Fabricating a sustained releaser of heparin using SBA-15 mesoporous silica†
Wen Juan
Qian
,
Mi Mi
Wan
,
Wei Gang
Lin
and
Jian Hua
Zhu
*
School of Chemistry and Chemical Engineering, Nanjing University, 22 Hankou Road, Nanjing, Jiangsu, China. E-mail: jhzhu@nju.edu.cn
First published on 16th October 2013
Abstract
The sustained release of heparin in sufficient amounts and over long time is a challenge to drive the development of functional materials. In this paper SBA-15 mesoporous silica is selected in the search for a favorable morphology and optimized surface state for the sustained release of heparin. In situ carbonization of the template in the as-synthesized sample enables SBA-15 to possess narrowed channels with rougher surfaces, while modification with (aminopropyl)triethoxysilane (APTES) through a one-pot synthesis offers SBA-15 with positive charges to attract heparin through electro-static interactions. The structure of modified SBA-15 samples is assessed with XRD (powder X-ray diffraction analysis), nitrogen adsorption–desorption and electron microscopy techniques, and their performance is evaluated in adsorption and release of heparin. These modifications improve the heparin adsorption in SBA-15 and thus promote its sustained release, prolonging the release-equilibrium time up to 60 days. Among them, the SBA-15 sample modified with APTES can trap three times as much heparin as the parent SBA-15, and the release ratio is elevated to 80% (that of SBA-15 is 38%), realizing the best performance of controlling heparin release to date.
Introduction
Heparin is a kind of biological macro-molecule formed by copolymerization of idose-hyaluronic acid and glucosamine. It is the sole effective drug for hemodialysis treatment and is mainly used in the prevention and control of diseases in heart blood vessels.1 As the world's best anticlotting drug for clinical laboratories, long-term controlled release of heparin is required, especially for the rapid development of artificial organ research on anticoagulants. As potential drug carriers, however, organic polymers are slowly fading from view because of certain side effects,2 thus inorganic materials have attracted a lot of attention for controlled release in recent years. Among them, mesoporous materials are outstanding due to their high thermal stability, mechanical strength and non-toxic characteristics. Macromolecular drugs can be wrapped into the mesoporous channels by immersion, and then released to the targeted place, which is controlled by a diffusion mechanism.3–5 The mechanism for drug release in mesoporous materials has been reported6–8 in which how the pore size, channel wall roughness and organic modifications influence the performance are described in detail.In order to achieve a long-term sustained release of heparin, it is crucial to build a suitable micro-environment in vessels where heparin can be stably trapped to retain the original conformation and biological activity. Also, an appropriate interaction is required to anchor the heparin, which should be weaker than chemical bonding but stronger than physical adsorption. For instance, the enhanced van der Waals force in a confined space has been found to be helpful for immobilization and controlled release of a large amount of heparin.9 In previous research, we found that suitably enlarged pores of MCM-41 are beneficial for the adsorption of heparin, and the residual CTAB template or introduced amino groups in the sample can promote heparin adsorption due to their special interactions.10 However, although the heparin encapsulated reaches 99 mg g−1, only about 40% of this is released. We also incorporated aluminum in SBA-15 and tailored the surface roughness of channels through forming plugs, and these composites adsorbed 2–4 times more heparin than the parent SBA-15 and released 60% to 130% more, maintaining the release up to 6 weeks;11 but the release ratio was still lower than 40%, which spurred us to seek new vessels for the sustained release of heparin. Based on these results, we chose SBA-15 to fabricate new vessels, not only because of its large pore size and rough wall surfaces,12 but also for its controllable synthesis including various additives for specific morphologies.13 In principle, release of heparin in solution from vessels is driven by the concentration difference between them, so that increasing the amount of heparin adsorbed in the vessel is important for the subsequent release procedure. Through controlling the channel length, surface curvature and organic functionality of mesoporous silica, we tried to establish an optimal environment for immobilization of heparin.
To promote the diffusion of heparin inside the channels of mesoporous silica, SBA-15 samples with uniform short primary particles13 or 3D-interconnected channels14 were chosen. The former have relatively short meso-channels to shorten the diffusion path of the protein and the latter offer the heparin more directions to diffuse. Monolithic SBA-15 sample was also synthesized since its net-linked morphology is efficient to intercept the target in fluids,15 which will be applied to increase the adsorption of heparin in SBA-15. Apart from these candidates with various morphologies, two modifications were utilized to build a suitable microenvironment inside the channels of SBA-15. One is the carbon modification of mesoporous silica with its inherent template micelles.16–18 These micelles have a spoke-like configuration to form numerous subnanospaces between the micelles and pore walls, and their softness and flexibility are changed with temperature.19In situ carbonization of template micelles in MCM-41 results in the efficient trapping of N-nitrosopyrrolidine,16 here the templates P123 in the as-synthesized SBA-15 will be carbonized in the range of 423–573 K, that is the decomposition temperature of P123,20 to subtly open the channels and regulate their roughness. Another modifier is the amine group –NH2,10 and thus (aminopropyl)triethoxysilane (APTES) was adopted to covalently link organosilane species (aminopropyl groups) through Si–O–Si bonds to the internal surface and rigidify the mesoporous framework,21 through direct co-condensation and post-synthesis grafting methods. The optimal amount of APTES and incorporation manner as well as the actual performance of SBA-15 in the adsorption and release of heparin have been carefully assessed.
