Formal anti-Markovnikov hydroamination of terminal olefins†
Sarah M.
Bronner
and
Robert H.
Grubbs
*
Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: rhg@caltech.edu; Fax: +1 626-564-9297
First published on 19th September 2013
Abstract
A new strategy to access linear amines from terminal olefin precursors is reported. This two-step, one-pot hydroamination methodology employs sequential oxidation and reduction catalytic cycles. The formal hydroamination transformation proceeds with excellent regioselectivity, and only the anti-Markovnikov product is observed. Up to 70% yield can be obtained from styrenes or aliphatic olefins and either primary or secondary aromatic amines. Additionally, the scope is broad with respect to the olefin and accommodates a variety of functionalities; we demonstrate that amines with removable aryl protecting groups may be utilized to allow access to a more diverse array of hydroamination adducts.
Introduction
Due to the prevalence of amines in therapeutics, as well as in the production of dyes, solvents, agrochemicals, and commodity and fine chemicals, the formation of carbon–nitrogen bonds is of tremendous importance.1 Hydroamination complements existing methods for the fabrication of carbon–nitrogen bonds and may also provide advantages – for example, catalytic hydroamination methodologies hold promise for being atom efficient and environmentally friendly,2 not requiring harsh conditions, and utilizing readily available and inexpensive amine and olefin (1 and 2 respectively, Fig. 1) starting materials. The construction of linear amines from terminal olefins represents a particularly significant and formidable transformation; selectivity is a consideration, as the amine may add at either of the two carbons of the alkene substrate (Fig. 1). Additions to give linear amines are generally more challenging, as Markovnikov addition is typically preferred. It should be noted that almost 20 years ago, the addition of amines to olefins in an anti-Markovnikov fashion was identified as one of the top ten challenges to be addressed by catalysis.3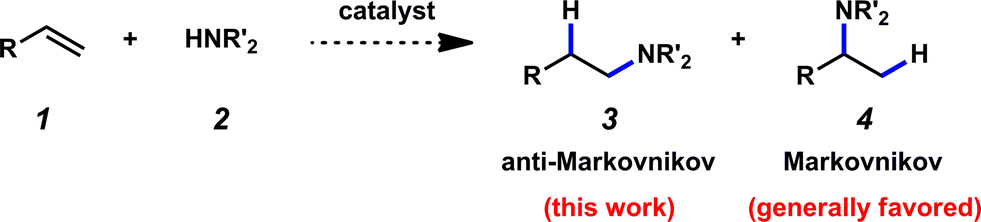 | ||
| Fig. 1 Olefin hydroamination. | ||
Although substantial progress has been made, a general catalytic method of anti-Markovnikov hydroamination of olefins remains to be developed.4,5 Previous approaches towards anti-Markovnikov hydroamination have traditionally involved activated olefins or intramolecular transformations.6–11 However, in 1999 the Beller group disclosed the first example of transition metal-catalyzed anti-Markovnikov hydroamination of olefins; this seminal study accomplished a low-yielding hydroamination of styrenes with secondary aliphatic amines using rhodium catalysis.12 More recently, some success in the metal-catalyzed intermolecular anti-Markovnikov hydroamination of unactivated olefins has been reported. For example, the Hultzsch and Beller groups have both performed the base-catalyzed anti-Markovnikov hydroamination of styrenes using LiN(SiMe3)2 and TMEDA13 or n-BuLi14 catalytic systems. Similarly, the Hartwig group has reported the rhodium-catalyzed anti-Markovnikov hydroamination of styrenes with secondary amines,15 while the Marks group has demonstrated that organolanthanide-catalysis is conducive to the anti-Markovnikov hydroamination of styrenes and two additional substrates bearing directing groups.16 Recently, Hill and coworkers disclosed the hydroamination of styrenes, dienes, and alkynes utilizing [Ca{N(SiMe3)2}2]2 or [Sr{N(SiMe3)2}2]2 precatalysts,17 and the Lalic group has developed a one-pot, two-step hydroboration/amination approach for the synthesis of tertiary alkyl amines from aliphatic olefins.18 However, these metal-catalyzed hydroamination methodologies have various limitations such as harsh basic conditions, the requirement of a directing group, and limited substrate scopes. Two additional approaches from the Studer19 and Beauchemin20 groups accomplish this transformation through free radical and pericyclic additions, respectively, although selectivity is substrate-dependent in the latter case.21 Despite considerable advances in the area of metal-catalyzed anti-Markovnikov hydroaminations of olefins, a general method remains an elusive goal.4c
Considering the importance of carbon–nitrogen bond forming processes, we sought to develop a new, mild hydroamination methodology. In this manuscript, we present a two-step, one-pot hydroamination protocol that tolerates a variety of olefin substrates, including aliphatic olefins as well as electronically biased systems such as styrenes. Both the olefin and the amine substrate scope are explored, and this report demonstrates how the use of cleavable N-protecting groups can give access to a diverse library of hydroamination adducts.
Results and discussion
It was envisioned that anti-Markovnikov hydroamination could occur through a one-pot, two-step Wacker oxidation/transfer hydrogenative reductive amination approach (1 → 3, Fig. 2) that would proceed via an aldehyde intermediate (5). Key challenges associated with this methodology include: (a) regioselectivity of the initial oxidation (1 → 5), as Wacker chemistry typically favors formation of the Markovnikov product, (b) catalyst compatibility during the reductive amination step, and (c) chemoselectivity of the reduction step to favor formation of the hydroamination adduct (3) over the hydration product (7). We recently reported the aldehyde-selective Wacker oxidation of styrenes,22 and it was expected that these optimized conditions, in which t-BuOH enhances anti-Markovnikov selectivity,23 could be utilized in hydroamination to access the key aldehyde intermediate (Fig. 2). In order to execute the reductive amination, we expected that the addition of an amine (2) would provide an imine/iminium intermediate, which could subsequently be reduced in the presence of a transfer hydrogenation catalyst [M] and an appropriate hydride source (6). It should be noted that the proposed one-pot methodology is advantageous over a two-pot technique because in addition to allowing for direct formation of the linear amine, isolation of the unstable aldehyde is bypassed; in our previous report of the aldehyde-selective oxidation of styrenes, the high reactivity of the aldehyde product necessitated isolation as the hydrazone derivative.22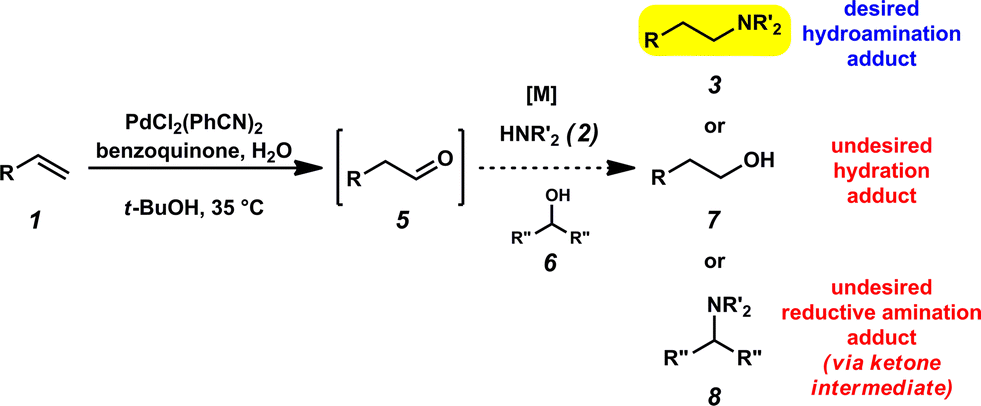 | ||
| Fig. 2 Challenges of the formal anti-Markovnikov hydroamination methodology. | ||
Previously, our group has demonstrated the compatibility of Shvo's catalyst (9, Table 1) with Wacker oxidation conditions;24 thus Shvo's catalyst, which is also well known to be effective in the transfer hydrogenation of imines,25 was chosen for our initial hydroamination studies. p-Methylstyrene (1a) and N-methylaniline (2a) were selected as the initial substrates for preliminary hydroamination studies. While p-methylstyrene was chosen for methodology development because this substrate yields exceptionally high anti-Markovnikov selectivities in Wacker oxidations when t-BuOH is used as a solvent,22N-methylaniline was selected because of the known compatibility of aryl amines with Pd(II)-catalyzed oxidations.26
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | [M] | Mol% [M] | [H] | Additives (step) | Equiv. amine | Equiv. H2O | Yielda |
| a Yield determined from analysis of the 1H NMR spectrum using 1,4-dioxane as an external standard. | |||||||
| 1 | 9 | 10% | Isopropyl alcohol | CuCl2 (ii) | 2.5 | 1 | 15% |
| 2 | 9 | 10% | 2,4-Dimethyl-3-pentanol | Mol. sieves (i); CuCl (ii) | 2.5 | 0 | 25% |
| 3 | 10 | 10% | 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 HCO2H/TEA :2 HCO2H/TEA |
— | 2.5 | 1 | 63% |
| 4 | 10 | 10% | 5:2 HCO2H/TEA |
— | 2.5 | 2 | 59% |
| 5 | 10 | 1% | 5:2 HCO2H/TEA |
— | 2.5 | 1 | 65% |
| 6 | 10 | 10% | 5:2 HCO2H/TEA |
— | 1.3 | 1 | 66% |
| 7 | 10 | 1% | 5:2 HCO2H/TEA |
— | 1.3 | 1 | 59% |
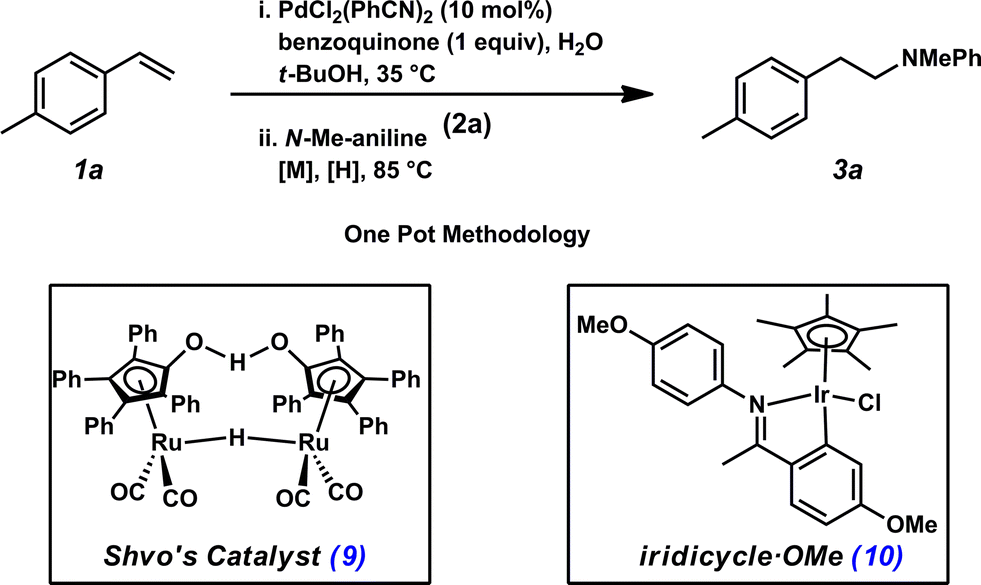
Using our one-pot, two-step hydroamination approach,27 a solution of p-methylstyrene (1a) in t-BuOH was treated with PdCl2(PhCN)2, the terminal oxidant benzoquinone, and H2O (Table 1, entry 1). After allowing the oxidation to progress for 4 hours at 35 °C, a solution of N-methylaniline (2a), Shvo's catalyst (9), and hydride source isopropanol was added to the reaction vessel, which was subsequently heated at 85 °C. In addition to providing desired hydroamination adduct 3a in 15% yield by 1H NMR spectroscopy, the hydration product (e.g., 7) was also observed, suggesting unselective reduction. Additionally, significant quantities of 8 were observed, resulting from reductive amination between the oxidized hydride donor and amine 2a. In order to minimize these undesired byproducts, a bulkier hydride source was used, and water was eliminated from the reaction conditions and replaced with molecular sieves (entry 2). Under these reaction conditions, hydroamination adduct 3a was obtained in 25% yield.
To address chemoselectivity issues in the reduction step, we found it necessary to replace Shvo's catalyst (9) with commercially available Ir-complex 10,28 which was developed by the Xiao group and has been demonstrated to be selective for transfer hydrogenative reduction of imines in the presence of carbonyls.29 Also necessary for this reduction, a 5:2 formic acid:triethylamine azeotropic mixture29,30 was utilized as the hydride source. Operationally, a solution of the hydride source, the Ir catalyst, and the amine was added to the reaction mixture after Pd-catalyzed oxidation had completed. Under these new conditions, in which formation of undesired hydration product 7 is significantly disfavored because of the inherent chemoselectivity of Ir-catalyst 10 for imine reduction, linear amine 3a was obtained in 63% yield by 1H NMR spectroscopy (entry 3).31 Importantly, no Markovnikov hydroamination product was detected.32 Brief attempts to further optimize the reaction conditions found no significant improvement in yield (e.g., entry 4), although it was found that either Ir catalyst loading or amine equivalents could be reduced without negatively impacting yields (entries 5 and 6, respectively). However, reduction of both Ir-catalyst loading and amine equivalents resulted in a slight decline in yield (entry 7).
The styrene substrate scope was examined using the optimized conditions (Table 2). Hydroamination of aryl-substituted styrenes 1a–j afforded desired linear amines 3a–j in good to moderate yield and with excellent regioselectivity; in all cases, no Markovnikov hydroamination product was isolated or detected in 1H NMR spectra of the unpurified reaction mixtures.32 The hydroamination methodology was found to accommodate a variety of aryl-substitution patterns, including ortho-substitution (entries 4 and 8). In addition to alkyl substituents (entries 1, 3, and 4), several functional groups were tolerated including ether (entry 5), aryl halide (entries 6–9), and alkyl halide (entry 10) groups. Yields substantially declined when these hydroamination conditions were applied to aliphatic olefins, and in the case of 4-phenyl-1-butene (1k), amine adduct 3k was isolated in 24% yield (entry 11).33
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yielda |
| a Yields determined by isolation (0.6 mmol scale). b Yield determined from analysis of the 1H NMR spectrum using 1,4-dioxane as an external standard. c Yield obtained with 10 mol% 10. | |||
| 1 |
|
3a | 61% (66%b) |
| 2 |
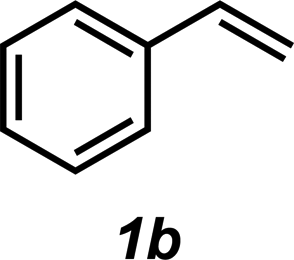
|
3b | 55% |
| 3 |
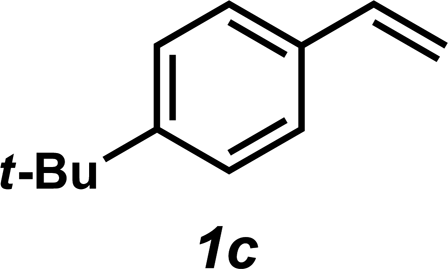
|
3c | 55%c |
| 4 |
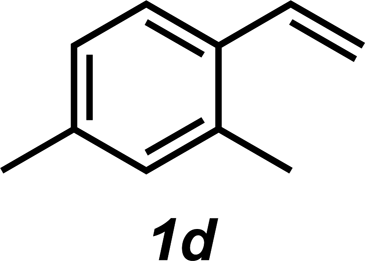
|
3d | 62% |
| 5 |
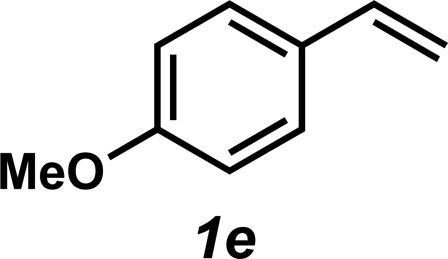
|
3e | 65% |
| 6 |
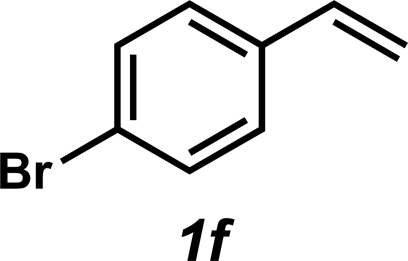
|
3f | 52% |
| 7 |
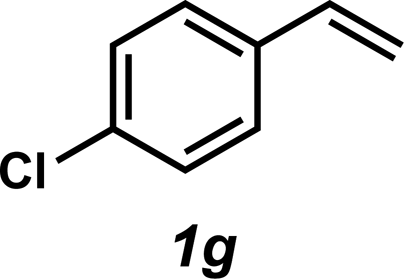
|
3g | 57%c |
| 8 |
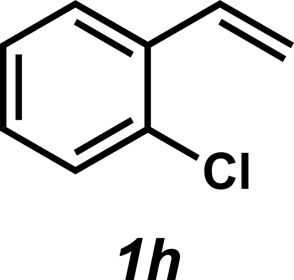
|
3h | 43%c |
| 9 |
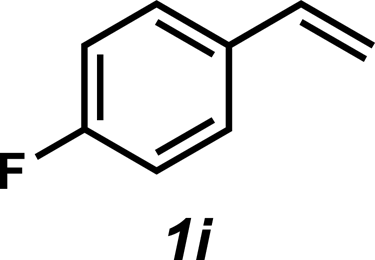
|
3i | 70% |
| 10 |
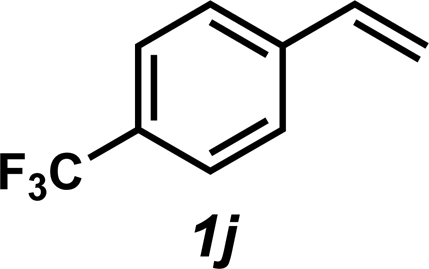
|
3j | 50%c |
| 11 |
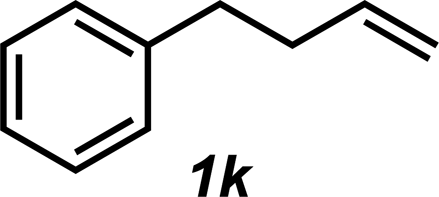
|
3k | 24%c |
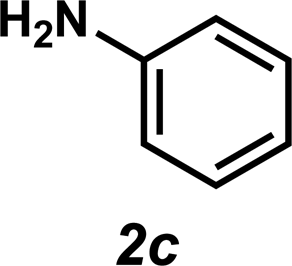
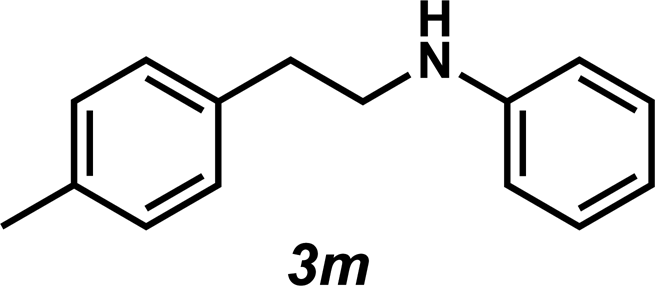
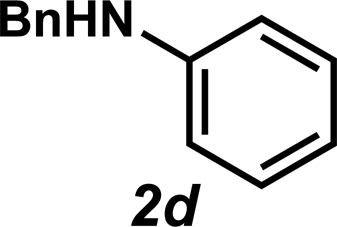
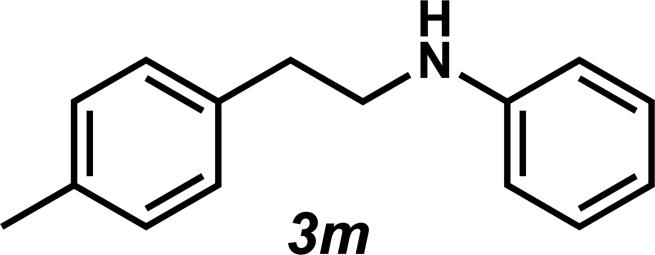
Next, the amine substrate scope was investigated, and efforts initially focused on aryl amines (e.g., 2a–f), which are prevalent in drug substances.34 Hydroamination of 1a with N-methylnaphthalene-2-amine (2b) gave a satisfactory 60% yield of desired product 3l (Table 3, entry 2). Aniline (2c) was a more challenging substrate (entry 3), but interestingly, when N-benzylaniline (2d) was used as the nucleophile, N-(4-methylphenethyl)aniline (3m), resulting from tandem hydroamination and hydrogenolysis reactions, was isolated in 46% yield (entry 4) – an improvement from entry 3's yield of 32% of the same adduct. Although primary aryl amines often do not participate in high yielding hydroamination transformations, this result demonstrates that one possible tactic for maximizing yields is to use benzyl-protected derivatives. Furthermore, whereas a number of previous anti-Markovnikov intermolecular hydroamination strategies do not accommodate aryl amines,17 our approach is best suited for this class of compounds; thus, our methodology offers a complementary hydroamination approach.
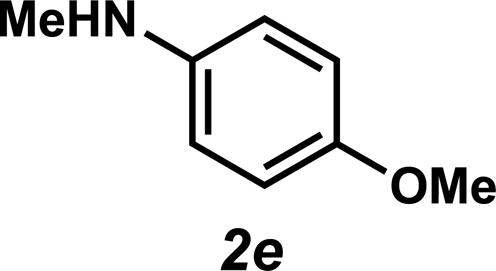
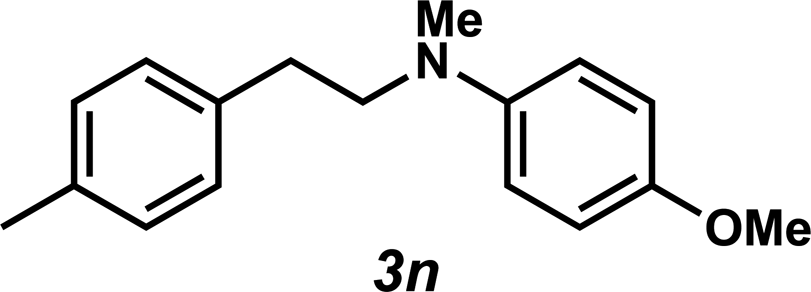
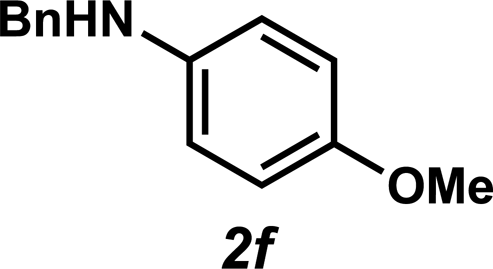
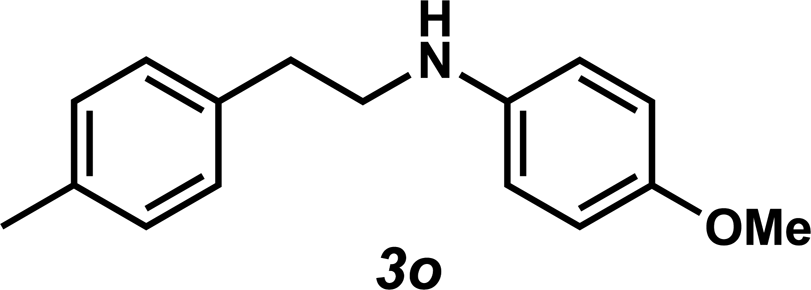
Unfortunately, employing the optimized conditions gave only decomposition mixtures when applied to hydroamination using aliphatic amines. This shortcoming prompted us to examine hydroamination with amines possessing removable aryl protecting groups. Hydroamination of p-methylstyrene (1a) with N-methylanisidine (2e) proceeded in 60% yield (entry 5). Similarly, N-benzylanisidine (2f) proved to be a suitable substrate, providing the hydroamination adduct 3o in 61% yield (entry 6). These two transformations furnished the linear amine product bearing a PMP (p-methoxyphenyl) protecting group, which can be readily cleaved by the action of dilute acid35 (e.g.3n → 11, Fig. 3); thus, the use of amines with removable aryl protecting groups allows access to a diverse array of hydroamination products.
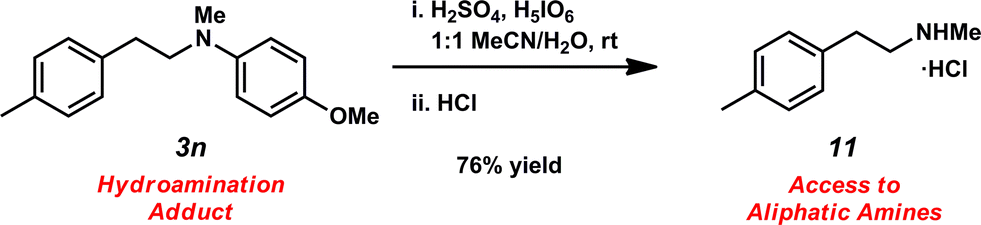 | ||
| Fig. 3 Cleavage of removable aryl protecting group. | ||
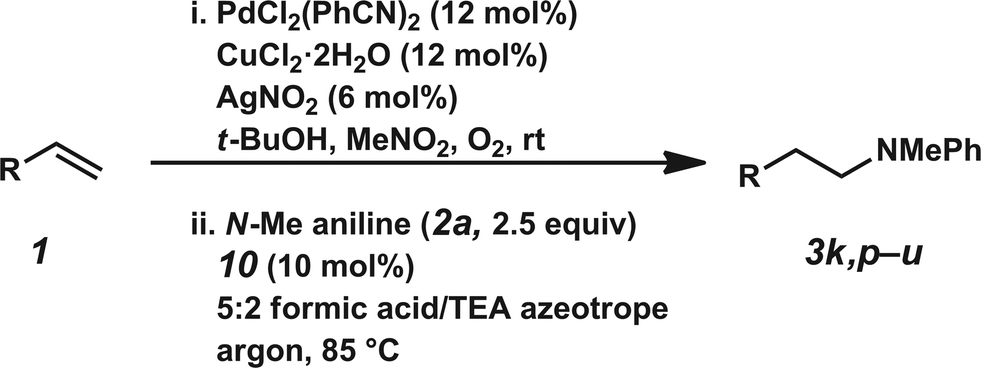
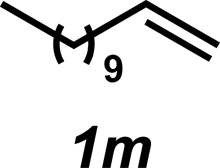
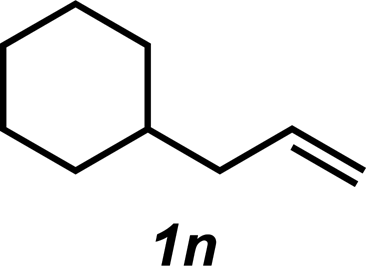
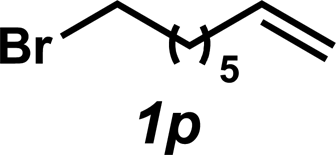
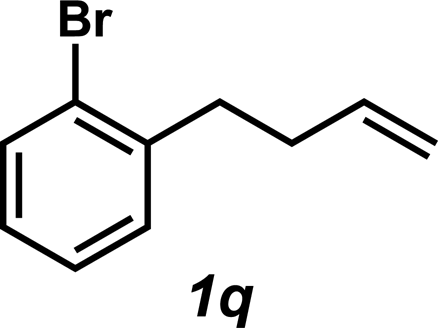
The hydroamination approach described thus far is not best suited for aliphatic olefins (e.g., Table 2, entry 11). Our group's olefin hydration research previously demonstrated that the initial oxidation proceeds in moderate to poor yield and with regioselectivity favoring the Markovnikov (ketone) product.24 However, during the course of our studies, coworkers developed a new catalytic process for the aldehyde-selective Wacker oxidation of aliphatic olefins.36 To our delight, it was found that the new Wacker oxidation conditions, which utilize AgNO2, could be applied to our hydroamination methodology in order to offer an entry into the anti-Markovnikov hydroamination of aliphatic olefins. As shown in Table 4, hydroamination of 4-phenyl-1-butene (1k) with N-methylaniline (2a) furnished adduct 3k in 64% yield (Table 4, entry 1), whereas our initial approach delivered 3k in 24% yield (Table 2, entry 11). Anti-Markovnikov hydroamination of 1-dodecene (1m), a transformation that notably cannot be substrate-controlled, proceeded in 40% yield to provide 3p (entry 3). The more sterically demanding allyl cyclohexane (1n) also proved to be a good substrate (entry 4), delivering 3r in 56% yield. Examination of the substrate scope revealed that this transformation could accommodate a variety of functional groups, including nitro (entry 2), ester (entry 5), alkyl halide (entry 6), and aryl halide (entry 7) groups. To the best of our knowledge, this methodology represents the first metal-catalyzed approach to the intermolecular anti-Markovnikov hydroamination of an unbiased olefin with an aryl amine. Furthermore, it should be noted that this catalytic system allows access to elusive linear amine adducts through a one-pot technique, thus avoiding isolation of less stable aldehyde intermediates.

Conclusions
In summary, a one-pot methodology for the intermolecular anti-Markovnikov hydroamination of olefins with aryl amines has been developed. The scope of the methodology is broad with respect to the olefin, and although the amine substrate scope is more limited, the use of amines with removable aryl protecting group expands the suite of accessible linear amines. This mild methodology complements existing literature and contributes to less developed areas of hydroamination research, namely the anti-Markovnikov intermolecular hydroamination of aliphatic olefins, as well as the use of aryl amines.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number F32GM102984. NMR spectra were obtained by instruments supported by the NIH (RR027690). We are grateful to J. S. Cannon, B. Morandi, and Z. K. Wickens for helpful discussions.Notes and references
-
The Chemistry of the Amino Group, ed. S. Patai, Interscience, New York, 1968 Search PubMed
.
-
(a)
Green Chemistry: Challenging Perspectives, ed. P. Tundo and P. T. Anastas, Oxford Science, Oxford, 1999 Search PubMed
- J. Haggin, Chem. Eng. News, 1993, 71, 23 Search PubMed
-
(a) K. D. Hesp and M. Stradiotto, ChemCatChem, 2010, 2, 1192–1207 CrossRef CAS
- In contrast, intermolecular anti-Markovnikov hydroamination of alkynes is a more precedented transformation. For pertinent reviews, see:
(a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–704 CrossRef PubMed
- S. Zhang, Y. Wei, S. Yin and C.-t. Au, Catal. Commun., 2011, 12, 712–716 CrossRef CAS PubMed
- R. Corberán, S. Marrot, N. Dellus, N. Merceron-Saffon, T. Kato, E. Peris and A. Baceiredo, Organometallics, 2009, 28, 326–330 CrossRef
-
(a) C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493 CrossRef CAS
- G. V. Shanbhag, S. M. Kumbar and S. B. Halligudi, J. Mol. Catal. A: Chem., 2008, 284, 16–23 CrossRef CAS PubMed
- T. Joseph, G. V. Shanbhag, D. P. Sawant and S. B. Halligudi, J. Mol. Catal. A: Chem., 2006, 250, 210–217 CrossRef CAS PubMed
-
(a) D. C. Leitch, P. R. Payne, C. R. Dunbar and L. L. Schafer, J. Am. Chem. Soc., 2009, 131, 18246–18247 CrossRef CAS PubMed
- M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller and O. R. Thiel, Chem.–Eur. J., 1999, 5, 1306–1309 CrossRef CAS
- P. Horrillo-Martínez, K. C. Hultzsch, A. Gil and V. Branchadell, Eur. J. Org. Chem., 2007, 3311–3325 CrossRef
- K. Kumar, D. Michalik, I. Garcia Castra, A. Tillack, A. Zapf, M. Arlt, T. Heinrich, H. Böttcher and M. Beller, Chem.–Eur. J., 2004, 10, 746–757 CrossRef CAS PubMed
-
(a) M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608–5609 CrossRef CAS PubMed
- J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584–12605 CrossRef CAS PubMed
-
(a) A. G. M. Barrett, C. Brinkmann, M. R. Crimmin, M. S. Hill, P. Hunt and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 12906–12907 CrossRef CAS PubMed
- R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, J. Am. Chem. Soc., 2012, 134, 6571–6574 CrossRef CAS PubMed
-
(a) J. Guin, C. Mück-Lichtenfeld, S. Grimme and A. Studer, J. Am. Chem. Soc., 2007, 129, 4498–4504 CrossRef CAS PubMed
-
(a) J. Moran, S. I. Gorelsky, E. Dimitrijevic, M.-E. Lebrun, A.-C. Bedard, C. Seguin and A. M. Beauchemin, J. Am. Chem. Soc., 2008, 130, 17893–17906 CrossRef CAS PubMed
- Very recently, the Nicewicz group disclosed a report of the anti-Markovnikov hydroamination of alkenes by an organic photoredox system. Their findings include two intermolecular examples. T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed
- P. Teo, Z. K. Wickens, G. Dong and R. H. Grubbs, Org. Lett., 2012, 14, 3237–3239 CrossRef CAS PubMed
-
(a) T. T. Wenzel, J. Chem. Soc., Chem. Commun., 1993, 862–864 RSC
- G. Dong, P. Teo, Z. K. Wickens and R. H. Grubbs, Science, 2011, 333, 1609–1612 CrossRef CAS PubMed
-
(a) J. S. M. Samec and J.-E. Bäckvall, Chem.–Eur. J., 2002, 8, 2955–2961 CrossRef CAS
-
(a) L. S. Hegedus, G. F. Allen and E. L. Waterman, J. Am. Chem. Soc., 1976, 98, 2674–2676 CrossRef CAS
- Initial attempts to design a one-pot, one-step hydroamination methodology were unsuccessful. Reactions utilizing Shvo's catalyst gave decomposition mixtures, while reactions involving iridicycle·OMe 10 resulted in the reduction of Pd(II) to Pd(0).
- Iridicycle catalyst 10 is commercially available from Strem. Catalogue number: 77-0418; CAS number 1258964-48-5.
- C. Wang, A. Pettman, J. Basca and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548–7552 CrossRef CAS PubMed
- Formic acid/triethylamine complex is commercially available through Sigma-Aldrich. Catalogue number: 06561; CAS number: 15077-13-1.
- The modest yield is largely attributed to the instability of the aldehyde intermediate. The oxidation reaction was monitored by GC analysis, and yields of the corresponding aldehyde were observed to peak between 2–4 hours of reaction time, and then declined thereafter. Minimal starting material was observed in the 1H NMR spectra of the unpurified reaction mixtures.
- Although the Markovnikov hydroamination product was not observed, our previous publications have shown that aldehyde-selective oxidations of styrenes give minor amounts of ketone products. See ref. 22 and 24.
- Although the Markovnikov hydroamination product was not observed, our previous publication suggests that oxidation of terminal aliphatic olefins using these conditions gives substantial amounts of ketone products. See ref. 24.
- J. Njardarson, Top 200 Brand Name Drugs by US Retail Sales in 2011, http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster (accessed June, 2013).
- J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, P. L. Alsters, F. L. van Delft and F. P. J. T. Rutjes, Tetrahedron Lett., 2006, 47, 8109–8113 CrossRef CAS PubMed
- Z. K. Wickens, B. Morandi and R. H. Grubbs, Angew. Chem., Int. Ed., 2013 DOI:10.1002/anie.201306756
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc51897c |
| This journal is © The Royal Society of Chemistry 2014 |