 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How phenyl makes a difference: mechanistic insights into the ruthenium(II)-catalysed isomerisation of allylic alcohols†
Simone
Manzini
a,
Albert
Poater
b,
David J.
Nelson
a,
Luigi
Cavallo
cd and
Steven P.
Nolan
*a
aEaStCHEM, School of Chemistry, University of St. Andrews, Purdie Building, North Haugh, St. Andrews, Fife, KY16 9ST, UK. E-mail: snolan@st-andrews.ac.uk
bInstitut de Química Computacional i Catàlisi, Departament de Química, University of Girona, Campus de Montilivi sn, 17071, Girona, Catalonia, Spain
cDepartment of Chemistry and Biology, University of Salerno, Via Ponte don Melillo, Fisciano I-84084, Italy
dKAUST Catalyst Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 18th October 2013
Abstract
[RuCl(η5-3-phenylindenyl)(PPh3)2] (1) has been shown to be a highly active catalyst for the isomerisation of allylic alcohols to the corresponding ketones. A variety of substrates undergo the transformation, typically with 0.25–0.5 mol% of catalyst at room temperature, outperforming commonly-used complexes such as [RuCl(Cp)(PPh3)2] and [RuCl(η5-indenyl)(PPh3)2]. Mechanistic experiments and density functional theory have been employed to investigate the mechanism and understand the effect of catalyst structure on reactivity. These investigations suggest a oxo-π-allyl mechanism is in operation, avoiding intermediate ruthenium hydride complexes and leading to a characteristic 1,3-deuterium shift. Important mechanistic insights from DFT and experiments also allowed for the design of a protocol that expands the scope of the transformation to include primary allylic alcohols.
Introduction
The redox isomerisation of allylic alcohols catalysed by transition metal complexes represents a powerful, elegant and green method to prepare carbonyl compounds where otherwise a two-step sequence of oxidation and reduction reactions would be required (Scheme 1), potentially using toxic and/or harsh reagents.1–4 Numerous complexes of Fe, Os, Ru, Rh, Co, Ni, Mo, Ir, and Pt have been shown to catalyse this rearrangement; however, most of them show restricted scope with regard to reaction conditions and substitution at the 1- and 3-positions (R1 and R2).3–5 Primary allylic alcohols are typically the most challenging substrates.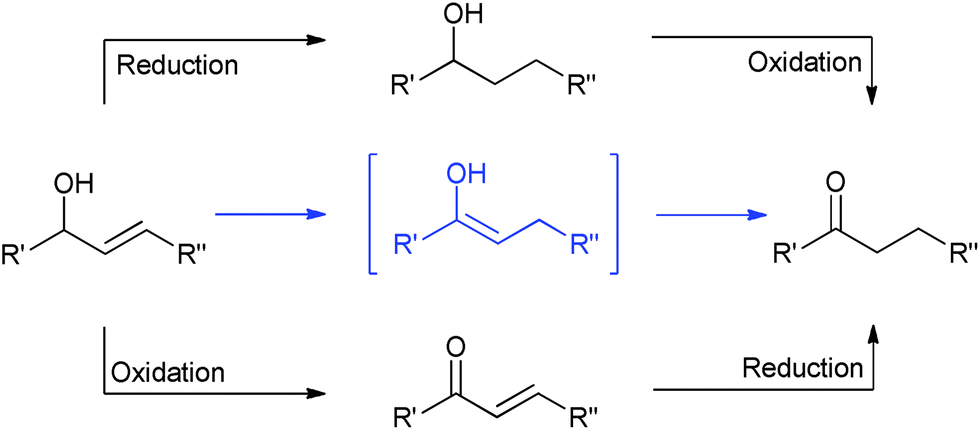 | ||
| Scheme 1 | ||
Various Ru complexes have been reported to date in the literature for this transformation.1,6–8 The first RuII–cyclopentadienyl-like complex employed in this reaction was [RuCl(Cp)(PPh3)2] by Kulawiec and Trost in the 1990s.9 This complex, in combination with [Et3NH][PF6] in dioxane at 100 °C, is able to catalyse the isomerisation of allylic alcohols, with the exception of primary alcohols and unsubstituted vinyl groups. Slugovc et al. improved the activity, reaching high conversions at 57 °C using a well-defined cationic complex of the form [Ru(Cp)(MeCN)2(PR3)][PF6] in CDCl3; notably this system did not require the use of base.10 Another relevant step forward was shown by Cadierno and Gimeno using a series of RuIV complexes such as [Ru(η3:η2:η3-C12H18)Cl2] and derivatives, which show high activity in aqueous media and in ionic liquids at 75 °C.11,12 However, all of these complexes display activity with only a limited substitution pattern and still require high temperatures to perform the isomerisation. In 2005, Bäckvall reported the use of [RuCl(1-methyl-2,3,4,5-tetraphenylcyclopentadienyl)(CO)2], which is able to isomerise substituted allylic alcohols at room temperature. However, this catalyst shows activity at relatively high loadings (5 mol%), which increases the cost of the chemistry and can lead to purification issues.13 In the same year, Ikariya introduced a series of Ru(Cp*)(P–N) complexes that were very active at 30 °C with 1 mol% of catalyst.14
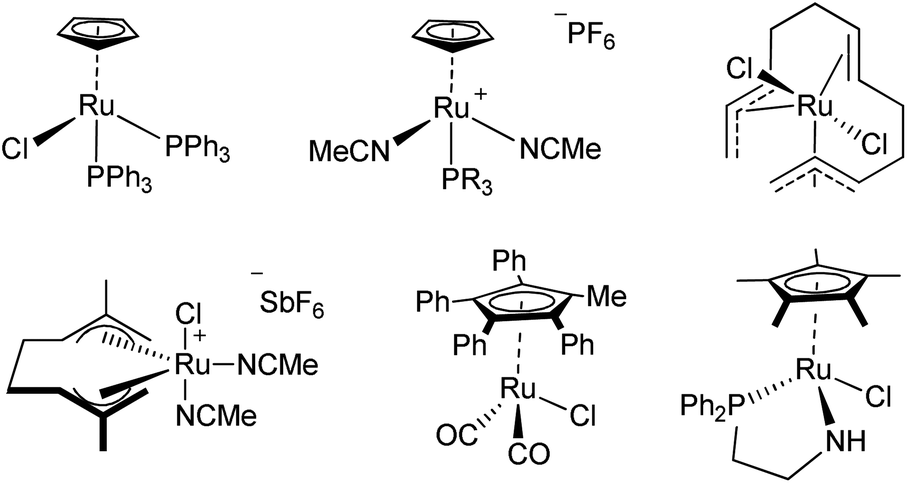
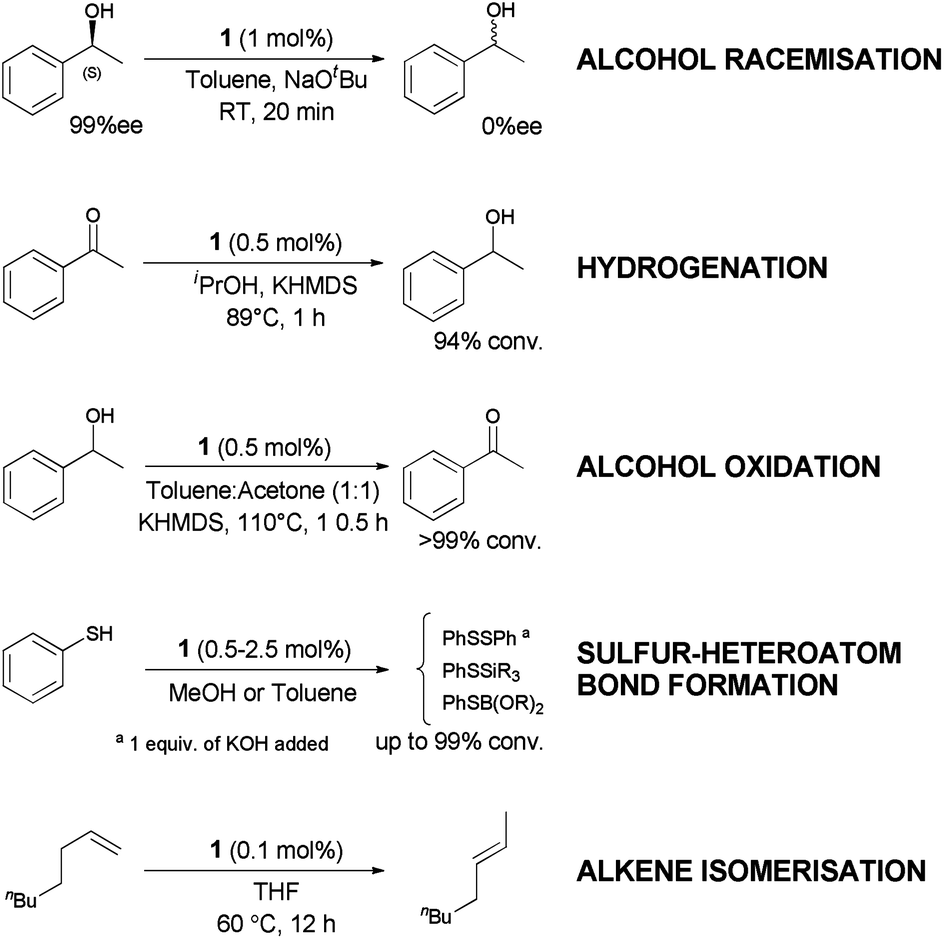
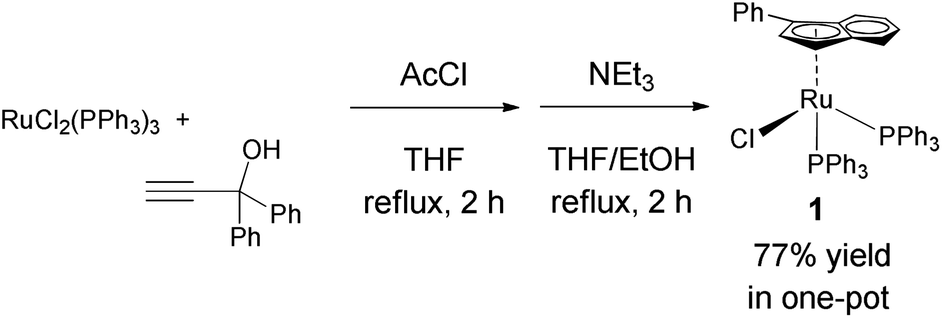
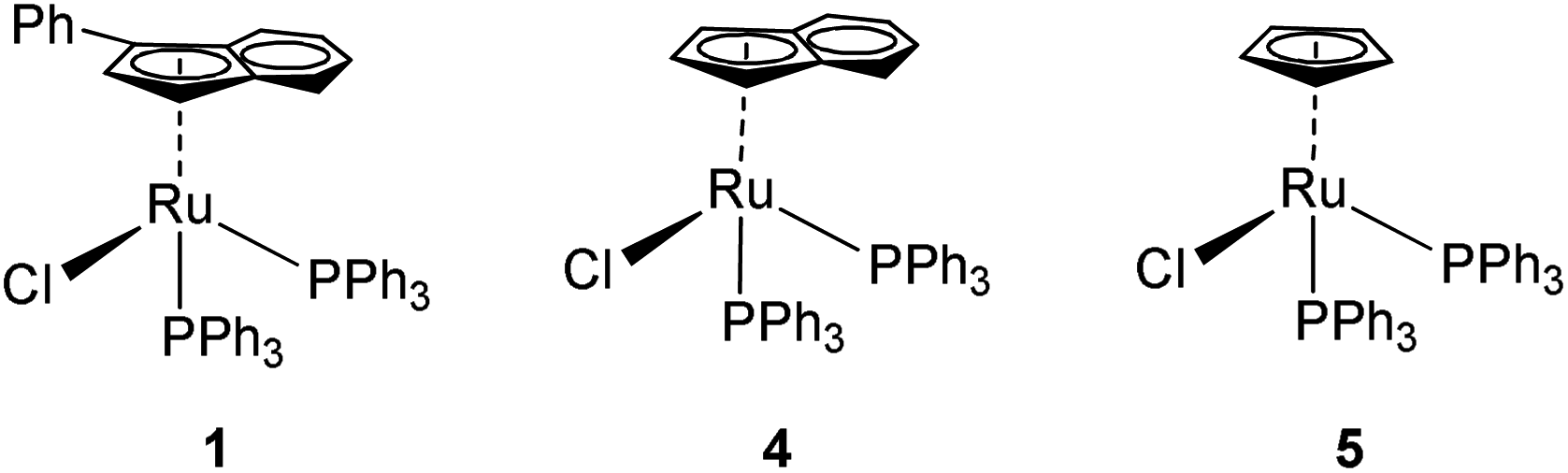
Ruthenium is an attractive metal for these reactions, due to its excellent activity, often at low loadings, and its relatively low cost when compared to many other late transition metals, such as iridium, that can also perform the reaction. We decided to evaluate the activity of [RuCl(η5-3-phenylindenyl)(PPh3)] (1), recently discovered in our laboratory, in allylic alcohol isomerisation.15 This complex has emerged as a highly active multi-tasking complex (Scheme 2); such multi-purpose complexes are highly valuable, as they minimise the number of precious metal catalysts that must be held in stock in synthetic chemistry laboratories. Complex 1 has already shown activity in the racemisation of chiral alcohols,15 the hydrogenation of ketones, aldehydes and imines,16 the chemoselective oxidation of secondary alcohols,17 the formation of S–S, S–Si and S–B bonds,18 and the isomerisation of alkenes19 (Scheme 2). Moreover, the straightforward synthesis of the complex starting from RuCl2(PPh3)3 and propargylic alcohol (Scheme 3) makes it a practical and cost effective complex.15 Importantly, the study of this reaction allows the reactivity of 1 to be further explored, yielding insights into the reactivity of complexes of this type. Simple phenyl substitution of the indenyl ligand has previously been shown to affect reactivity in this type of complex, so we wished to examine whether this extended to the allylic alcohol isomerisation. Understanding the effects of catalyst structure is of critical importance when designing the next generation of complexes.
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Optimisation, scope and limitations
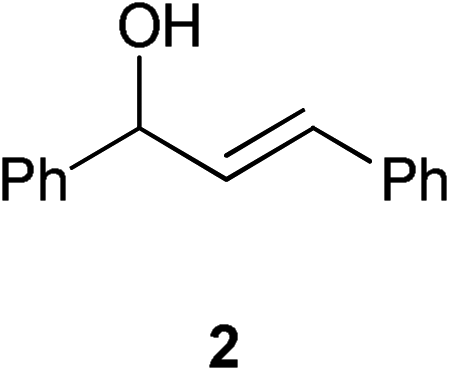
Using the isomerisation of (E)-1,3-diphenylpropen-1-ol 2 to the ketone 3 as a benchmark reaction, complex 1 was tested using the same reaction conditions that were used for alcohol racemisation. Substrate 2 is typically one of the more challenging allylic alcohols.1–3 Pleasingly, it was discovered that 1 is very active at room temperature, achieving total isomerisation in just 1 h with only 0.25 mol% of catalyst and 4 mol% base (Scheme 4). While the use of a base has an impact on the atom economy of the process, only a very low loading is required here, presumably to generate the active ruthenium species (vide infra).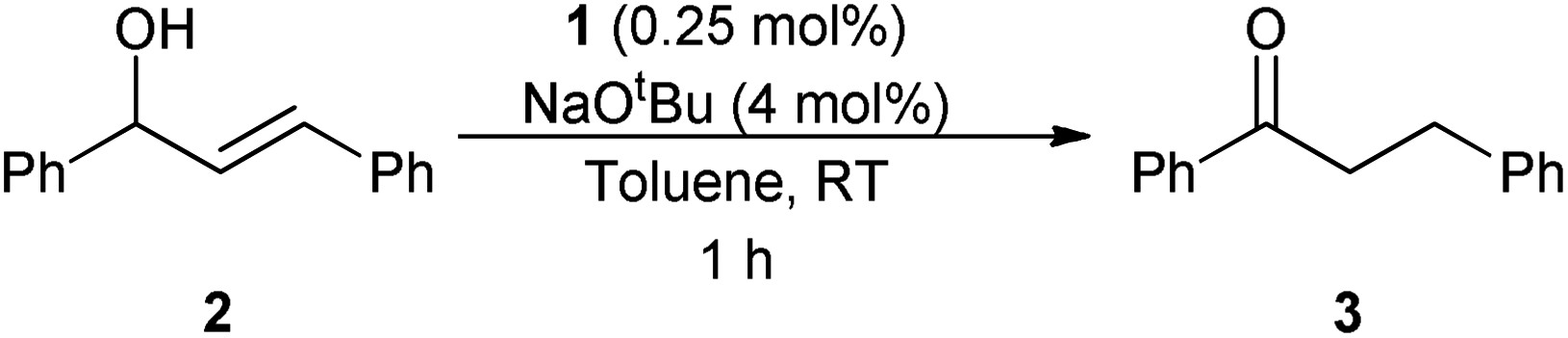 | ||
| Scheme 4 | ||
To evaluate the effect of the phenylindenyl ligand on reactivity, complex 1 was compared with analogues [RuCl(η5-indenyl)(PPh3)2] (4) and [RuCl(Cp)(PPh3)2] (5), which have also been employed for these types of reactions, under the same reaction conditions (Table 1). Surprisingly, complexes 4 and 5 show practically no activity under these conditions, meaning that the phenyl ring in complex 1 makes a significant difference in activity. This result was intriguing, and stimulated us to investigate the reaction in more detail (vide infra).
|
|
||||
|---|---|---|---|---|
| Entry | [Ru] | Catalyst loading | Time (h) | Conv. %b |
| a Conditions: substrate (0.5 mmol), base (0.02 mmol), catalyst (1.25 μmol), and toluene (1 mL) added. Reactions stirred at ambient temperature for 1 h. b Conversion determined from 1H NMR analysis average of at least two experiments. | ||||
| 1 | 1 | 0.25 mol% | 1 | >95 |
| 2 | 4 | 0.25 mol% | 1 | <5 |
| 3 | 5 | 0.25 mol% | 1 | <5 |
The reaction conditions that were employed initially are already highly effective, and therefore further optimisation would be of little use. Instead, the use of milder conditions and reagents was explored, in order to render this chemistry accessible to as wide a range of synthetic chemists.
A number of bases were screened for use in this reaction; while these conditions allow for rapid and excellent conversions at room temperature, the use of a strong base such as NaOtBu could potentially be incompatible with some substrates, and therefore milder bases are attractive. The results (Table 2) show that the choice of counterion (K or Na) makes no difference to the reaction, while milder bases such as KOH can still allow for complete conversion. Much weaker bases, such as K2CO3, are not suitable for this transformation. These results suggest that there is a pKa threshold, below which the base is ineffective.
| Entry | Base | Conversion %b |
|---|---|---|
| a Conditions: substrate (0.5 mmol), base (0.02 mmol), 1 (1.25 μmol) and toluene (1 mL) added. Reactions stirred at ambient temperature for 1 h. b Conversion determined from 1H NMR analysis; average of at least two experiments. | ||
| 1 | NaOtBu | >99 |
| 2 | KOtBu | >99 |
| 3 | KHMDS | 96 |
| 4 | KOH | >99 |
| 5 | K2CO3 | 31 |
The system is compatible with some of the most common industrially accepted solvents,20 reaching quantitative (or close to quantitative) conversion in 1 h reaction time in each case. While toluene is still acceptable on a large scale, more sustainable solvents such as cyclopentyl methyl ether, 2-methyltetrahydrofuran (2-MeTHF) and isobutylmethyl ketone are also compatible with this chemistry.20 This renders this transformation scalable, and further enhances its green credentials.
To understand the substrate compatibility of the catalytic system, systematic variation of the substrates has been considered (Table 3). It is important to note that optimisation of the reaction conditions has taken place using one of the more challenging substrates, however “easier” substrates, such as un-substituted secondary allylic alcohols were evaluated.
| Entry | Substrate | 1 (mol%) | t (h) | Conv. (yield)b,c |
|---|---|---|---|---|
| a Conditions: substrate (1 mmol), base (4 mol%) and toluene (1 mL) added. Reactions stirred at ambient temperature for 1 h. b Conversion determined from 1H NMR analysis. c Yields are isolated yields. | ||||
| 1 |
|
0.25 | 1 | >99% (97%) |
| 2 |
|
0.25 | 1 | >99% (95%) |
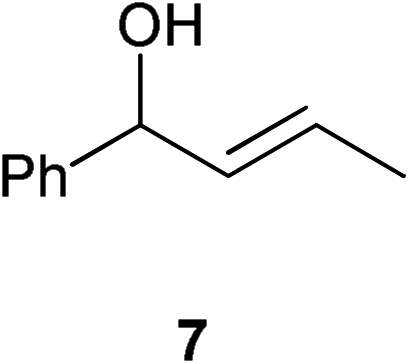
| 3 |
|
0.25 | 1 | >99% (97%) |
| 4 |
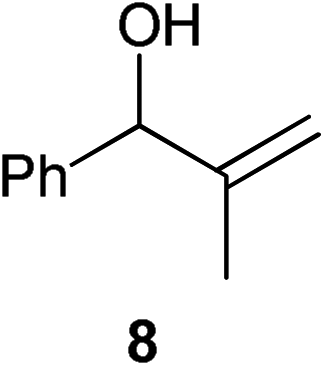
|
0.25 | 1 | 90% (89%) |
| 5 |
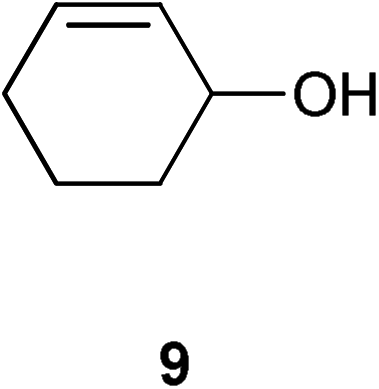
|
0.5 | 1.5 | >99% (81%) |
| 6 |
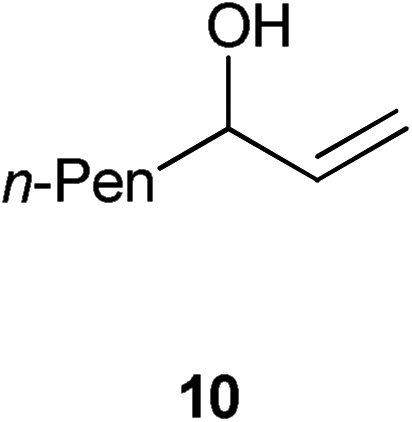
|
0.5 | 2 | >99% (94%) |
| 7 |
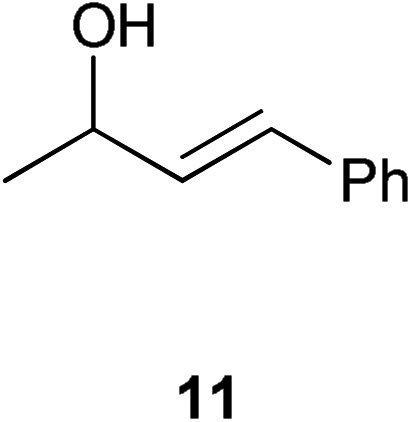
|
0.5 | 6 | >99% (96%) |
| 8 |
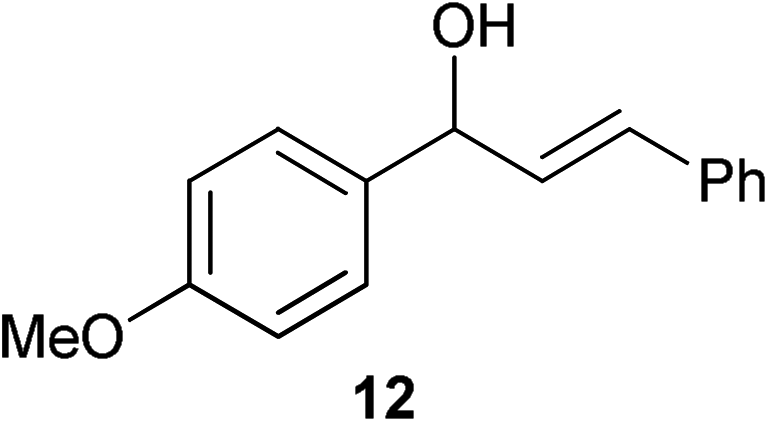
|
0.25 | 2.5 | 96% (92%) |
| 9 |
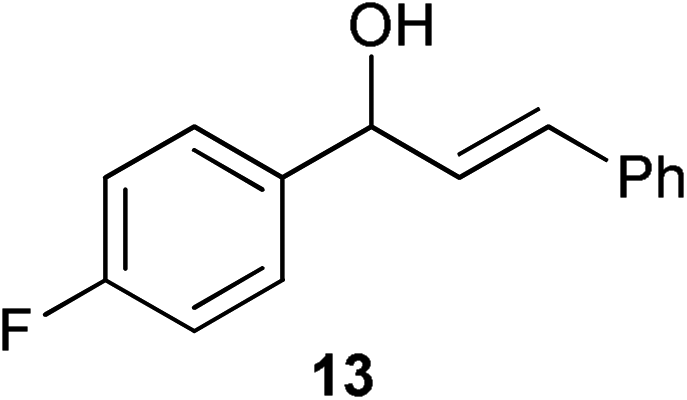
|
0.25 | 1 | >99% (92%) |
Complex 1 shows high compatibility with differently substituted substrates (Table 3). 1 allows isomerisation of internal and terminal benzyl allylic alcohols in only 1 h with 0.25 mol% of catalyst. Electronic effects were found to play an important role in the isomerisation. Indeed, substrates bearing electron-donating groups (e.g.12) are more difficult to isomerise than the corresponding substrates bearing electron-withdrawing groups (e.g.13). This suggests that the pKa of the alcohol or the proton α- to the alcohol may be a key determinant of reaction efficiency; electron-withdrawing groups would stabilise a developing negative charge on the benzylic carbon and therefore might be responsible for the observed difference in reactivity.
With alkyl allylic alcohols it was necessary to increase the catalyst loading to 0.5 mol% to obtain the same conversions. With regard to the degree of substitution on the vinyl group, it is easy to isomerise monosubstituted alkenes and 1,1- or 1,2-disubstituted alkenes; trisubstituted alkenes cannot be isomerised using the system considered here, presumably due to the degree of hindrance around the double bond making co-ordination of the vinyl group to the metal difficult. However, this methodology can potentially be used as a selective system for internal and 1,1- and 1,2-disubstituted allylic alcohols, which should render it of interest to, for example, natural product chemists working on highly functionalised scaffolds. To our disappointment, primary allylic alcohols cannot be isomerised using 1.
The scope and limitations have been explored at room temperature, and represent the most accessible conditions for synthesis. However, the performance of the system was evaluated at lower catalyst loadings and higher temperatures in order to probe the limits of the catalyst. 1 can catalyse the reaction of 2, selectively producing 3 (77% conversion) at 100 °C with a catalyst loading of 100 ppm in 15 minutes. This represents a turnover number (TON) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and a turnover frequency (TOF) of 36000 h−1, which are highly competitive with the state-of-the-art. Notably, this is in fact a conservative estimate of TOF when compared to alternative measurements at low conversion, as discussed by Kozuch and Martin.21 This result is also made more remarkable by the use of 2 in this TON/TOF determination, which is typically a more challenging substrate for ruthenium complexes.1–3
000, and a turnover frequency (TOF) of 36000 h−1, which are highly competitive with the state-of-the-art. Notably, this is in fact a conservative estimate of TOF when compared to alternative measurements at low conversion, as discussed by Kozuch and Martin.21 This result is also made more remarkable by the use of 2 in this TON/TOF determination, which is typically a more challenging substrate for ruthenium complexes.1–3
The role of additives
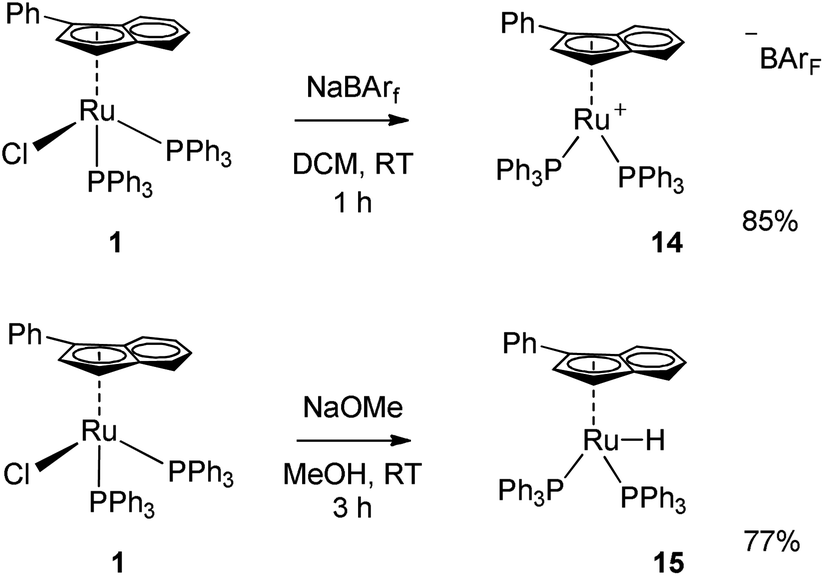
In order to gain a better understanding of the action of complex 1 in this reaction, the effect of the base was investigated. In fact, the role of the base was rather unclear, whether it acts as chloride abstractor to generate the active species or promotes the activation of the allylic alcohol through deprotonation of the hydroxyl moiety. A series of exploratory reactions were conducted, where the concentration of base and [nBu4N][Cl] (TBACl, added as a chloride source) were varied.As shown in Fig. 1, there is clearly an optimum loading of base (4 mol%); the base is necessary for the reaction to proceed, yet at high loadings the substrate completely decomposes in the reaction medium. A more subtle effect was observed with [nBu4N][Cl]: a slight inhibition of the reaction is observed, with only 50% conversion when 1 equiv. TBACl was present. The results suggest that either chloride abstraction is involved in the initiation of the catalyst, or that one of the catalytic intermediates is sensitive to chloride. In order to have a clearer understanding as to the need to remove the chloride in order to activate the catalyst, two further complexes were prepared: cationic 14 and hydride 15. The former complex has been shown to be active in the isomerisation of terminal alkenes (Scheme 5).19
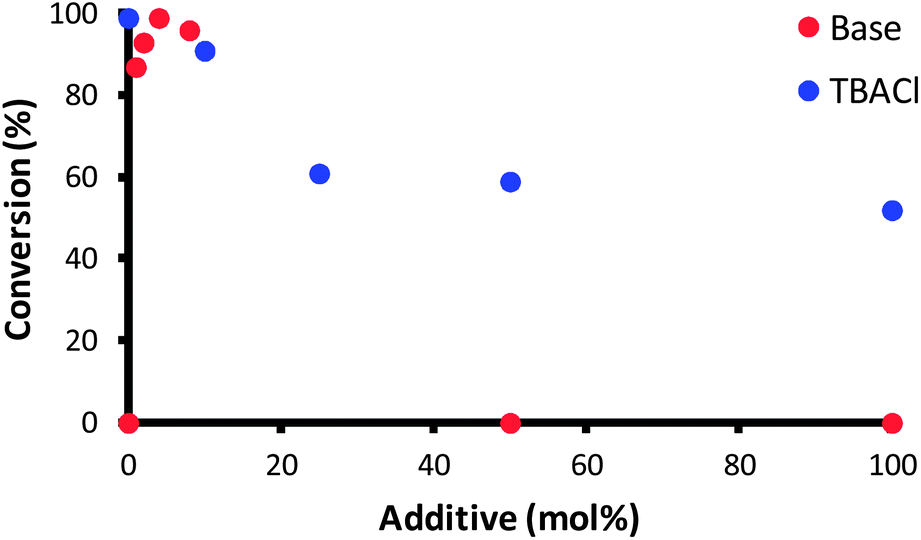 | ||
| Fig. 1 Isomerisation of 2 in the presence of various equivalents of NaOtBu (red points) and TBACl (blue points). Reaction conditions were otherwise as per Scheme 4. | ||
 | ||
| Scheme 5 | ||
As shown in Table 4, complexes 1 and 14 are both active in the presence of base, while complex 15 is inactive. In the absence of base, 14 remains active, albeit at a slightly higher catalyst loading (0.5 mol%), confirming the necessity of the chloride abstraction in order to generate the active species.
| Entry | Complex | Catalyst loading (mol%) | Base (mol%) | Conv. %b |
|---|---|---|---|---|
| a Conditions: substrate (0.5 mmol), base (0.02 mmol) and catalyst (125 μmol) and toluene (1 mL) added. Reactions stirred at ambient temperature for 1 h. b Conversion determined from 1H NMR; average of at least two experiments. | ||||
| 1 | 1 | 0.25 | 4 | >99 |
| 2 | 14 | 0.25 | 4 | >99 |
| 3 | 15 | 0.5 | 4 | 0 |
| 4 | 1 | 0.5 | 0 | 0 |
| 5 | 14 | 0.5 | 0 | >99 |
| 6 | 15 | 0.5 | 0 | 0 |
Mechanistic study
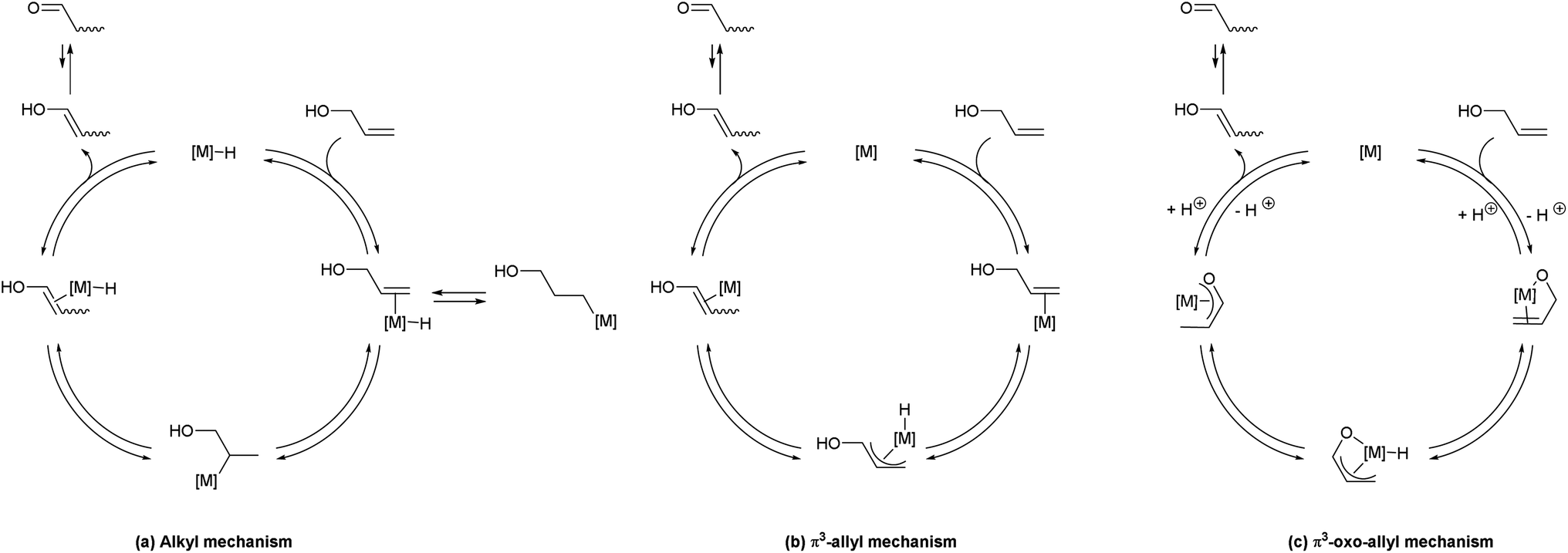
A detailed understanding of the reaction mechanism was desired, in order to rationally design the next generation of catalysts and fully understand the interplay of structure and reactivity.Three pathways have been suggested in the literature for this class of transformations (Scheme 6).9,12,22 In each case the mechanism leads to an enol, which will tautomerise to the thermodynamically-favoured ketone. Trost has shown that complex 4 does not operate via a metal-hydride addition/elimination sequence, via deuterium-labelling crossover experiments;9 attention was also drawn to the significant improvement in activity when the cyclopentadienyl ligand was replaced with an indenyl moiety. A particularly detailed study was presented by Cadierno et al., in which DFT calculations (B3LYP/6-311+G(d,p)//B3LYP/6-31G(d,p)) were used to compare reaction mechanisms for the isomerisation reaction using a Ru(IV) complex, favouring a π3-oxo-allyl mechanism.12 McGrath et al. used a simple Ru(II) system for allylic alcohol isomerisation, and suggested a metal-hydride addition/elimination mechanism on the basis of deuterium labelling and crossover experiments.22
 | ||
| Scheme 6 | ||
In the alkyl mechanism (Scheme 6(a)), in which a metal hydride complex is the active species, the reaction proceeds via reversible addition of metal hydride across the alkene moiety, followed by reversible β-hydride elimination to effectively move the double bond down the chain. The net result is a 1,3-hydrogen shift, but the reversible nature of the reaction means that a net 1,2-hydrogen shift may also result. This type of mechanism has been shown to occur in metathesis reactions, where ruthenium hydrides are generated via decomposition of the active metathesis catalyst.23,24 Alternatively, a η3-allyl mechanism might be in operation (Scheme 6(b)), which proceeds via abstraction of an allylic proton by a metal which is bound to the alkene via η2-coordination. The hydride is then deposited at the terminus of the allyl complex, to yield a new η2-complex. The η3-oxo-allyl mechanism (Scheme 6(c)) is similar, but takes advantage of the presence of an alcohol in the allylic position. The alcohol co-ordinates to the metal, which is followed by η2-complexation of the alkene, abstraction of the allylic proton, and generation of a new η3-allyl complex by depositing the hydride on the terminus.
A number of experiments were therefore conducted to probe the mechanism of this transformation.
Isotopic labelling studies
Selectively-labelled substrate 2-d was prepared by reduction of the corresponding ketone using NaBD4 in THF, according to the literature procedure.16 Two reactions were conducted with this substrate. First, the allylic isomerisation was conducted using the same conditions as for the synthetic experiments (Scheme 7(a)). The reaction showed only the 1,3-deuterium shift, suggesting that the reversible metal hydride addition/elimination shown in Scheme 6(a) does not occur; if it did, both 1,2- and 1,3-deuterium shifts would be expected.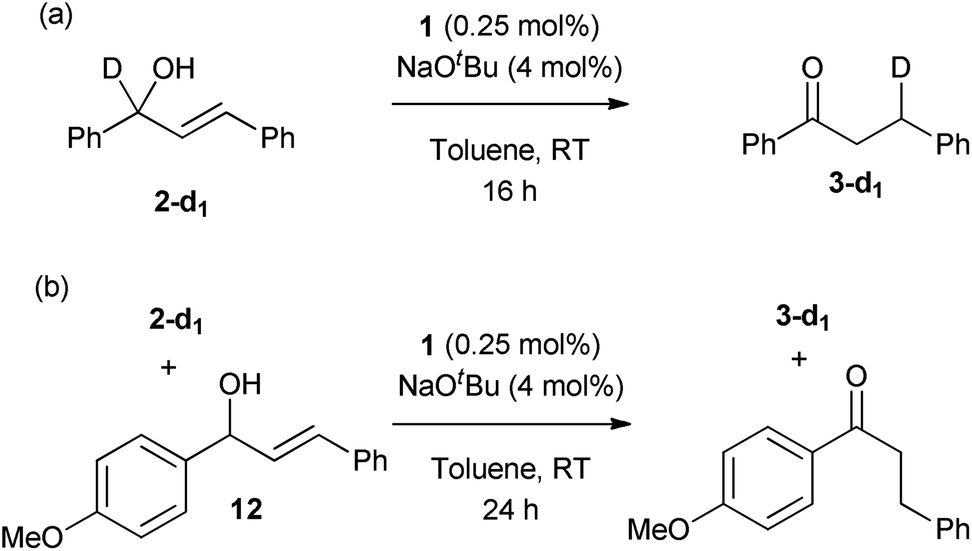 | ||
| Scheme 7 | ||
To further probe the potential presence of metal hydrides, a crossover experiment was performed (Scheme 7(b)) in which 2-d and 12 were exposed to the same charge of 1 (0.25 mol%, based on the sum of substrates) for 24 h. There was no transfer of deuterium between compounds, ruling out the presence of an unbound metal hydride (or deuteride) in the reaction. These experimental results permit the alkyl mechanism to be ruled out (Scheme 6(a)).
Hydroxyl effect
In an attempt to discriminate between a mechanism involving a simple η3-allyl intermediate (Scheme 6(b)) and one in which the oxygen contributes to the reaction to form an η3-oxo-allyl intermediate (Scheme 6(c)), the isomerisation was performed using O-methylated allylic alcohol 16 (Scheme 8). If the reaction involves only an η3-allyl intermediate, the isomerisation of 16 should yield enol ether 17, which is thermodynamically favoured over the substrate by ca. 1.7 kcal mol−1 (see ESI† for computational details). However, the reaction showed no conversion, even after 24 h, while the corresponding alcohol shows complete conversion in only 1 h. Assuming that the limit of detection of 1H NMR spectroscopy is at worst ca. 5%, this corresponds to a greater than twenty-fold difference in turnover number, and a ca. 500-fold difference in turnover frequency. While this is a negative proof, rather than a positive proof, this result suggests a role for the alcohol function, lending some support to the η3-oxo-allyl mechanism (Scheme 6(c)).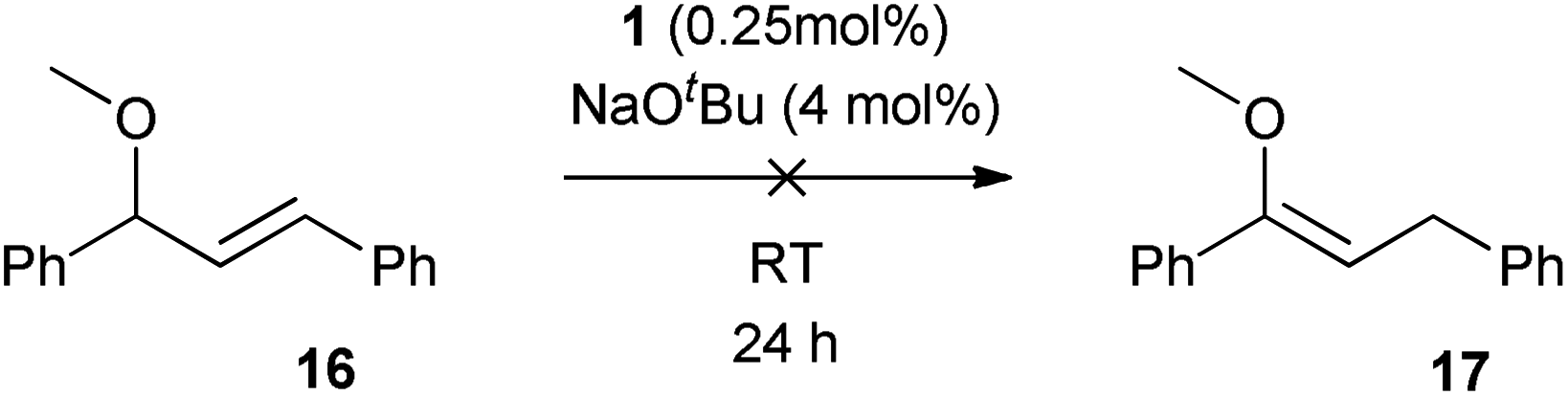 | ||
| Scheme 8 | ||
Density functional theory calculations
In order to tie experimental observations together, thus understanding fully the mechanism and setting the scene for further catalyst development, we embarked on density functional theory studies of the reaction mechanism (at the M06//TZVP//BP86/SVP level of theory).Potential energy surfaces (PES) were evaluated for the isomerisation of three model substrates using 1: allyl alcohol, but-1-ene-3-ol, and α-vinyl benzyl alcohol. In each case, the η3-oxo-allyl mechanism in Scheme 6(c) was found to be the most favourable. In addition the isomerisation of but-1-ene-3-ol was explored with [RuCl(indenyl)(PPh3)2] 4 and [RuCl(Cp)(PPh3)2] 5. All PESs are normalised to pre-catalyst plus substrate plus base, which is termed intermediate A for 1, A′ for 4, and A′′ for 5.
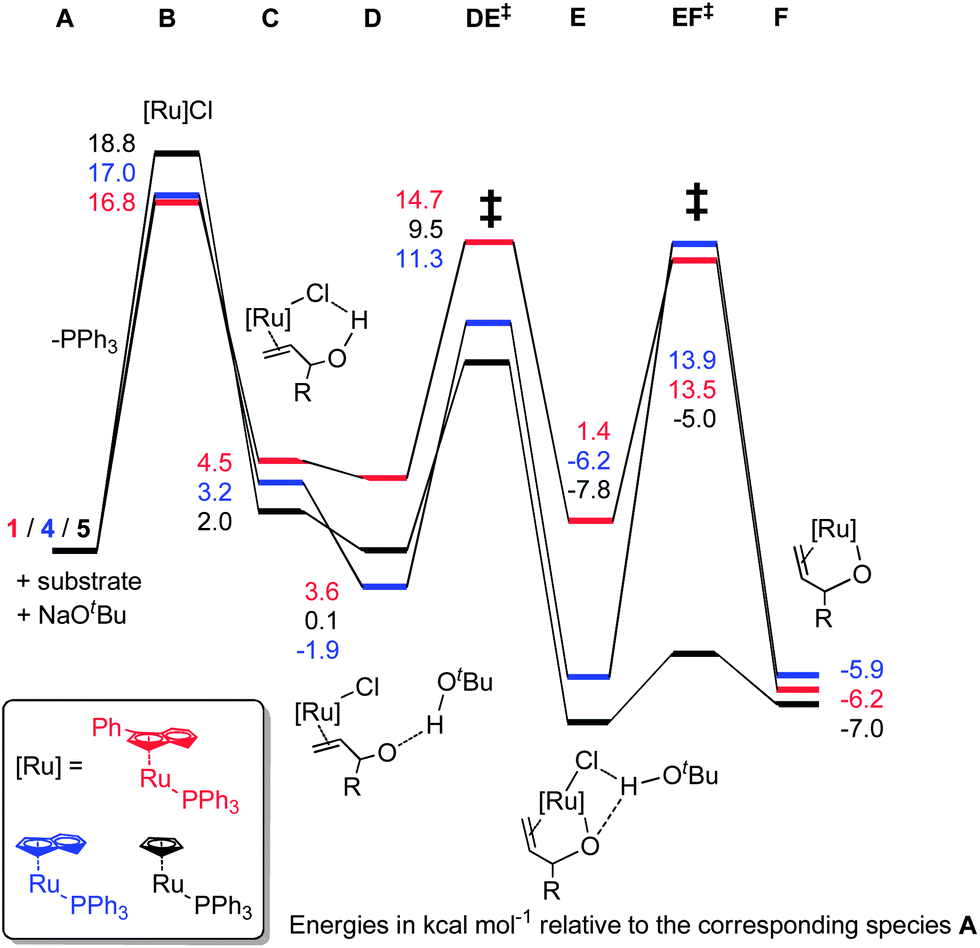
Pre-catalyst 1 undergoes a series of steps to form the active species, with the energetics somewhat dependent on the substrate (Scheme 9). Phosphine dissociation is relatively facile (16.8 kcal mol−1), and is followed by coordination of the substrate and deprotonation by the base. Chloride abstraction leads to intermediate F, and thus into the catalytic cycle. Calculations on alternative mechanisms showed that the base is necessary at this point to remove the HCl produced by initiation of the pre-catalyst. In addition, removal of the second phosphine (from C) leads to much higher energy intermediates (Grel = 25–35 kcal mol−1) on the PES of the catalytic cycle itself, so this possibility was discounted.
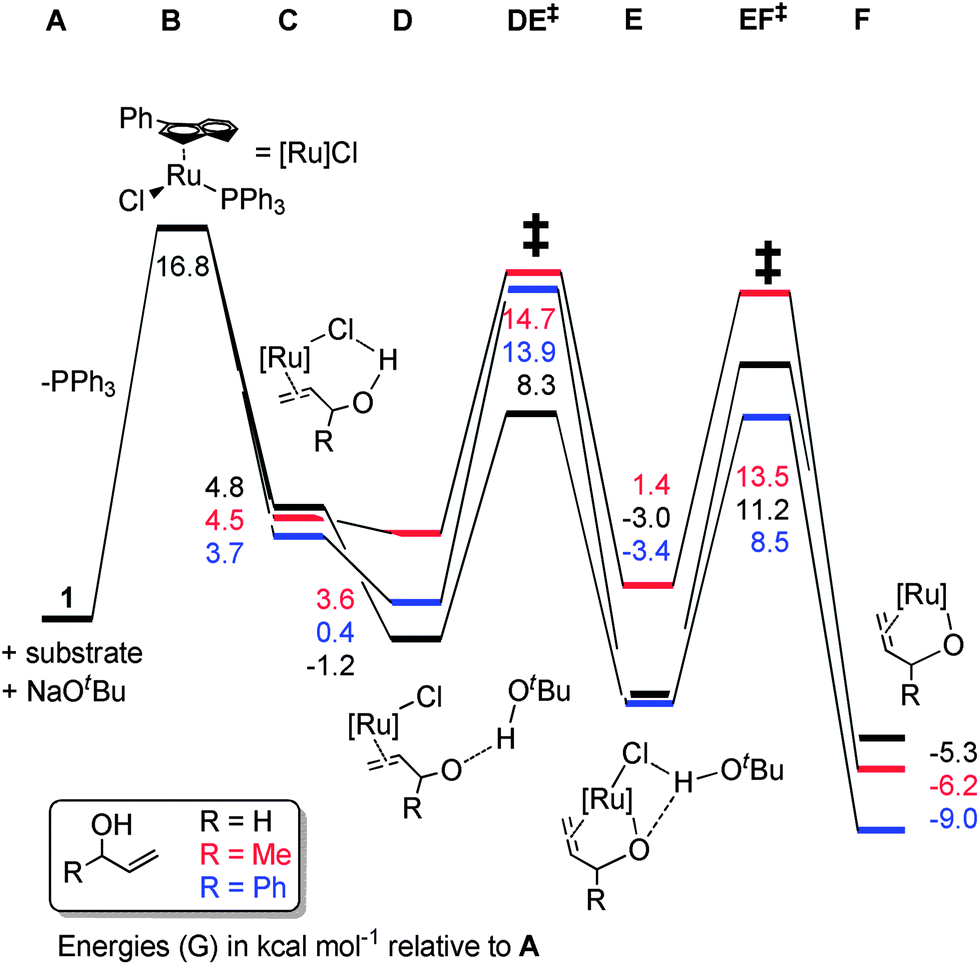 | ||
| Scheme 9 | ||
The reaction proceeds via abstraction of the second allylic proton to yield the enone η2-coordinated to a ruthenium hydride species (G) in a potential energy well (Scheme 10). Rotation of this η2-ligand has a considerable (20 kcal mol−1) barrier viaGH‡, yet presents the alkene terminus for delivery of the hydride via a rather facile process, with H to HI‡ being only 2–3 kcal mol−1. The complex then rearranges to bind the enol product via the oxygen, setting the scene for a series of steps to liberate the enol product and co-ordinate a subsequent molecule of substrate. Notably, the energetics of the isomerisation of primary and secondary allylic alcohols are rather similar, suggesting that the inability of 1 to isomerise primary allylic alcohols is not a consequence of high kinetic barriers. The energetic spans of the catalytic cycles can be calculated by subtracting the energy of G (the turnover-determining intermediate, or TDI) from that of LF‡ (the turnover-determining transition state, or TDTS) according to the work of Kozuch and Shaik (eqn (1)).25
| δE = TTDTS − ITDI | (1) |
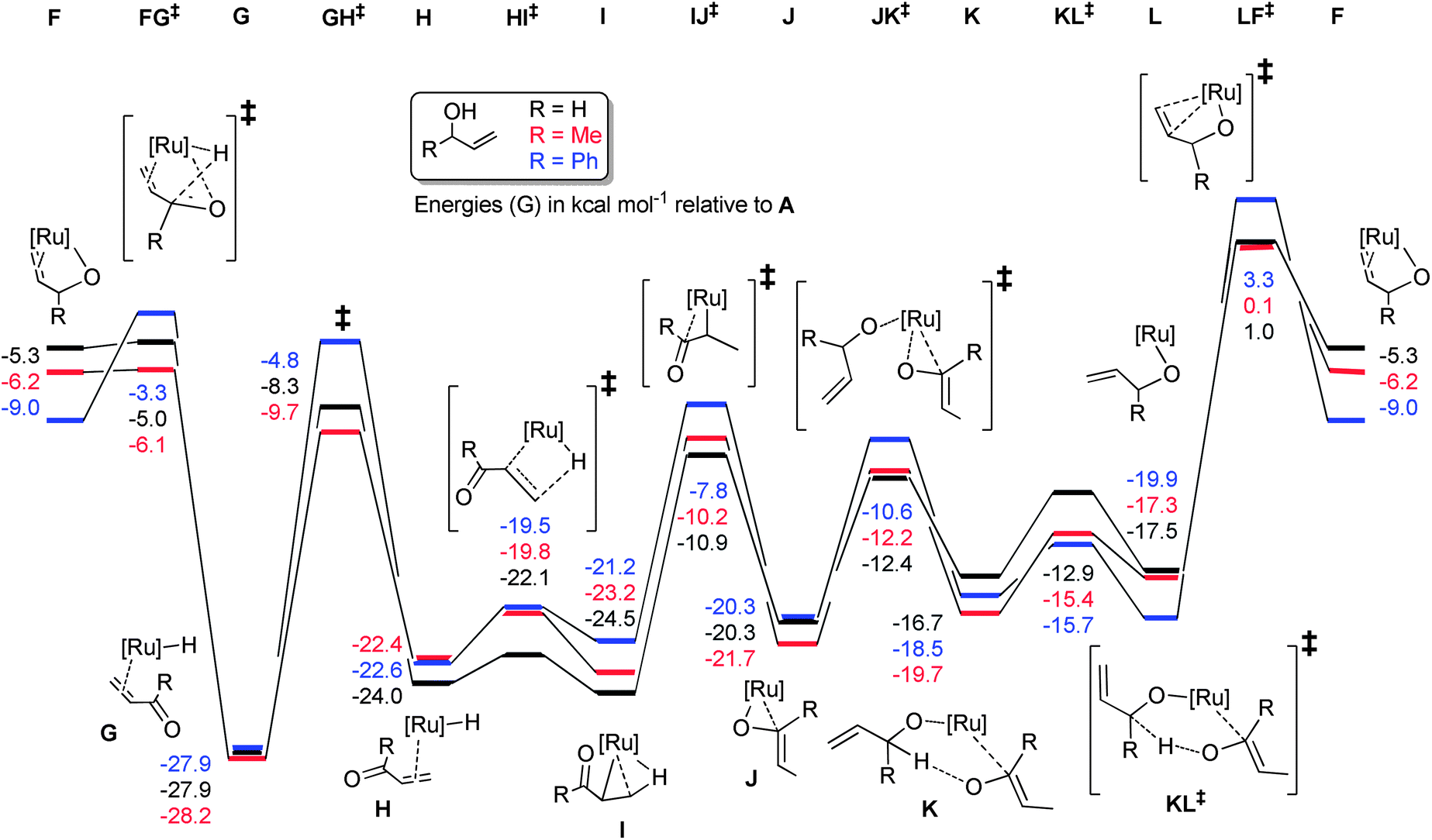 | ||
| Scheme 10 | ||
Values of 28.0, 31.5 and 28.9 kcal mol−1 can be obtained for allyl alcohol, but-1-ene-3-ol and α-vinyl benzyl alcohol, respectively. Therefore, it would be expected that primary alcohol isomerisation should be faster than that of the secondary alcohols (see the following section for further investigations into this discrepancy).
With calculated intermediates and transition states in hand, we examined the effect of catalyst structure on reactivity (Schemes 11 and 12). The energetics of the initiation sequence are quite different. Small differences in phosphine dissociation and substrate binding energies were calculated. Surprisingly, even though the phosphine dissociation is favoured for 1, the deprotonation and chloride abstraction steps exhibited the highest barriers for complex 1, despite the experimental observations that 1 is a more efficient catalyst than 4 or 5.
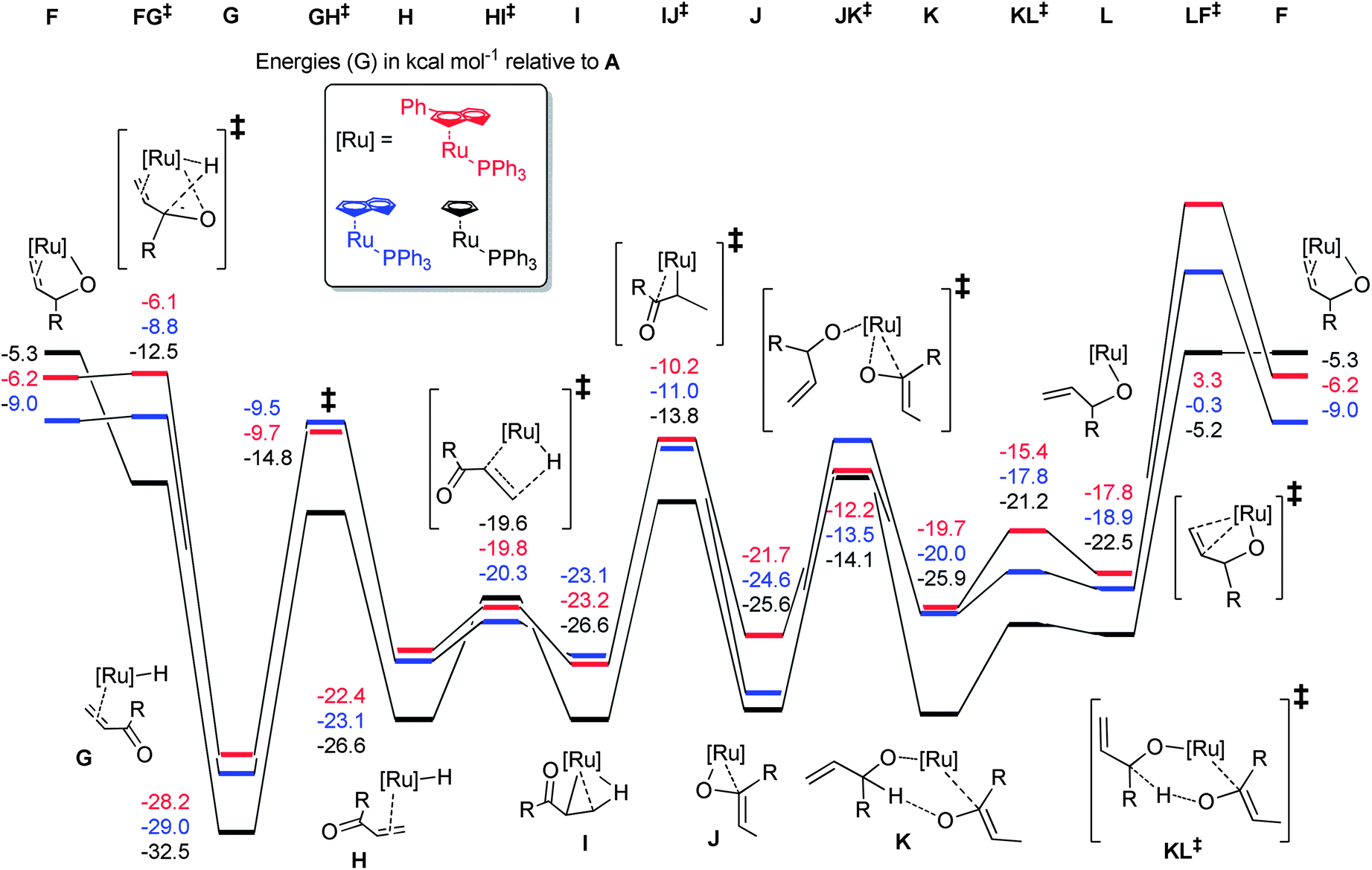 | ||
| Scheme 11 | ||
 | ||
| Scheme 12 | ||
The calculated energetics of the catalytic cycle (Scheme 12) show that, in general, the intermediates on the reaction pathway catalysed by the cyclopentadienyl analogue 4 are much lower in energy, although the overall energetic span (δE) is similar (31.5, 27.3 and 28.7 kcal mol−1 for 1, 4 and 5, respectively). However, the barriers to each of the individual steps for 1 are typically smaller, compared to 4 and 5, e.g.J to JK‡versusJ′ to JK′‡. Alternatively, decomposition of 1 (or intermediates derived from 1) may be slower under the reaction conditions than for 4 and 5.
Isomerisation of primary allylic alcohols
One of the aims of the DFT study was to understand why primary allylic alcohols were not successfully converted to the corresponding aldehydes. Surprisingly, calculations indicated that this transformation should be both kinetically and thermodynamically feasible. ΔG values for the overall reactions were calculated to be 6.4, 7.1 and 7.2 kcal mol−1 for allyl alcohol, but-3-en-1-ol, and α-vinyl benzyl alcohol, respectively.In an attempt to reconcile experiments and theory, it was hypothesised that either the base present is consumed in a side-reaction, or that the presence of the base itself might promote side reactions with one of the ruthenium intermediates. The second alternative seems to be most likely, as even in the presence of ca. 16 equivalents of base, no reaction occurs. This suggested that a base-free protocol should be developed to catalyse isomerisation of primary allylic alcohols. In addition, the use of a base has implications for the overall atom economy of a process; simple addition of a catalyst to a substrate is both more convenient and atom-efficient than protocols requiring the use of extra reagents such as bases.
This was explored using complex 14, which is already capable of isomerising secondary allylic alcohols. In addition, DFT calculations were performed to explain the inactivity of 15 in this transformation. These calculations suggested that phosphine dissociation from 15 presents a considerable barrier (30.4 kcal mol−1), yielding a series of intermediates that were much higher in energy than those in the schemes presented above (see the ESI† for details).
Initial experiments were conducted at room temperature with four model substrates (in NMR tubes) (Table 5), and showed the ability of 14to isomerise primary allylic alcohols. Allyl alcohol was isomerised rapidly, yielding complete conversion to propanal within 1.5 h with only 0.25 mol% of catalyst (Entry 1). The addition of steric hindrance at the γ-position (crotyl alcohol) resulted in a significant decrease in reactivity, with only very modest conversion, even after extended periods of time (Entries 2–4). An increase of catalyst loading to 1 mol% furnished complete conversion overnight (Entry 5). Methylation at the alternative position had a less dramatic effect on reactivity, but several hours were needed to allow complete conversion (Entries 6 and 7). γ,γ-Dimethylation effectively shuts down the reaction, with no reaction detected within 12 h at rt (Entries 8–10). The catalyst system is therefore clearly very sensitive to substitution pattern, particularly γ- to the alcohol, which is in accordance with the reactivity of 1.
| Entry | Substrate | [14] (mol%) | Time (h) | Conv.b |
|---|---|---|---|---|
| a Substrate solution (0.6 mL of a 0.5 mol L−1 solution in toluene-d8) added to 14 in an J. Young type NMR tube and mixed at room temperature. b Conversion determined from 1H NMR; average of at least two experiments. | ||||
| 1 |
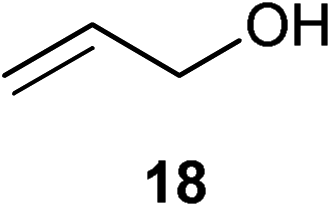
|
0.25 | 1.5 | >95% |
| 2 |
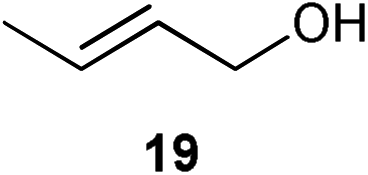
|
0.25 | 1.5 | <5% |
| 3 | 17.5 | 24% | ||
| 4 | 43.5 | 34% | ||
| 5 | 1 | 16 | >95% | |
| 6 |
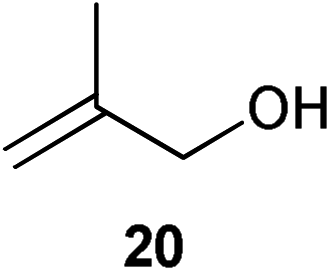
|
0.25 | 1.5 | 33% |
| 7 | 6 | 94% | ||
| 8 |
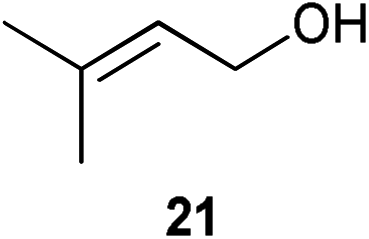
|
0.25 | 1.5 | <5% |
| 9 | 0.25 | 6 | <5% | |
| 10 | 0.25 | 12 | <5% | |
A number of cinnamyl alcohols were tested in the reaction, to explore whether these conditions using 14 were synthetically useful (Table 6). A catalyst loading of 1 mol% was employed, in light of the challenging nature of primary allylic alcohol substrates. While cinnamyl alcohol could be isomerised in a straightforward manner, additional steric hindrance precluded reaction (Entries 1 and 2). Electron-withdrawing groups rendered the reaction more challenging, although this could be mitigated by increasing the reaction time (Entries 3–5). Interestingly 14 shows reduced reactivity in the isomerisation of 27, probably due to the combination of steric hindrance and coordinating effect of the NMe2 on the aryl ring.
| Entry | Substrate | [14] (mol%) | Time (h) | Conv.b |
|---|---|---|---|---|
| a Conditions: substrate (1 mmol), 14 and toluene (1 mL) added. Reactions were stirred at ambient temperature for 1 h. b Conversion determined from 1H NMR; average of at least two experiments. | ||||
| 1 |
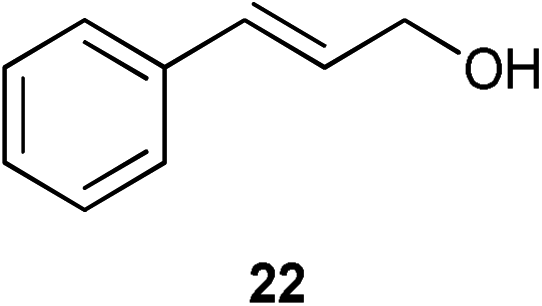
|
1 | 4 | >95% |
| 2 |
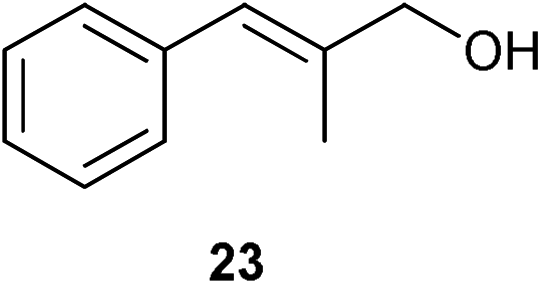
|
1 | 4 | <5% |
| 3 |
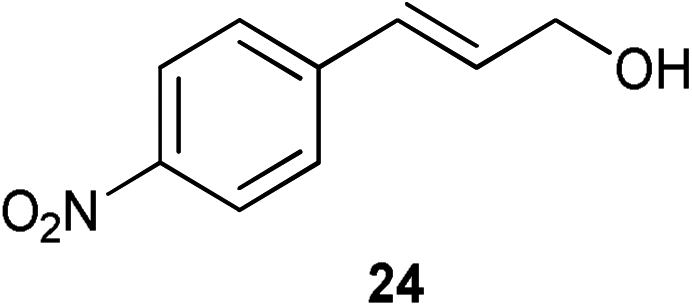
|
1 | 4 | 70% |
| 1 | 7 | >95% | ||
| 4 |
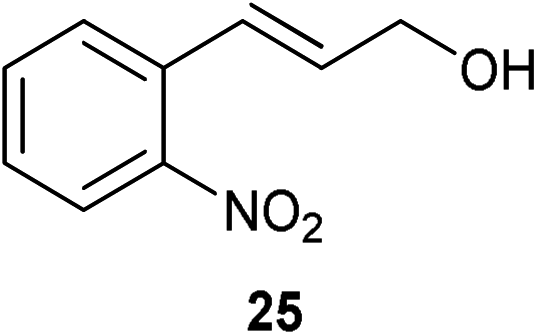
|
1 | 7 | >95% |
| 5 |
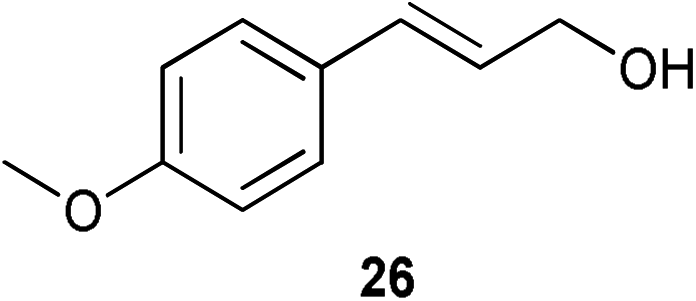
|
1 | 4 | >95% |
| 6 |
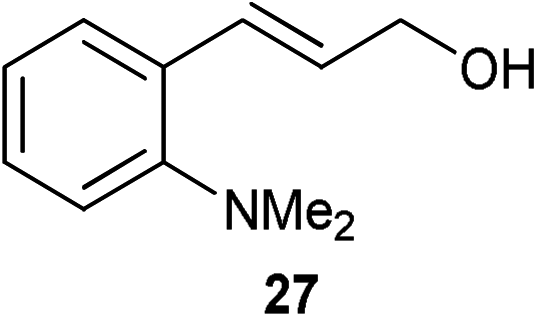
|
1 | 4 | >28% |
| 1 | 7 | 32% | ||
Catalysts based on the (3-phenylindenyl)bis(triphenyl phosphine)ruthenium motif are therefore capable of isomerising primary alcohols, when a base is not present. Here, this has been achieved via use of a cationic ruthenium species.
Conclusions
Complex 1 has been evaluated in the isomerisation of allylic alcohols, showing high activity and broad compatibility with substituted allylic compounds. Indeed, 1 can be considered one of the most active ruthenium complexes reported in the literature for this reaction, requiring only 0.25–0.5 mol% ruthenium and short reaction times at room temperature. The potential energy surface for this reaction has been explored, and compared to that of the analogous reactions with similar catalysts that show far less activity.In addition, it was shown that the use of cationic species 14 can remove the need for added base, and widen the scope of the reaction to include challenging primary allylic alcohol substrates.
Further investigations into the applications of 1, the synthesis and study of a systematic series of variants, and their further application are currently underway in our laboratories.
Acknowledgements
We thank the ERC (Advanced Investigator Award ‘FUNCAT’ to SPN) and the EPSRC for funding. Umicore is thanked for gifts of materials. SPN is a Royal Society Wolfson Merit Award holder. Melanja Smith and Dr Tomas Lebl are thanked for assistance with NMR spectroscopy facilities. Dr César A. Urbina-Blanco is acknowledged for useful discussions. LC thanks the HPC team of Enea for using the ENEA-GRID and the HPC facilities CRESCO in Portici (Italy) for access to remarkable computational resources. AP thanks the Spanish MICINN for a Ramón y Cajal contract (RYC-2009-05226) and European Commission for a Career Integration Grant (CIG09-GA-2011-293900).Notes and references
- R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2002, 103, 27–52 CrossRef PubMed.
- S.-I. Murahashi, Ruthenium in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- R. C. van der Drift, E. Bouwman and E. Drent, J. Organomet. Chem., 2002, 650, 1–24 CrossRef CAS.
- S. Bellemin-Laponnaz and J.-P. Le Ny, C. R. Chim., 2002, 5, 217–224 CrossRef CAS.
- L. Mantilli, D. Gérard, C. Besnard and C. Mazet, Eur. J. Inorg. Chem., 2012, 2012, 3320–3330 CrossRef CAS.
- A. Azua, S. Sanz and E. Peris, Organometallics, 2010, 29, 3661–3664 CrossRef CAS.
- A. P. da Costa, J. A. Mata, B. Royo and E. Peris, Organometallics, 2010, 29, 1832–1838 CrossRef.
- R. García-Álvarez, F. J. Suárez, J. Díez, P. Crochet, V. Cadierno, A. Antiñolo, R. Fernández-Galán and F. Carrillo-Hermosilla, Organometallics, 2012, 31, 8301–8311 CrossRef.
- B. M. Trost and R. J. Kulawiec, J. Am. Chem. Soc., 1993, 115, 2027–2036 CrossRef CAS.
- C. Slugovc, E. Rüba, R. Schmid and K. Kirchner, Organometallics, 1999, 18, 4230–4233 CrossRef CAS.
- V. Cadierno, S. E. Garcia-Garrido and J. Gimeno, Chem. Commun., 2004, 232–233 RSC.
- V. Cadierno, S. E. García-Garrido, J. Gimeno, A. Varela-Álvarez and J. A. Sordo, J. Am. Chem. Soc., 2006, 128, 1360–1370 CrossRef CAS PubMed.
- B. Martín-Matute, K. Bogár, M. Edin, F. B. Kaynak and J.-E. Bäckvall, Chem.–Eur. J., 2005, 11, 5832–5842 CrossRef PubMed.
- M. Ito, S. Kitahara and T. Ikariya, J. Am. Chem. Soc., 2005, 127, 6172–6173 CrossRef CAS PubMed.
- S. Manzini, C. A. Urbina-Blanco, A. Poater, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 1042–1045 CrossRef CAS PubMed.
- S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 3036–3044 CrossRef CAS.
- S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Organometallics, 2013, 32, 660–664 CrossRef CAS.
- J. A. Fernandéz-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 5829–5831 RSC.
- S. Manzini, D. J. Nelson and S. P. Nolan, ChemCatChem, 2013, 5, 2848–2851 CrossRef CAS.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
- D. V. McGrath and R. H. Grubbs, Organometallics, 1994, 13, 224–235 CrossRef CAS.
- I. W. Ashworth, I. H. Hillier, D. J. Nelson, J. M. Percy and M. A. Vincent, Eur. J. Org. Chem., 2012, 2012, 5673–5677 CrossRef CAS.
- F. C. Courchay, J. C. Sworen, I. Ghiviriga, K. A. Abboud and K. B. Wagener, Organometallics, 2006, 25, 6074–6086 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2010, 44, 101–110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation data. See DOI: 10.1039/c3sc52612g |
| This journal is © The Royal Society of Chemistry 2014 |