Experimental section
Materials and reagents
TEOS (tetraethylorthosilicate) and P123 (EO20PO70EO20) were purchased from Shanghai Wulian (China) and Aldrich, respectively. APTES was bought from Nanjing Chemical Reagent (China). All of the reagents and solvents were of A.R. grade and used as received. Phosphate buffer solution (PBS) (pH = 7.1) was prepared by dissolving Na2HPO4·12H2O and NaH2PO4·2H2O in deionized water.11Preparation of samples
SBA-15 was synthesized according to the literature.22,23 In a typical synthesis, 2 g P123, 60 g HCl (2 mol L−1) and 15 g H2O were mixed at room temperature. After P123 was dissolved, 4.25 g TEOS was added into the mixture at 313 K, followed by vigorous stirring for 24 h, and then this solution was transferred into a Teflon bottle and heated statically at 373 K for another 24 h. The solid product was recovered by filtration, washed with distilled water, air-dried and finally calcined at 773 K for 5 h to obtain SBA-15. Monolithic SBA-15 was synthesized in a similar way with HCA (citric acid) and TMOS (tetramethyl orthosilicate),24 and the initial reaction composition was 9.3 g TMOS : 3 g P123 : 0.105 g HCA : 112.5 g H2O, but the aging temperature was changed to 333 K. The resulting sample was named as SM. Sample SE was prepared in the same way as SBA-15 except that 1 min ultrasonication was utilized after addition of TEOS.13 For the preparation of SBA-15 with 3D-interconnected channels, a high-temperature hydrothermal process was used14 to produce the sample S3D, while trimethylbenzene (TMB) was introduced into the meso-structured materials to enlarge the pore size, forming the samples named S3D-T.To utilize the template micelles inherently occluded inside the channels of mesoporous silica,16,18 some as-synthesized SM composites were carbonized in the range of 423–573 K, that is the decomposition temperature of P123,20 for a given time as listed in Table 1, giving the samples SC-n. For the samples treated at high temperature, 1.0 g as-synthesized SM and 0.23 g H2SO4 (98%) were mixed with 5 g H2O, then ultrasonicated and heated at 373 K or 433 K,18 and finally carbonized in a nitrogen flow at 673 K or 773 K for 5 h to get the composites SC-a or SC-b. In another experiment, APTES was incorporated in SBA-15 through a one-pot synthesis,21 that is, by the co-condensation of TEOS and APTES in the preparation. APTES was introduced 2 h after the addition of TEOS, rather than added immediately,25 with the different molar ratios as listed in Table 2, and the resulting samples were refluxed with ethanol to remove template and were denoted as SA-n. In a similar way the SMA was prepared using the synthesis procedure of SM instead of SBA-15. Sample SA-P is the SBA-15 post-modified with APTES:26 0.5 g powder-like SBA-15 was mixed with a 25 mL chloroform solution of APTES (0.2 M) and stirred for 12 h at room temperature, and then the precipitate was filtered and washed with plenty of chloroform, and air-dried to give the final product. For comparison, SBA-15 mixed with chloroform was also prepared in the same way to give sample SA-s, in order to check the solvent effect on the adsorption and release properties of SBA-15.
| Sample | Temperature (K) | Time (h) | Content of carbon (%) |
|---|---|---|---|
| SC-1 | 423 | 2 | 25.8 |
| SC-2 | 473 | 2 | 22.5 |
| SC-3 | 523 | 2 | 21.9 |
| SC-4 | 573 | 2 | 10.3 |
| SC-5 | 573 | 5 | 6.8 |
| Sample | Initial APTES/TEOS molar ratio | Initial APTES content (%, a) | Final APTES content (%, b) | b/a |
|---|---|---|---|---|
| SA-1 | 0.05 | 15.9 | 3.3 | 0.21 |
| SA-2 | 0.10 | 27.4 | 5.0 | 0.18 |
| SA-3 | 0.13 | 32.0 | 6.2 | 0.19 |
| SA-4 | 0.20 | 36.6 | 16.4 | 0.45 |
| SA-P | 0.10 | 27.4 | 3.3 | 0.12 |
| SMA | 0.10 | 27.4 | 4.0 | 0.15 |
Characterization of samples
The XRD patterns of samples were recorded on an ARL XTRA diffractometer with Cu Kα radiation in the 2θ range of 0.5–6°.27 Nitrogen adsorption isotherms were measured at 77 K on a Micromeritics ASAP 2020 volumetric adsorption analyzer, and the samples were degassed for 4 h prior to measurement. The BET specific surface areas (SBET) of samples were calculated using adsorption data acquired in the relative pressure (P/P0) range of 0.05–0.22, and the total pore volumes (Vp) were determined from the amount adsorbed at a relative pressure of about 0.99. The pore size distribution (PSD) curves were calculated from the analysis of the adsorption branch of the isotherm by using the improved Kruk–Jaroniec–Sayari (KJS) method,28 and the microporous volumes of samples (Vmic) were calculated by the t-plot method. Scanning electron microscopy (SEM) images were obtained by using a HITACHI S4800 microscope, and the samples were coated with Au films to improve the conductivity prior to imaging. Transmission electron microscopy (TEM) images were acquired on a JEM-2100 (JEOL) electron microscope, for which the sample was ground and suspended in ethanol, and then dispersed on a copper net by ultrasonication. Combustion chemical analyses (C, H and N) of the silicate-containing organic materials were carried out in a CHN-O-rapid elemental analyzer, and the APTES contents of samples were calculated according to nitrogen content. Thermo-gravimetric and differential scanning calorimetric (TG-DSC) analyses of samples were performed on a NETZSCH STA449C apparatus in oxygen from 293 K to 1073 K with a heating rate of 10 K min−1.Adsorption and release of heparin
Detection of heparin with an improved colorimetric toluidine blue method and preparation of standard heparin solution were performed as reported previously.11,29 Adsorption of heparin by mesoporous samples was carried out as follows: 100 mg sample was added into a tube containing 5 mL heparin solution (10 mg g−1) and kept at 277 K for 72 h. Afterward the sample was separated and washed with 5 mL PBS solution three times, and then the residual heparin amount in solution was measured and the difference between the original and the residual amounts of heparin was determined as the adsorption amount. The TG-DSC method was also used to detect the adsorbed amount of heparin in samples SM, SE, S3D and SBA-15, as reported previously.10 To assess the release of heparin from the mesoporous composite, the washed solid was put into 10 mL PBS solution at 310 K. Then, the release amounts of heparin at different times were determined by the toluidine blue method, and release profiles were obtained by plotting the time vs. the release amount.9–11Results
Characterization of SBA-15 composites
Different morphologies of SBA-15 and its analogues are displayed in the SEM images shown in Fig. 1 and S1.† SBA-15 is wheat-like and composed of rod-like primary particles whose lengths vary from several to tens of micrometers. Monolithic sample SM has a special 3D net-linked morphology in which lots of fibers are randomly cross-connected, forming a complicated net framework.24 Sample SE contains many rod-like primary particles with the unit size of about 10 μm, obviously shorter than that of SBA-15 (Fig. 1). S3D sample has the morphology similar to that of SBA-15, but there are many small fragments around the wheat-like particles as seen in Fig. 1. S3D-T sample shows a similar appearance as that of S3D (Fig. S1†), with more tiny fragments. SC-2 is a filamentous linked network, SA-2 has some small blocks, while SMA shows an approximately hexagonal net-linked morphology. The TEM image in Fig. S2† reveals the pore structure of S3D. This sample has an ordered hexagonal pore arrangement and the distances between the centers of adjacent channels are around 11 nm, in accordance with the a0 value (10.9 nm) detected with the XRD method (Table 3). Moreover, some meso-tunnels with the size of 2–3 nm are randomly distributed on the silica walls of S3D sample to connect the channels. Addition of TMB in the synthesis of S3D-T made the sizes of these tunnels enlarge to the cylindrical channel size (8 nm), forming completely interconnected hexagonal channels (Fig. S2†). The TEM images of SA-2 and SMA samples are also illustrated in Fig. S2.† SA-2 sample still has ordered cylindrical channels, validating the existence of the p6mm symmetric mesophase, but its pore walls become distorted and some parts of the channel walls seem to be broken. In contrast, the order of the mesopore structure of SMA decreases, possessing a worm-like pore structure. | ||
| Fig. 1 The SEM images of SBA-15 series samples. | ||
| Sample | S BET (m2 g−1) | S mic (m2 g−1) | V p (cm3 g−1) | V mic (cm3 g−1) | D KJS (nm) | a 0 (nm) | Adsorbed hep (mg g−1, a) | Released hep (mg g−1, b) | b/a (%) |
|---|---|---|---|---|---|---|---|---|---|
| a SM, sample of monolithic SBA-15; SE, sample which is made of ultra-short nanorods; S3D, sample SBA-15 with 3D- interconnected channels; S3D-T, sample S3D synthesized with TMB to enlarge the interval size; SC-n, samples carbonized in nitrogen at different temperatures (see Table 1); SA-n, sample SBA-15 modified with APTES by one-pot (n = 1–4, see Table 2) and post-loaded (SA-P) method; SMA, sample SM modified with APTES by one-pot method. b S BET, BET surface area; Smic, micropore area; Vp, total pore volume; Vmic, micropore volume; DKJS, KJS mesopore diameter calculated from the adsorption branch; a0, the lattice parameter calculated from a0 = d100 × 2/31/2. Release amount is the total released heparin within 30 days. | |||||||||
| SBA-15 | 887 | 159 | 1.0 | 0.07 | 7.7 | 11.4 | 30.4 | 11.6 | 38.2 |
| SM | 554 | 244 | 0.46 | 0.11 | 6.1 | 11.3 | 44.5 | 13.0 | 29.2 |
| SE | 523 | 54.6 | 0.86 | 0.02 | 7.7 | 10.9 | 41.4 | 10.2 | 24.7 |
| S3D | 514 | 17.2 | 1.02 | 0 | 8.7 | 10.9 | 68.8 | 7.7 | 8.7 |
| S3D-T | 557 | 28.2 | 1.37 | 0 | 10.5 | 11.5 | 36.3 | 6.6 | 18.1 |
| SC-1 | 18 | 0 | 0.02 | 0 | 6.1 | 11.5 | 77.8 | 8.8 | 11.3 |
| SC-2 | 22 | 0 | 0.03 | 0 | 6.1 | 11.7 | 64.1 | 18.8 | 29.4 |
| SC-3 | 103 | 0 | 0.16 | 0 | 6.1 | 11.7 | 50.7 | 16.2 | 31.9 |
| SC-4 | 339 | 32 | 0.38 | 0.01 | 6.2 | 11.7 | 42.1 | 14.1 | 38.5 |
| SC-5 | 490 | 152 | 0.44 | 0.07 | 6.3 | 10.8 | 39.3 | 8.1 | 20.6 |
| SA-1 | 641 | 71.4 | 0.90 | 0.03 | 7.6 | 11.3 | 80.1 | 57.1 | 71.2 |
| SA-2 | 544 | 41.2 | 0.80 | 0.01 | 7.6 | 11.4 | 114.8 | 75.8 | 66.1 |
| SA-3 | 518 | 27.3 | 0.75 | 0 | 7.5 | 10.4 | 86 | 69.5 | 80.8 |
| SA-4 | 207 | 32.3 | 0.19 | 0.01 | 6.8 | 11.3 | 73 | 5.7 | 7.7 |
| SA-P | 420 | 35.0 | 0.65 | 0.01 | 7.3 | 10.4 | 52.1 | 4.7 | 9.1 |
| SA-s | 748 | 114 | 0.90 | 0.04 | 7.7 | 11.4 | 33.8 | 10.5 | 31.1 |
| SMA | 335 | 4.81 | 0.59 | 0 | 7.6 | 12.0 | 68.7 | 28.8 | 41.9 |
Powder X-ray diffraction patterns of amine-modified SBA-15 are exhibited in Fig. 2, in comparison with that of parent SBA-15 and its analogues (Fig. S3†). Common SBA-15 shows three well-resolved XRD peaks in the region of 2θ = 0.9–1.8°, which are indexed to the 100, 110 and 200 diffractions of the 2D hexagonal symmetry (p6mm).30 There is only one single peak of 100 diffraction in the patterns of SM samples, due to the less ordered structure formed in weakly acidic conditions.24 Three well-resolved peaks can also be observed in the patterns of SE and S3D samples (Fig. S3†) and they are indexed as 100, 110 and 200 diffractions respectively,31 but their intensity declines more or less, mirroring the less ordered mesoporous structures of these two samples. The similar decrease in the intensity of S3D and SE implies a minor effect of ultrasonic treatment and aging temperature on the order degree of the meso-structure of the resulting samples. Sample S3D-T seems to have a more disordered mesoporous structure since two weak peaks of 110 and 200 diffractions are connected as shown in Fig. S3a.† Fig. S3b† shows the low-angle XRD patterns of SC-n samples. These composites all have only one 100 diffraction peak in their patterns, like their parent SM (Fig. S3a†) due to the relatively disordered structure of monoliths.24 Besides, the carbonaceous modifier in these samples seems not to badly affect the structure, in agreement with the previous report on carbon modified MCM-41.16 The low-angle XRD patterns of SA-n composites are shown in Fig. 2, in which all samples have three obvious peaks, except the SA-4 sample. However, the intensities of their XRD peaks are weakened as more APTES additives are added in the synthesis, which reflects the less ordered structure. When the initial ratio of APTES/TEOS in the synthesis is increased to 0.2, the mesopore structure of the resulting sample SA-4 seems to be almost destroyed because there was only one small peak of 100 diffraction remaining, since too much APTES is not beneficial for the hydrolysis and coagulation of silica.31,32 On the other hand, such a decrease of XRD peak intensity implies the successful introduction of amino groups into the channels of SA-n composites, since the reflection intensities relate to the extent of pore filling and the scattering contrast between the pore walls and the inside of the pores.33 The XRD patterns of SA-P and SA-s samples are also illustrated in Fig. 2, in which the intensity of XRD peaks of the SA-P sample is weaker than that of SA-2. Since the same APTES/TEOS ratio of 0.20 (Table 2) is used in the preparation of these two samples, this difference is caused by different modification procedures. Again, the unit cell parameter a0 of SA-P is obviously decreased in comparison with SBA-15 (Table 3), owing to the grafting of amine groups inside the pore.
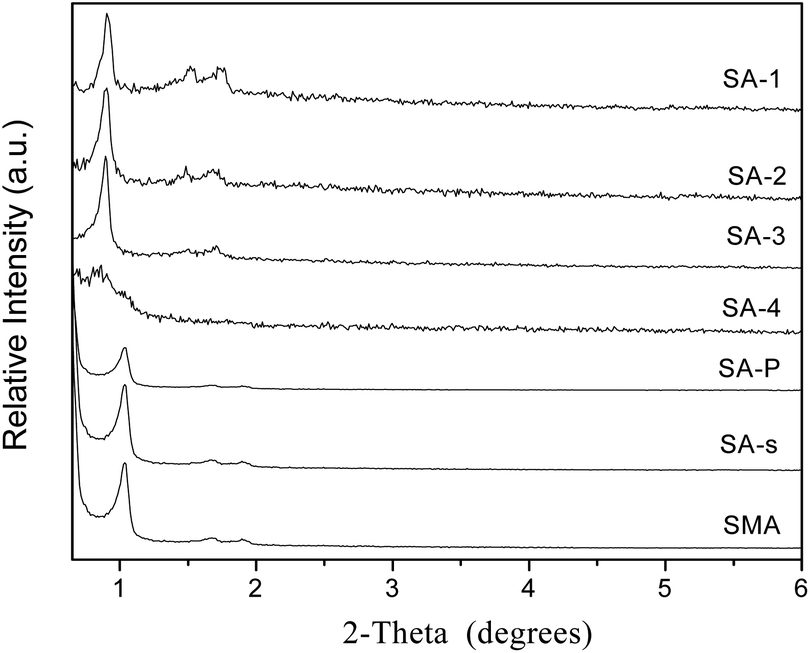 | ||
| Fig. 2 Low-angle XRD patterns of amine-loaded (SA-n) samples. | ||
Fig. S4a and b† present the nitrogen adsorption–desorption isotherms and pore size distribution of SBA-15 and its analogues with different morphologies. All of these composites show the typical IV isotherms with large type H1 hysteresis loops and relatively narrow pore size distributions, except SM sample whose hysteresis loop is slightly shifted toward lower values of P/P0 (Fig. S4a†). According to the pore size distributions shown in Fig. S4b,† these composites all have relatively narrow mesopores, and the average pore size varies from 6.1 to 10.5 nm (Table 3); among them S3D and S3D-T samples have pore sizes (8.7 and 10.5 nm) larger than SBA-15 (7.7 nm), but their cell parameters (10.9, 11.5 nm) are smaller, which implies the interconnection of channels.14 Fig. S4c and d† illustrate the adsorption–desorption isotherms of nitrogen at 77 K and the pore size distribution of carbonized SBA-15 samples SC-n. The samples carbonized at above 523 K (SC-3, SC-4, SC-5) show distorted isotherms with the hysteresis loops located in the low range P/P0 between 0.4 and 0.7, indicating structurally disordered pores and an ill-defined pore shape.34 Again, more nitrogen is trapped by the samples carbonized at higher temperature, indicating the larger pore volume of the composite. As demonstrated in Fig. S4d† and Tables 1 and 2, all SC-n samples have pore sizes (6.1–6.3 nm) smaller than SBA-15 (7.7 nm), because of the carbonaceous modifier inside the channels.16 Besides, the carbon content of SC-n decreases gently as the carbonization temperature is increased from 423 K to 523 K but dramatically declines as the temperature rises to 573 K. As the carbonization time is increased from 2 h to 5 h, the carbon content of the resulting SC-5 sample is lowered from 10.3% to 6.8% (Table 1).
 | ||
| Fig. 3 The N2 adsorption–desorption isotherms (a) and pore size distribution (b) of the amine-loaded SBA-15 (SA-n) composites. | ||
Fig. 3a and b display the nitrogen adsorption–desorption isotherms and pore size distribution of amine-loaded SBA-15 samples SA-n. Most of the composites, except SA-4, possess the typical IV isotherms with H1 hysteresis loops. However, less N2 is adsorbed on the samples incorporated with more APTES, and when the ratio of APTES/TEOS is up to 0.2 (SA-4 sample), the isotherm almost disappears to illustrate the absence of mesoporous structure, which is also confirmed by its XRD pattern (Fig. 2). The isotherm of SA-s sample (Fig. 3b) is almost the same as that of SBA-15 (Fig. S4a†), indicating the almost negligible effect of organic solvent such as chloroform on the structure of SBA-15 during the modification with APTES. However, the average pore size of SA-P sample is reduced from 7.6 to 6.8 nm in comparison with that of parent SBA-15 (Table 3) due to the introduction of APTES through the post-modification, since the whole modifier will be coated on the channel walls of the mesoporous host.35 In contrast, SMA sample has an expanded average pore size of 7.6 nm compared with parent SM (6.1 nm, Table 3), accompanied with a better ordered structure (Fig. 2), since the preparation of SM is more sensible for the acid concentration24 and its pore structure strongly depends upon the assembly kinetics of the silica hydrolysis/condensation. Addition of APTES changes the acidity of the solution and affects the hydrolytic condensation,31 so that a more ordered mesoporous structure is observed on SMA than that of SM (Fig. S3a† and 2). The final APTES contents of samples are listed in Table 2, in which the value of SA-n increases as more APTES additives are used in the synthesis but these is no linear relationship. Sample SA-P has a value (3.3%) smaller than SA-2 (5.0%) though they have the same initial APTES content (10%), but the same as SA-1 (3.3%). This difference reveals the strong influence of the synthesis procedure on the incorporation of APTES in SBA-15, which is confirmed by sample SAM which has an APTES content (4.0%) higher than SA-P but lower than SA-2.
Adsorption of heparin by mesoporous composites
The textural properties, adsorption and release performance of various mesoporous composites are summarized in Table 3. Compared with the parent SBA-15, its analogues with different morphologies have reduced surface area and pore size. The average pore size of SM sample is reduced from 7.7 nm (SBA-15) to 6.1 nm, meanwhile the ratio of micropores increases distinctly (Table 3); this hierarchical structure seems helpful for heparin adsorption, and it adsorbs 50% more heparin than SBA-15 with the assistance of the special 3D-net like morphology that is beneficial to intercept the heparin in solution. SE sample has a 41% reduced surface area in comparison with SBA-15, but it still has a higher adsorption capacity because of its relatively short channels to shorten the diffusion path. S3D sample also has 42% reduced surface, but it adsorbs 68 mg g−1 of heparin (Table 3), 130% more than SBA-15. The elevated adsorption capacity probably results from the existence of those narrow channels with the size of 2–3 nm that are randomly distributed between the 2D channels. However, S3D-T sample traps slightly more heparin than SBA-15 though its surface area and pore volume are larger than those of S3D (Table 3), and this difference may relate to its interconnected channels with larger size (about 8 nm, Fig. S2†) than S3D (2–3 nm), because large pores and channels of mesoporous host are not beneficial to immobilize heparin.10 These results of heparin adsorbed on the unmodified samples are also confirmed by TG-DSC tests as shown in Fig. S5.†Carbonizing the template P123 in silica pores to form carbon–silica composites can change the surface curvature of silica walls, at the expense of partially blocking channels.16 As the carbonization temperature increases from 423 K to 573 K, the surface area of SC-n samples increases from 18 to 490 m2 g−1 while the carbon content decreases from 25% to 6% (Table 1) and the pore size is also slightly increased. However, the actual adsorption capacities of heparin decrease from 77.8 to 39.3 mg g−1. In particular, these samples carbonized below 573 K can adsorb more heparin than the parent SM (44.5 mg g−1, Table 3) though they have surface areas smaller than 150 m2 g−1 and lack micropores. Carbonization at 573 K makes the surface area of SC-4 reach 339 m2 g−1 and micropores appear (Table 1), but this sample adsorbs less heparin (42.1 mg g−1) than sample SM. Prolonging the carbonization time from 2 h to 5 h at 573 K further increases the surface area of the SC-5 sample and the ratio of micropores in the structure (Table 3), but causes a lower adsorption capacity of heparin, which implies the minor effect of micropores on heparin adsorption.
Addition of APTES in the synthesis successfully introduced amine groups into the framework of SBA-15, and the final APTES contents in SA-n are shown in Table 2. The BET surface areas of SA-n composites gently decrease as more APTES additives are used, meanwhile the pore size is decreased and the amount of micropores declines distinctly because of the grafting of functional groups on the internal surface of SBA-15. As expected, SA-n samples adsorb far more heparin than parent SBA-15, among them SA-2 traps 114.8 mg g−1 which is almost 3 times more than SBA-15 (30.4 mg g−1), thanks to the powerful promotion effect of amino groups on the adsorption of heparin.10 However, as the initial ratio of APTES/TEOS in the synthesis is up to 0.2, the surface area of the resulting sample SA-4 is 23% of that of SBA-15, and the pore size is reduced to about a half of that of SBA-15, hence the amount of immobilized heparin declined to 73 mg g−1 though this value is still larger than that of SBA-15. SA-P and SMA samples are prepared with the same initial ratio of APTES/TEOS as that for SA-2, but their final APTES contents are lower than SA-2 (Table 2) hence they adsorb much less heparin (52.1, 68.7 mg g−1) than SA-2 under the same conditions. On the other hand, a similar amount of heparin is adsorbed on SBA-15 (30.4 mg g−1) and SA-s (33.8 mg g−1), and this result indicates the minor influence of solvent chloroform on the heparin adsorption by SBA-15.
Release of heparin from mesoporous composites
The release procedure of heparin from mesoporous composites usually consists of three stages:10,36 eruptible, reposeful and equilibrium stages and the last two stages can persist for rather long times to ensure a sustained heparin release. An initial release burst occurs in the first few days (1–2 days), owing to desorption of the heparin located on the external surface or near the channel mouth in the host. After that, the release becomes relatively slower because heparin needs to diffuse from channels, which plays a significant role in the surgical treatment of wound healing to keep a constant concentration of heparin.9 The release in the final stage, after 10 days or longer, is rather slow and persists for a long period since the heparin deep within the channels needs time to diffuse out, which is inevitable affected by the interaction between heparin and the pore walls of the adsorbent.Fig. 4a shows the release profiles of heparin on the SBA-15 with different morphologies, in which all samples exhibit a significant “burst effect”. Although SM, SE, S3D and S3D-T composites trap more heparin than SBA-15, only SM releases more heparin within 30 days (Table 3). Moreover, their releases reach equilibrium on the 10th day, more rapidly than that of SBA-15 (20th day). It seems that either shortened or interconnected channels fail to improve the sustained release of heparin from SBA-15; in contrast, the diffusion of heparin is accelerated. Moreover, SM sample has a lower heparin release ratio (29.2%) than SBA-15 (38.2%), indicating the disadvantage of net-linked morphology for heparin diffusion and release.
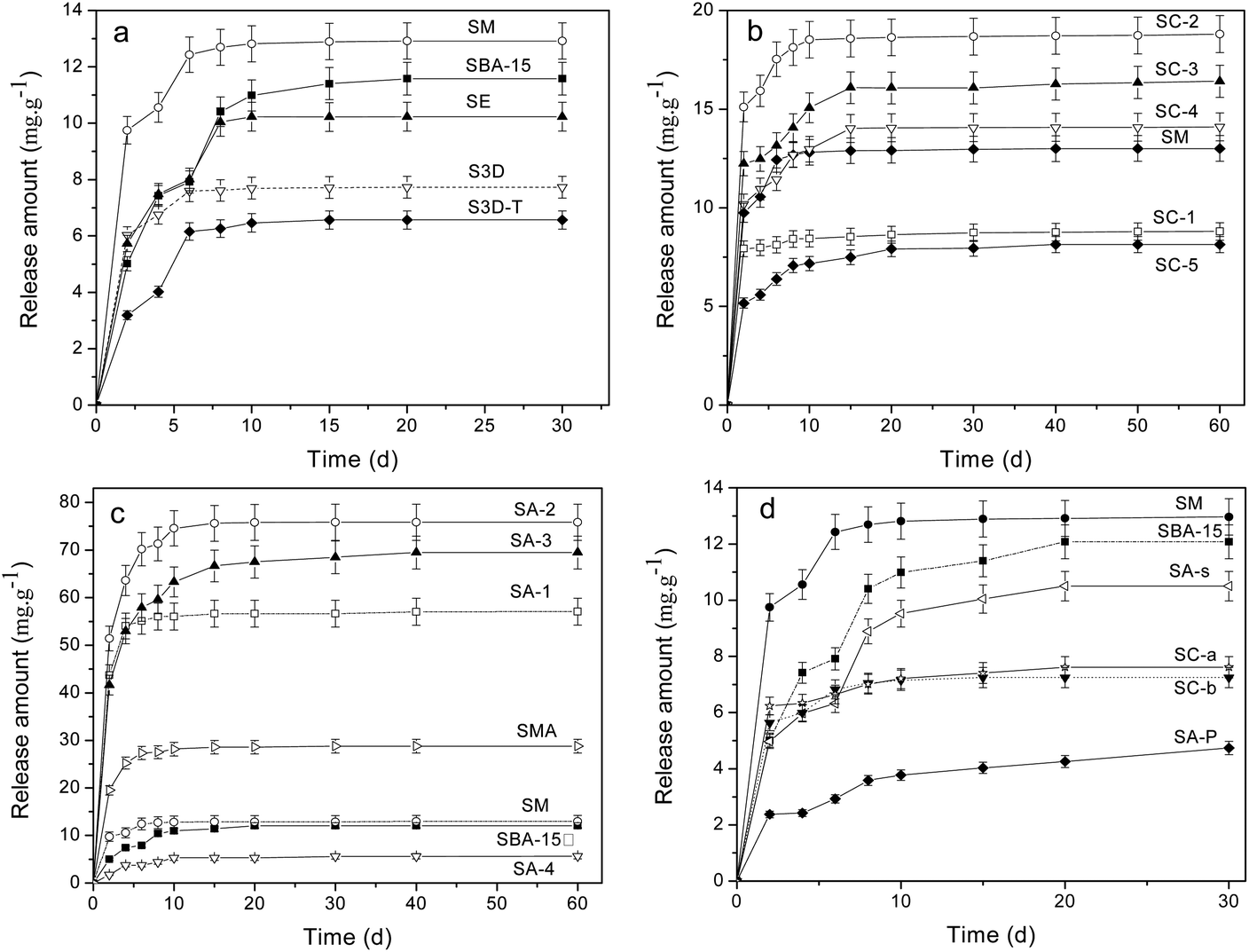 | ||
| Fig. 4 Release profiles of heparin on (a) SBA-15 with different morphologies, (b) SC-n samples, (c) amine-loaded SBA-15 prepared by one-pot synthesis and (d) SM samples treated with sulfuric acid (SC-a, SC-b) and the SBA-15 with post-amine modification (SA-P). | ||
The release profiles of heparin on SC-n composites are illustrated in Fig. 4b. The carbonized template in SC-n composites significantly promotes the adsorption of heparin because of physical interception (Table 3). Since the release of heparin is driven by the concentration difference between the vessel and the medium, SC-n samples release more heparin than SM except SC-1. Also, their release procedure becomes continuously longer to 15 days than that of common SM (10 days). Among these samples, SC-2 is the best to trap (64.1 mg g−1) and liberate (18.8 mg g−1) heparin, 110% and 62% more than the parent SM (30.4 and 11.6 mg g−1). However, the release ratios of heparin adsorbed in SC-n (n = 2, 3, 4) are slightly more than that in SM (29%), but still less than SBA-15 (38%).
Fig. 4c and d illustrate the release profiles of heparin on amine-loaded SBA-15. The release amount follows the order: SA-4 < SBA-15 < SM < SMA < SA-1 < SA-3 < SA-2, indicating the complex effect of organic modification on the release of heparin. SA-2 composites is the best one to release 75.8 mg g−1 heparin, 5 times more than that by SBA-15 (11.6 mg g−1), and 66% of the heparin adsorbed is released. And this ratio is much higher than that of SBA-15 (38%). On the other hand, the second release procedure of SC-2 can persist for 20 days though it is inferior to that of SA-3 (40 days), offering a high concentration of heparin during release progress. A higher release ratio of heparin is found in SA-1 (71.2%) and SC-3 (80.8%), demonstrating the elevated use-ratio of heparin. As expected, SA-4 composite releases the least among SA-n because it traps the least, but its release ratio is incredibly low (7.7%). A similar result is observed on SA-P sample from which only 4.7 mg g−1 heparin is released and the release ratio is 9.1% (Table 3). More heparin (28.8 mg g−1) is released from SMA composite and the equilibrium state is reached in 10 days, mirroring the limited promotion of amino groups on the release of heparin in the mesoporous silica with net-like morphology. SA-s sample exhibits a release performance similar to parent SBA-15, excluding the possible influence of solvent such as CHCl3 on the heparin release from mesoporous silica. Both SC-a and SC-b samples show a poor performance of release and about 7 mg g−1 of heparin is detected within 30 days (Fig. 4d). This indicates the disadvantage of high temperature carbonization of as-synthesized SBA-15 on the release of heparin.
The release profiles of heparin from SC-2, SA-2 and SA-3 along with SM and SBA-15 are fitted to theoretical models such as the Higuchi model37 and the Peppas model38 in order to study the models of heparin release on SBA-15 samples. Both models are short time approximations and limited to be applied to the first 60% of the release.10,11 Consequently, the release profiles in 10 days are fitted with these two models, and the fitted results are shown in Table 4. Fig. 5 illustrates the fitted curves of these samples. Most of the R2 values are above 0.930 indicating good fitting and relative correlation. The “a” values of SA-2 and SA-3 samples approach 0.5 (Table 4) so that their release mechanisms are similar to the Higuchi model, which indicates the uniform pores of the samples. The Peppas model is more suitable for other samples to fit their release profiles, and this phenomenon implies the existence of a small amount of heterogeneous pores on these samples.39
| a | b | R 2 | |
|---|---|---|---|
| SBA-15 | 0.311 | 0.486 | 0.969 |
| SM | 0.652 | 0.189 | 0.930 |
| SC-2 | 0.723 | 0.135 | 0.960 |
| SA-2 | 0.555 | 0.154 | 0.940 |
| SA-3 | 0.593 | 0.229 | 0.962 |
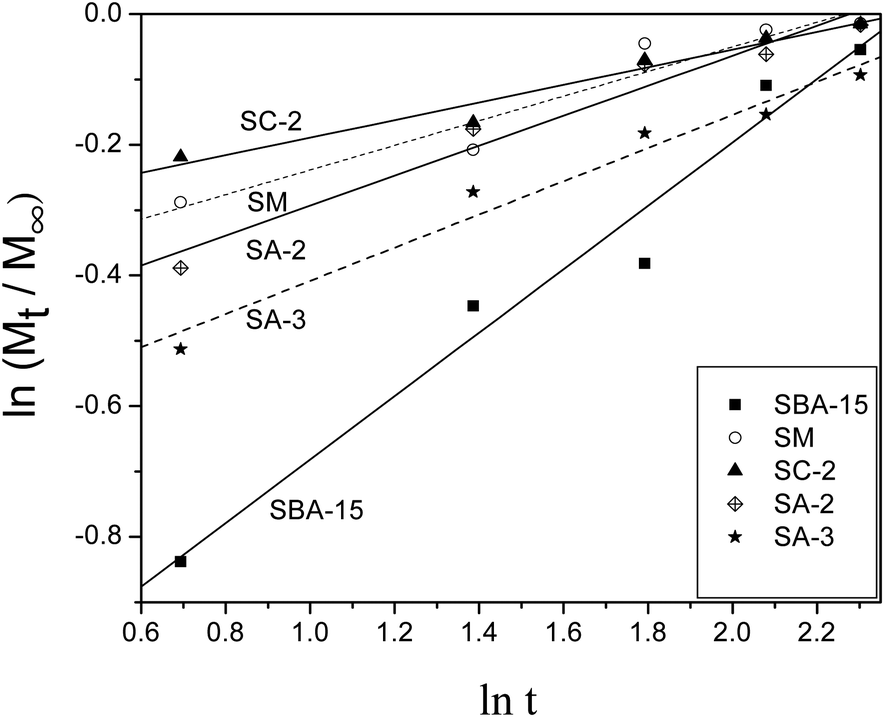 | ||
| Fig. 5 Release data fits to the Peppas model for samples. | ||
Discussion
Release of heparin from a vessel into medium solution is driven by the concentration difference between them so it is necessary to adsorb sufficient heparin prior to release. Heparin has special long chain structure feature of 1–1.5 nm in width and several dozens of nanometres in length, hence it should be a slow procedure for the chain-like heparin to diffuse into the channels of mesoporous silica, which inevitably hinders the subsequent adsorption more or less. Thus, favourable morphology of SBA-15 is sought to shorten the diffusion path: the net-like morphology of SM sample increases the contact probability of heparin with the vessel in solution; the short channels of SE sample cut the diffusion path and provide more channel mouths in the same mass. S3D sample has many narrow (2–3 nm) connections and tunnels between microchannels for the convenience of heparin diffusion, which enables 19–126% more heparin to be adsorbed in SBA-15 (Table 3), and its better performance (126%) than that of S3D-T (19%) demonstrates the importance of narrow mesopores. Those short interconnected channels with 2–3 nm size seem to be beneficial for the accommodation of heparin, because of the close geometric match content.10 However, these morphologies have not promoted the heparin release of SBA-15; rather, S3D and S3D-T even release less heparin, probably the long-chain molecule is anchored in the interconnections of channels so the host–guest interaction needs to be adjusted.Coating carbon into mesoporous silica to change the surface curvature of channels is a physical modification to elevate the adsorption amount of the target and to slow the desorption rate.16 The carbonaceous species formed by in situ carbonization of P123 in SBA-15 really improve the heparin adsorption through enlarging its collision probability with channel walls as illustrated in Scheme 1, and their performances depend on their softness and flexibility that are affected by carbonization temperature.16 P123 itself does not decompose at 455 K and shows a notable weight loss of ∼80% at ∼483 K and ∼20% in the range of 483–643 K.40 For the P123 in SBA-15, it will be decomposed in the range of 373–923 K31 or more accurately, in 423–873 K41 and finally forms carbon at 1073 K.18 Some reactions will have occurred during the carbonization, including the decomposition and desorption of the polymeric template and, to a smaller extent, the release of water formed from the condensation of silanols in the silica framework.31 Accordingly, the SC-1 sample carbonized at 423 K hardly has carbon precursors but residual micelles inside channels and these residues are soft.19 They are located inside the channels of SBA-15 like weeds in an alley to retard the movement of heparin. The heparin molecules can break through these barriers to enter the channels during adsorption process because of the large concentration difference between the original solution and the vessel. However, the heparin concentration difference between the vessel and the blank solution in the release procedure is smaller than that in adsorption, so that there is not enough driving force to promote the heparin adsorbed to break through these barriers again. Thus, SC-1 adsorbs 156% more heparin than SBA-15 but releases 25% less (Table 3). As the carbonization temperature is increased to 573 K, the formed sample adsorbs less heparin than SC-1 but the released ratio rises from 11% to 38%, since the P123 micelles are dramatically carbonized near 600 K,31 and the colour of the sample changes from white to yellow, at the same time, micropores appear in SC-4 sample (Table 3). Prolonging the heating time at 573 K to 5 h makes the carbon content of SC-5 decline further due to carbonization of micelles while more micropores are formed. Based on the worse performance of SC-5 along with those of SC-a and SC-b in adsorption and release of heparin, it is clear that the residual soft species in channels have a larger influence than micropores on the heparin.
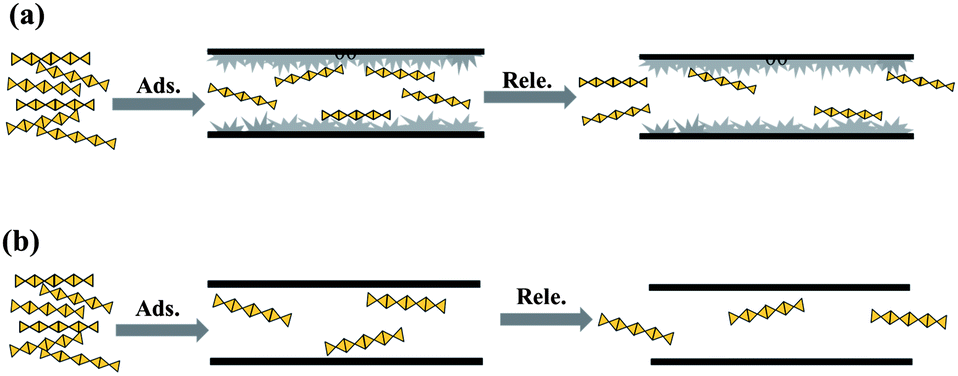 | ||
| Scheme 1 Possible adsorption–release procedure of heparin on (a) SC-n and (b) SM samples. | ||
The existence of flexible micelle-like species in SC-2 and SC-3 markedly improves the adsorption and release of heparin, revealing the importance of suitable physical barriers within mesoporous channel: these flexible residual micelles imperceptibly impede the movement of heparin adsorbed, retarding its desorption to reach the sustained release so that SC-2 and SC-3 exhibit a longer release time of heparin (15–50 days) than that of the parent SM sample (10 days). However, elaborate chemical interaction is required to synergize the physical modification in order to further enhance the adsorption and release of heparin in SBA-15.11
Heparin consists of repeating disaccharide units, in which the main functional groups are negatively charged, such as COO−, NH–SO3−, O–SO3−, CO–NH–CH3, and –OH;42,43 thus, modifying the mesoporous vessel with electro-positive organic groups should be very efficient to improve its adsorption.10 What's more, heparin can be trapped by silanol groups on the channel walls that will provide the confined space and van der Waals force.9,44 When the negatively charged groups of heparin are close to the silicate wall of adsorbent, the total energy of the system will decrease due to intermolecular interactions such as electrostatic and hydrogen bonding interactions, reaching tens of kJ mol−1.45 The incorporated amine groups in SBA-15 will increase the positive charge on channel walls, but slightly reduce the channel size. Since the van der Waals force is a function of distance, the pore configuration of the adsorbent like the size and the curvature, including space steric hindrance, length and connection of channels, will strongly affect the adsorption of heparin. Scheme 2 is the assumed adsorption process of heparin by the SA-n samples where the protein is attached by the positive charge of amino groups in channels but its diffusion is retarded by the rough channels. Under the synergy of electrostatic interaction and geometric confinement, more heparin molecules are trapped inside the mesoporous vessels and then undergo sustained release, up to 114.8 and 75.8 mg g−1, respectively (Table 3). Here the two important factors, suitable electrostatic interaction and channel structure, are related to the incorporation of APTES in SBA-15, since the former is depended on the amount and distribution of amino groups while the latter is affected by the synthesis of SA-n composite. There is an optimal amount of APTES additive since too much APTES would interrupt the formation of the mesoporous structure. For instance, SA-2 sample containing 5 wt% of APTES adsorbs and releases more heparin than the SA-4 consisting of 16.4 wt% APTES. On the other hand, its one-pot synthesis enables APTES to participate in the formation of the mesoporous assembly, being incorporated inside the framework forming the Si–C structure31 or on the channel walls offering electro-static attractions toward heparin. Post-modification of APTES makes most of the modifiers left in the dissociated state and coated on the channel wall, blocking the pores to obstruct subsequent adsorption. Furthermore, their stronger attraction force makes the heparin molecules adsorbed very difficult to release (Fig. 4d). Conversely, one-pot synthesis also endows the monolith sample SMA to have an improved ordered structure in comparison with the parent SM (Fig. 2 and S3a†), and thus SMA exhibits a better capability in the adsorption and release of heparin (Table 3).
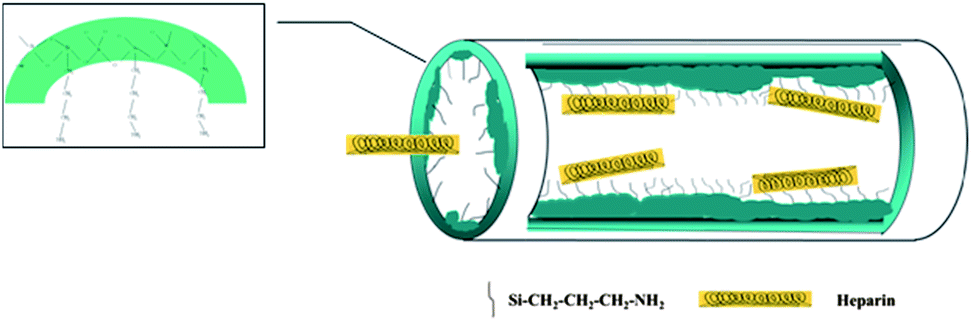 | ||
| Scheme 2 The possible adsorption process of heparin in SA-n samples. | ||
The optimal micro-environment should be established inside the mesoporous vessel where heparin is attached by relatively weak electrostatic interaction while the rugged surface of the channels makes the movement of heparin difficult, which should function like a special swing door that is easily opened by a stream of people but laborious for a single person. Toward this goal, we prudently utilize APTES additive to tailor the channel state of SA-n vessels. The amine groups of APTES are easily protonated under acidic conditions and cross-link with silanol groups of the silicate species, forming zwitterions (–NH3+⋯OSi) and resulting in local disruptions of the silicate walls.31 Accordingly, we add APTES after 2 h of adding TEOS in the synthesis solution to limit the disturbance of APTES, ensuring the formation of mesoporous SBA-15 silica under acidic conditions through a mechanism of (S0H+)(X−I+); at the same time, the protonated terminal amine groups can interact with some silanol groups to prevent direct interactions of surfactant P123 with these silicates, leading to a less ordered pore structure.31 Consequently, rough surfaces are formed inside the channels of SA-n composites and the pore walls of SA-2 become distorted (Fig. S2†). Unlike the pore-expanded and APTES incorporated MCM-41 composites whose pore size is below 4 nm with worm-like pore channels,10 SA-n (n = 1–3) samples keep the structural characteristics of SBA-15 (Fig. S3a and S4a†) and their pore size exceeds 5 nm (Table 3), which facilitates the entrance and diffusion of heparin so that the excellent performance of the SA-2 composite is observed in the adsorption and release of heparin.
Conclusion
The adsorption and release of heparin in mesoporous material SBA-15 are influenced by the morphology, pore size, channel surface state (surface curvature, organic group modification) of the vessel. A special morphology such as a 3D reticular structure can promote the diffusion of heparin in the channels and increase the adsorption amount obviously.The carbonized micelles inside SBA-15 change the surface curvature, and the sample with the proper carbon content (22%) promotes both immobilized and liberated amounts, extending the time of high-concentration release from 10 days to 15 days as well.
The SBA-15 sample modified with amino groups by one-pot synthesis has an excellent performance of sustained release in large amounts. The appropriate amount of APTES is the key for the performance, and the samples with APTES contents of 3.3–6.2 wt% have an outstanding trapping and high release ratio, as well as a sustained release time over 20 days. Among them SA-2 adsorbs almost 3 times (114.8 mg g−1) more heparin than SBA-15, and releases 5 times (75.8 mg g−1) more. A rough surface of silica walls and organic modification can boost the adsorption–release procedure of heparin, offering a candidate to fabricate highly efficient drug carriers.
Acknowledgements
NSF of China (21173117 and 21273106), Scientific Research Foundation of Graduate School as well as Analysis Center of Nanjing University financially supported this research.References
- S. Khan, J. Gor, B. Mulloy and S. J. Perkins, J. Mol. Biol., 2010, 395, 504–521 CrossRef CAS PubMed.
- M. Prabaharan and S. Gong, Carbohydr. Polym., 2008, 73, 117–125 CrossRef CAS PubMed.
- Z. L. Yang, Y. F. Lu and Z. Z. Yang, Chem. Commun., 2009, 2270–2277 RSC.
- S. Wang, Microporous Mesoporous Mater., 2009, 117, 1–9 CrossRef CAS PubMed.
- V. Meynen, P. Cool and E. F. Vansant, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS PubMed.
- M. