophenol host undergoes metal ion-induced co-operative folding from a more fluorescent, more linear structure into less fluorescent, more curved states, enabling easy classification of the bound metal ion guests as well as selective sensing of Cu2+ ions.
Helically or circularly folded aromatic foldamers have continuously attracted considerable interest over the past two decades since the pioneering work using multiple-center H-bonding systems to direct the intramolecular folding of aromatic pyridine-based backbones by Hamilton and co-workers in 1994.1 Concurrent with the elaboration of their wide-ranging structures, these H-bonded aromatic foldamers2 have been demonstrated to perform highly variable functions including reaction catalysis,3a controllable molecular motion,3b reactive sieving,3c–f solvent gelating,3g,h ion transportation,3i stabilization of G-quadruplex structures,3j and selective recognition of both ionic4 and neutral5 species (e.g., amines,4i–k water,5a–c methanol/dichloromethane,5d,e saccharides,5f tartaric acid,5g etc.). Nevertheless, the hitherto developed diverse classes of H-bonded aromatic foldamers mostly lack the structure-switching ability, and so perform their pre-designed functions without significant conformational changes in their molecular backbones. In other words, H-bonded aromatic backbones containing switchable units have been studied comparatively much less,4e,j,k and no study on the use of metal ions to trigger the conformational change of these H-bonded aromatic foldamers for selectively sensing certain metal ions has been reported. The H-bonded foldamer molecules endowed with “switchable” functions are undoubtedly of great interest in the creation of advanced adaptive bio- and nanomaterials and molecular sensing devices for such as environmental monitoring of toxic molecular species including metal ions. In this communication, we report our very first step toward developing specific sensing of metal ions using a carefully designed and optimized fluorescent folding molecule derived from phenol unit 1a, which can undergo dynamic folding/unfolding processes in response to the external stimuli such as pH and amines,4j,k,6 and methoxybenzene unit 2 for fine-tuning the curvature of the folding backbone (Fig. 1a).
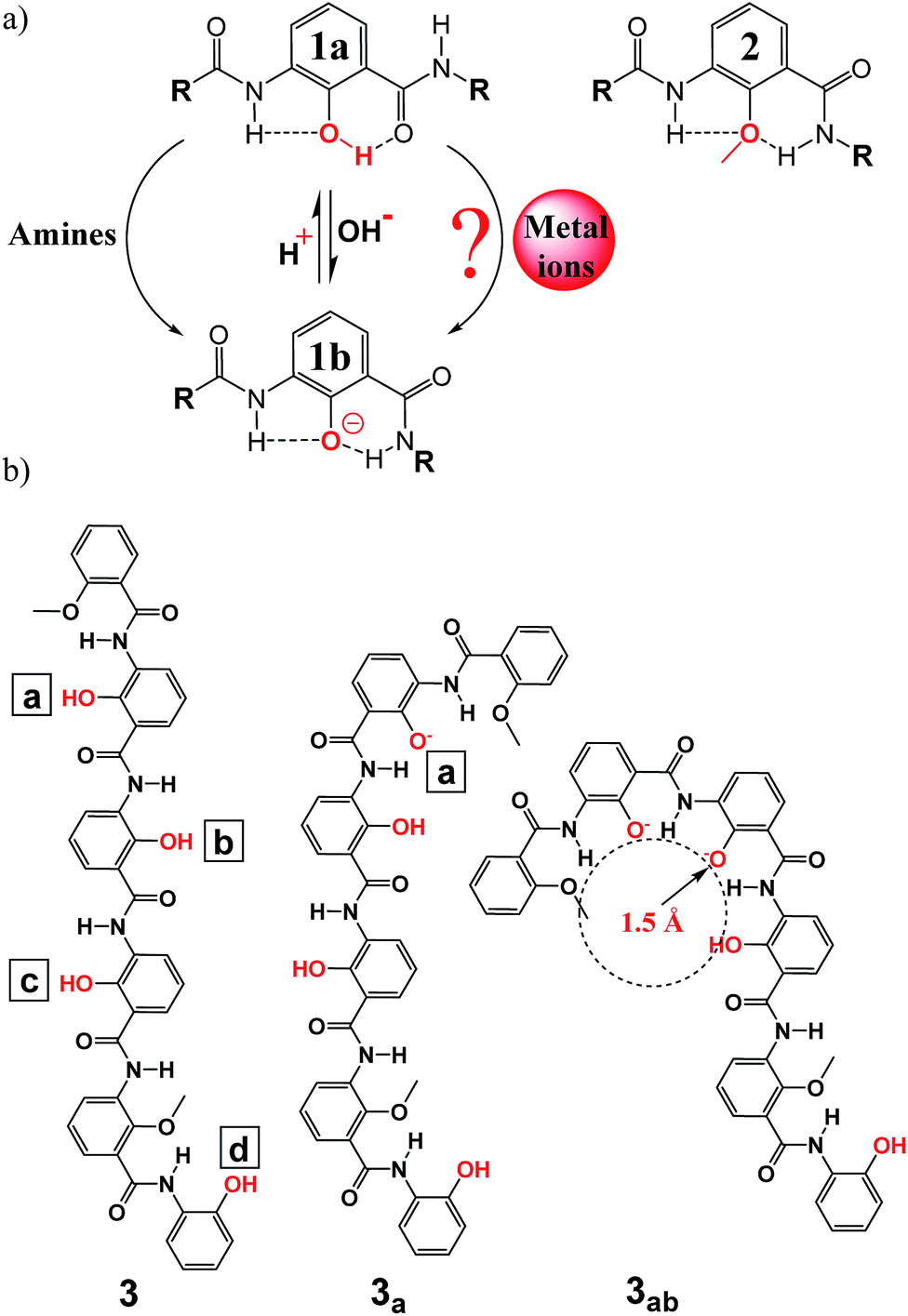 | ||
| Fig. 1 (a) Conformational switching between phenol-based amides 1 and 2 can be efficiently induced by primary amines4j,k or by deprotonation/protonation6 of the hydroxyl group; whether or not metal ions can induce the similar conformational switching is the focus of the current work. (b) Structure of a hexameric oligophenol foldamer 3 containing four deprotonable OH groups and its representative anionic oligomers such as 3a and 3ab obtained by selectively deprotonating some hydroxyl groups in 3 at positions a–d; for instance, anionic 3ab refers to the anionic oligomer where the two hydroxyl groups at positions a and b were deprotonated. Do note that mono-anionic oligomers such as 3a are moderately fluorescent, and di-, tri- or tetra-anionic oligomers are virtually non-fluorescent.4j,k | ||
As demonstrated recently by us4j,k and others,6 neutral phenol unit 1a makes the phenol-based oligomeric backbone more linear and possibly more planar while both phenolate unit 1b and methoxybenzene unit 2 result in a more curved or even helically folded conformation if the molecular backbone contains five or more repeating units 1b or 2.4j,k,6,7 Depending on the location and number of units 1a containing an OH group and 2 containing an OMe group, the hybrid foldamer molecules composed of units 1a and 2 in various ratios can thus differ dramatically in their backbone curvature.4j,k Moreover, these neutral oligophenol molecules as represented by 3 display very interesting conformation-dependent fluorescent properties roughly in line with the relative linearity of the hexameric backbones.4j The corresponding anionic versions such as 3a generated from 3 by deprotonating the OH group at position a (Fig. 1b) were also demonstrated to follow the same curvature-dependant fluorescent properties.4j Therefore, the fluorescent ability decreases in the order of strongly fluorescent linear 3 > moderately fluorescent less linear mono-anionic 3a, 3b and 3c > virtually non-fluorescent more crescent-shaped di-, tri- and tetra-anionic 3ab, 3abc, 3abcd, etc. Since the phenolic OH groups at positions a–d are sensitive to the basicity of the solutions, amines of different types cause the four OH groups to deprotonate in a defined sequence but to varying degrees, leading to a four-step sequential folding of 3 from a more fluorescent linear structure into an essentially non-fluorescent helically folded tetra-anionic state via mono-anionic 3b, di-anionic 3bc and tri-anionic 3abc. Experimentally, it was observed that non-branched primary amines and cyclic secondary amines produce crescent-shaped anionic oligomers to the largest extent, followed by branched primary amines and acyclic secondary amines with both tertiary amines and ammonium ions to the least, making possible the patterned recognition of varying amines and ammonium ions by oligophenol 3.4j,k
Encouraged by the amine-mediated conformational switching of 3 and the corresponding patterned recognition of amines and ammonium ions by 3, we further envisioned that 3 containing deprotonable phenolic OH groups possibly could be sensitive to the basic metal ions capable of removing the phenolic hydroxyl protons, subsequently forming strong coordination bonds with and stabilizing the resultant phenolate O-atoms. Under this hypothetical scenario, metal ions might be able to induce the folding of linear 3 into crescent-shaped or helically folded anionic oligomers with lessened fluorescence ability (Fig. 1b) to differential extents, possibly allowing an ease detection or classification of metal ions via changes in fluorescence intensity of 3. A selective detection of trace amount of certain metal ions against a background of other competing ions is highly preferred and of great interests given that the heavy metal pollutions are now a worldwide problem that will continue to grow in the future.
To test this hypothesis, a series of titration experiments using 22 metal ions were carried out. In the typical experimental set-up, metal ions were dissolved in water to prepare a stock concentration of 10 mM, followed by addition of 2 μL of the aqueous solution into 1 mL of 3 in THF containing 1% DMSO at 10 μM at room temperature. The resultant solution contains a 3–metal ion molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and the corresponding fluorescence spectra/data were obtained with an excitation wavelength at 351 nm. Following this procedure, treatment of 3 with various metal ions indeed gave rise to differential changes in fluorescence intensity of 3 that mostly depend on the nature of the metal ions rather than the charge-neutralizing anionic parts (Fig. 2) or fluorescence-quenching ability of the metal ions.8
:2, and the corresponding fluorescence spectra/data were obtained with an excitation wavelength at 351 nm. Following this procedure, treatment of 3 with various metal ions indeed gave rise to differential changes in fluorescence intensity of 3 that mostly depend on the nature of the metal ions rather than the charge-neutralizing anionic parts (Fig. 2) or fluorescence-quenching ability of the metal ions.8
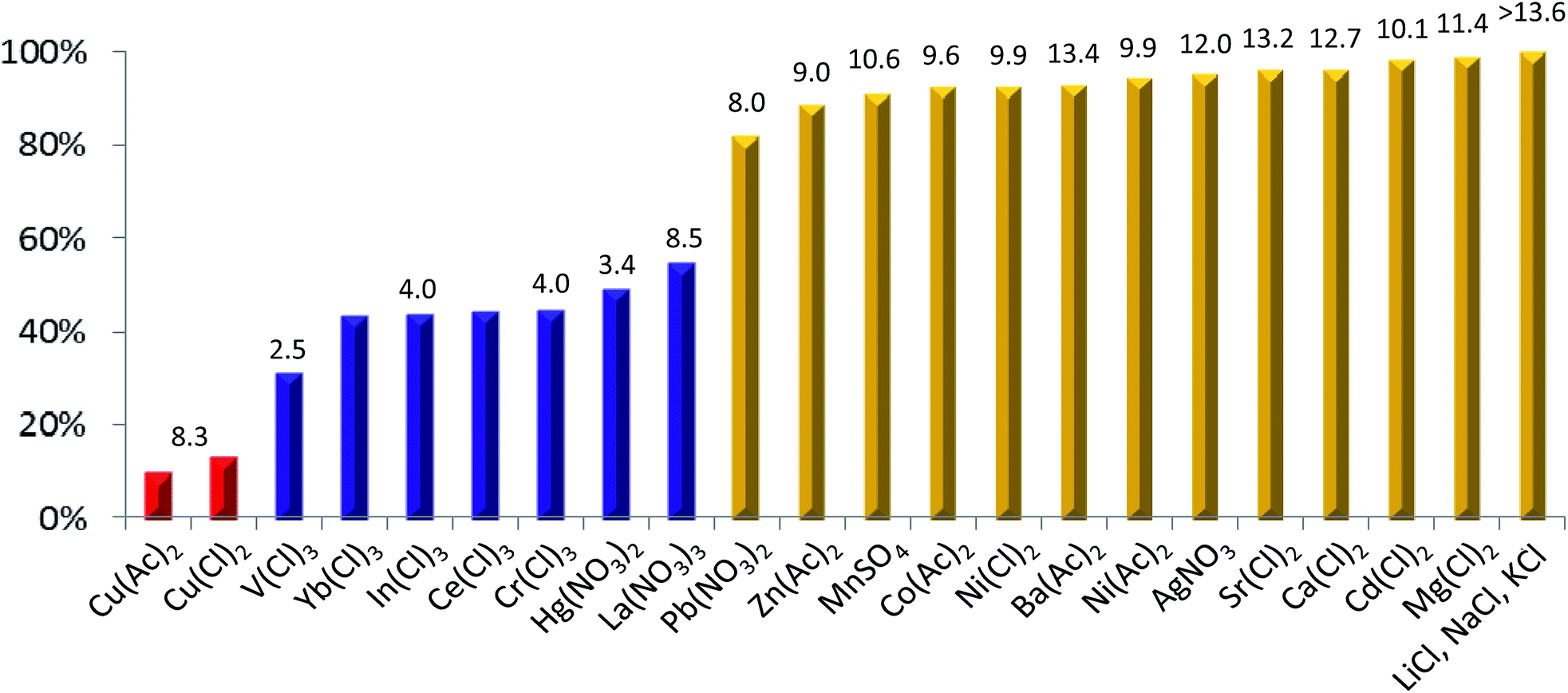 | ||
| Fig. 2 Patterned fluorescence quenching of 3 by two equivalents of metal ions of various types. The fluorescence spectra/data were obtained at 10 μM of 3 in THF containing 1% DMSO at room temperature with an excitation wavelength at 351 nm. The values above the columns are the hydrolysis constants (pKa's) of the respective metal ions.9a,b The fluorescence intensity of 3 was quenched to the highest extents of 90.1% and 86.6% by Cu(Ac)2 and CuCl2, respectively, while Li+, Na+ and K+ ions do not elicit any measurable quenching of 3 within the detection limit of the instrument. | ||
A few points can be noted in terms of correlation between the hydrolysis constant (pKa)9a,b of metal ions and fluorescence quenching of 3 by the metal ions: (1) mono- and divalent metal ions with a pKa value of 8 or larger generally effect a marginal quenching of 18% in the best scenario by Pb(NO3)2, (2) divalent metal ions such as Hg2+ with a much lower pKa or trivalent ions such as La3+ with a pKa as high as 8.5 still can produce significant quenching of about 50%, and (3) trivalent metal ions with lower pKa values quench the fluorescence intensity of 3 by as much as 69% in the case of VCl3. Similar to the hydrolytic reactions the hydrated metal ions may undergo in aqueous solution, these data strongly suggest that, in the presence of 3 containing deprotonable phenolic OH groups, metal ions might react preferentially with the more reactive phenolic OH groups with a pKa value of around 10, rather than with the water molecules with a pKa value of 15.7.9c Such deprotonation reactions produce phenolate anions that can be further stabilized by metal ions via the formation of coordination bonds, cooperatively enhancing the deprotonation extent of the phenolic OH groups and concurrently inducing very fluorescent linear 3 into its moderately or weakly fluorescent anionic structures of varying types by switching from H-bonding pattern in 1a to that in 1b (Fig. 1a).
Since the hydrolysis constant of the metal ions, pKa, is a parameter that has taken into consideration of metal ions' ionic radius and valence state, and the stability of metal hydroxide complexes, the fact that the quenching extent by various metal ions does not strictly follow their relative magnitude in pKa value suggests the ability of the metal ions to stabilize the phenolate anions via the formation of coordination bonds to be another key factor that determines the extent of conformational folding of the linear host into more curved weakly fluorescent anionic structure. Accordingly, the extent of metal ion-induced deprotonation and subsequent conformational switching of 3 is dependent on not only the hydrolysis constant of the metal ions but also the strength of metal–ligand coordination bonds that can more than compensate for the dehydration energy associated with the loss of a few water molecules around metal ions upon their binding to phenolate O-atoms. On these premises, even with similar pKa values, La3+ ions are possibly more able to stabilize the crescent-shaped anionic oligomers, subsequently induce linear 3 into curved anionic structures to higher extents and elicit more significant fluorescence quenching than metal ions such as Pb2+ and Zn2+. On the same grounds, the highest quenching of about 90% by Cu2+ ions can be explained on the basis of its expected highest ability to stabilize the phenolate-based anionic oligomers of certain structures, rather than its hydrolysis constant of as larger as 8.3.
Previously, by titrating 3 stepwise using strong organic base, tetrabutylammonium hydroxide, we have demonstrated that the fluorescence intensity of mono-anionic oligomer is about 40% of that of neutral 3, and di-/tri-/tetra-anionic oligomers are essentially non-fluorescent.4j The fluorescence quenching of as high as 91% by two equivalents of Cu(Ac)2 points to a high likelihood of Cu2+-assisted generation of di-anionic oligomers such as 3ab and 3bc as the predominant anionic forms10a with co-existence of neutral 3 and its other mono-/di-/tri-/tetra-anionic oligomers in minor forms in solution.10b In di-anionic 3ab or 3bc, a cavity of about 1.5 Å in radius excluding the atomic volume of O-atoms is clearly present that is formed by four O-atoms among which two are from the two immediately adjacent phenolate anions (Fig. 1b). Highly likely, it is the excellent ability of the Cu2+ ions to fit into the cavity that leads to surprisingly high quenching effect on 3 exerted by Cu2+ ions with respect to all the other metal ions studied in this work.
To support the above notion that the crescent-shaped electron-rich cavity of about 1.5 Å in radius is able to bind electron-deficient metal ions, various efforts to crystallize the metal–ligand complexes formed between metal ions and 3 or other oligomers such as trimer 4 and 5 have been attempted. Eventually, the neutral 4 and anionic 5 in complex with a Na+ ion were crystallized with their crystal structures illustrated in Fig. 3. Structural comparison between neutral 4 and anionic 5 once again substantiates our above statement that phenol unit 1a makes the backbone more linear via a six-membered H-bond of O–H⋯O type while phenolate unit 1b causes the backbone to bend more to enclose a sizable hydrophilic cavity in trimer or higher oligomers. The crystal structure of 5·Na+ demonstrates the cation-binding ability of the enclosed cavity in 5. Specifically, the Na+ ion forms two strong coordination bonds of 2.279 Å and 2.664 Å in length with phenolate and methoxy O-atoms, respectively, and another three bonds with one water molecule and the two amide O-atoms from the two adjacent molecules of 5. Apparently, the two methoxy groups in 5 force Na+ ion to stay above or below the near planar trimeric backbone.
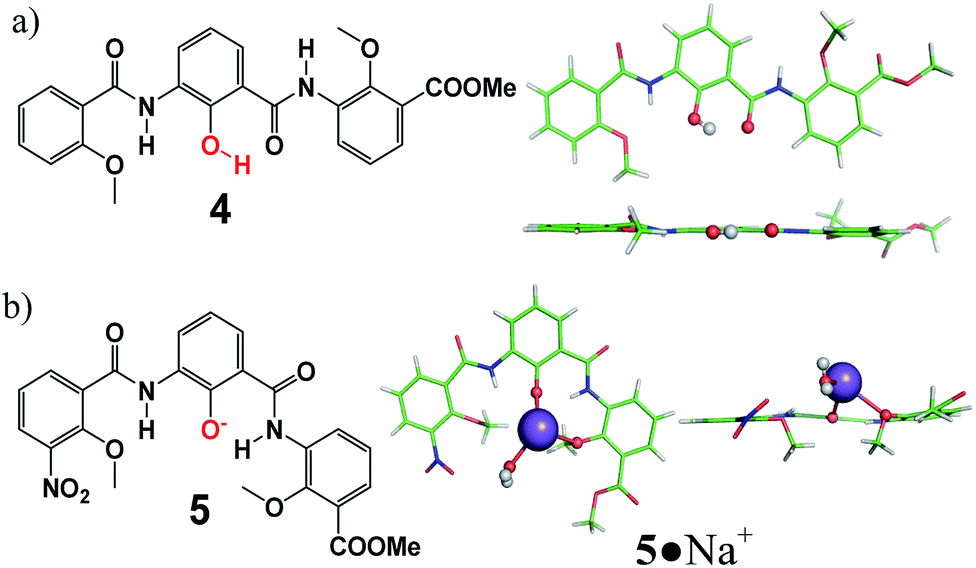 | ||
| Fig. 3 The chemical structures of (a) trimer 4 containing a phenol unit and (b) trimer 5 containing a phenolate unit as well as the corresponding crystal structures of 4 and 5·Na+. The large purple balls in (b) refer to Na+ ion, and a water molecule coordinated to the Na+ ion is also shown. | ||
First principle calculations at the level of B3LYP/6-31G(d,p) using THF as the explicit solvent were then carried out to deduce the possible complexes formed between Cu2+ and di-anionic oligomers 3ab or 3bc. Our calculations reveal complex 3ab·Cu2+ to be significantly more stable than 3bc·Cu2+ by 6.71 kcal mol−1 (Fig. S1†), leading us to favorably consider 3ab·Cu2+ as the predominant quenching species responsible for the observed quenching of 3 by Cu2+ ions. In the computationally determined structure for 3ab·Cu2+, Cu2+ ion is stabilized primarily by forming two strong coordination bonds with the two negatively charged phenolate O-atoms with respective bond lengths of 1.89 Å and 1.86 Å, and additionally by interacting with the adjacent amide N-atom (2.48 Å) (Fig. S1a†).
To summarize, the above observations demonstrate that metal ions can be used to induce a conformational change in phenol-based host 3 containing switchable phenolic hydroxyl groups. In most cases studied, such structure-switching ability exhibited by metal ions seems to be in good accord with the metal ions' hydrolysis constants in that metal ions with considerably smaller hydrolysis constants are more able to deprotonate the OH groups in 3 and thus switch 3 from a more linear more fluorescent structure to more curved less fluorescent states. Nevertheless, for La3+ and Cu2+ ions, the hydrolysis constant appears not to be the sole determining factor. In particular, the abnormally high quenching ability exhibited by Cu2+ ions suggests the efficient co-operative interactions between the in situ generated anionic hosts such as 3ab or 3bc and Cu2+ ions, rather than the metal ion's hydrolysis constants, to be the most influential factor that induces the more linear 3 into more curved anionic structures possibly with 3ab as the predominant anionic form in solution. In connection with the recently elucidated diverse functions by the H-bonded aromatic foldamers of varying types,2 it is quite unusual to note that there has been no use of metal ions to promote the conformational switching of these H-bonded aromatic foldamer molecules for possibly selective sensing of metal ions. The approach described here may promise more selective recognitions of metal ions by other analogous conformationally switchable phenol-based foldamers with elongated backbones and/or with the replacement of methoxy groups by other bulkier groups and the incorporation of electron-donating/withdrawing groups in the aromatic backbone.
Acknowledgements
Financial supports of this work to H.Z. by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore), the A*STAR Computational Resource Centre through the use of its high performance computing facilities and NRF CRP Grant (NRF-CRP7-2010-03).Notes and references
- Y. Hamuro, S. J. Geib and A. D. Hamilton, Angew. Chem., Int. Ed., 1994, 33, 446 CrossRef.
- (a) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS; (b) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (c) K. Yamato, M. Kline and B. Gong, Chem. Commun., 2012, 48, 12142 RSC; (d) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed; (e) W. Q. Ong and H. Q. Zeng, J. Inclusion Phenom. Macrocyclic Chem., 2013, 76, 1 CrossRef CAS; (f) H. L. Fu, Y. Liu and H. Q. Zeng, Chem. Commun., 2013, 49, 4127 RSC.
- (a) H. Q. Zhao, J. Shen, J. J. Guo, R. J. Ye and H. Q. Zeng, Chem. Commun., 2013, 49, 2323 RSC; (b) Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172 CrossRef CAS PubMed; (c) K. Srinivas, B. Kauffmann, C. Dolain, J. M. Leger, L. Ghosez and I. Huc, J. Am. Chem. Soc., 2008, 130, 13210 CrossRef CAS PubMed; (d) H.-Y. Hu, J.-F. Xiang, J. Cao and C.-F. Chen, Org. Lett., 2008, 10, 5035 CrossRef CAS PubMed; (e) B. Qin, L. Y. Jiang, S. Shen, C. Sun, W. X. Yuan, S. F. Y. Li and H. Q. Zeng, Org. Lett., 2011, 13, 6212 CrossRef CAS PubMed; (f) Z. Y. Du, B. Qin, C. Sun, Y. Liu, X. Zheng, K. Zhang, A. H. Conney and H. Q. Zeng, Org. Biomol. Chem., 2012, 10, 4164 RSC; (g) W. Cai, G. T. Wang, P. Du, R. X. Wang, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2008, 130, 13450 CrossRef CAS PubMed; (h) C. L. Ren, S. Y. Xu, J. Xu, H. Y. Chen and H. Q. Zeng, Org. Lett., 2011, 13, 3840 CrossRef CAS PubMed; (i) A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. H. Yuan, A. J. Clements, S. V. Harding, G. Szabo, Z. F. Shao and B. Gong, J. Am. Chem. Soc., 2008, 130, 15784 CrossRef CAS PubMed; (j) P. S. Shirude, E. R. Gillies, S. Ladame, F. Godde, K. Shin-Ya, I. Huc and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 11890 CrossRef CAS PubMed.
- (a) A. Petitjean, L. A. Cuccia, J.-M. Lehn, H. Nierengarten and M. Schmutz, Angew. Chem., Int. Ed., 2002, 41, 1195 CrossRef CAS; (b) A. R. Sanford, L. H. Yuan, W. Feng, K. Y. R. A. Flowersb and B. Gong, Chem. Commun., 2005, 4720 RSC; (c) C. Li, S.-F. Ren, J.-L. Hou, H.-P. Yi, S.-Z. Zhu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725 CrossRef CAS PubMed; (d) B. Qin, C. L. Ren, R. J. Ye, C. Sun, K. Chiad, X. Y. Chen, Z. Li, F. Xue, H. B. Su, G. A. Chass and H. Q. Zeng, J. Am. Chem. Soc., 2010, 132, 9564 CrossRef CAS PubMed; (e) J.-M. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938 CrossRef CAS PubMed; (f) C. L. Ren, V. Maurizot, H. Q. Zhao, J. Shen, F. Zhou, W. Q. Ong, Z. Y. Du, K. Zhang, H. B. Su and H. Q. Zeng, J. Am. Chem. Soc., 2011, 133, 13930 CrossRef CAS PubMed; (g) S. Tashiro, K. Matsuoka, A. Minoda and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 13123 CrossRef CAS PubMed; (h) L. J. Zhong, L. Chen, W. Feng, S. L. Zou, Y. Y. Yang, N. Liu and L. H. Yuan, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 367 CrossRef CAS; (i) W. Q. Ong, H. Q. Zhao, C. Sun, J. E. Wu, Z. C. Wong, S. F. Y. Li, Y. H. Hong and H. Q. Zeng, Chem. Commun., 2012, 48, 6343 RSC; (j) C. Sun, C. L. Ren, Y. C. Wei, B. Qin and H. Q. Zeng, Chem. Commun., 2013, 49, 5307 RSC; (k) C. Sun, Y. Liu, J. Q. Liu, Y.-J. Lu, L. Yu, K. Zhang and H. Q. Zeng, J. Org. Chem., 2014, 79, 2963 CrossRef CAS PubMed; (l) A.-M. Stadler and J.-M. P. Lehn, J. Am. Chem. Soc., 2014, 136, 3400–3409 CrossRef CAS PubMed.
- (a) J. Garric, J.-M. Leger and I. Huc, Angew. Chem., Int. Ed., 2005, 44, 1954 CrossRef CAS PubMed; (b) W. Q. Ong, H. Q. Zhao, X. Fang, S. Woen, F. Zhou, W. L. Yap, H. B. Su, S. F. Y. Li and H. Q. Zeng, Org. Lett., 2011, 13, 3194 CrossRef CAS PubMed; (c) H. Q. Zhao, W. Q. Ong, X. Fang, F. Zhou, M. N. Hii, S. F. Y. Li, H. B. Su and H. Q. Zeng, Org. Biomol. Chem., 2012, 10, 1172 RSC; (d) H. Q. Zhao, W. Q. Ong, F. Zhou, X. Fang, X. Y. Chen, S. F. Y. Li, H. B. Su, N.-J. Cho and H. Q. Zeng, Chem. Sci., 2012, 3, 2042 RSC; (e) F. Zhou, H. L. Fu, W. Q. Ong, R. J. Ye, W. X. Yuan, Y.-J. Lu, Y.-P. Huo, K. Zhang, H. B. Su and H. Q. Zeng, Org. Biomol. Chem., 2012, 10, 5525 RSC; (f) J. L. Hou, X. B. Shao, G. J. Chen, Y. X. Zhou, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2004, 126, 12386 CrossRef CAS PubMed; (g) Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grelard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2010, 132, 7858 CrossRef CAS PubMed.
- D. Kanamori, T. A. Okamura, H. Yamamoto and N. Ueyama, Angew. Chem., Int. Ed., 2005, 44, 969 CrossRef CAS PubMed.
- Y. Yan, B. Qin, Y. Y. Shu, X. Y. Chen, Y. K. Yip, D. W. Zhang, H. B. Su and H. Q. Zeng, Org. Lett., 2009, 11, 1201 CrossRef CAS PubMed.
- The intrinsic fluorescence-quenching ability of transition/lanthanide metal ions should not be the determining factor for the observed differential quenching by the metal ions as compiled in Fig. 2. This can be substrantiated by the demonstrated conformation-dependent fluorescence property of 3 and the facts that (1) moderate quenchers such as Zn2+, Mn2+, Co2+, Ni2+ and Cd2+ behave similarly with non-quenchers such as Ba2+, Sr2+, Ca2+, Mg2+, Li+, Na+, and K+, and they all exhibit insignificant or weak fluorescence quenching of 3, (2) the fluorescence quenching of 3 by strong quenchers such as Hg2+, Pb2+ and lanthanide ions mostly follows their trend in pKa values, and (3) Cu2+ ions as a moderate quencher nevertheless produce the largest quenching among all the metal ions studied.
- (a) C. F. Baes and R. E. Messmer, The Hydrolysis of Cations, John Wiley & Sons Inc, New York, 1976 Search PubMed; (b) J. Burgess, Metal Ions in Solution, Ellis Horwood Ltd, Chichester, England, 1978, p. 264 Search PubMed; (c) It has been observed that Yb3+ ions are able to dramatically lower down the pKa value of the aliphatic OH group from 16 to about 6.4 in water, see: M. Lelli, G. Pintacuda, A. Cuzzola and L. D. Bari1, Chirality, 2005, 17, 201 CrossRef CAS PubMed.
- (a) Di-anionic oligomer 3ac is unlikely to be produced by Cu2+ as the two phenolate units in 3ac are separated by a phenol group, and as a result, the complex 3ac·Cu2+ is expectedly less stable than complexes 3ab·Cu2+ and 3bc·Cu2+; (b) Although we speculate that it is least likely to produce tri-anioinic oligomers such as 3abc or tetra-anionic 3abcd as the dominant anionic forms by two equivalents of Cu2+ ions via simultaneous deprotonation of three or four phenolic OH groups in 3, such processes cannot be completely excluded on the basis of data we have.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1017708 for trimer 4 and 1017709 for trimer 5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11706a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |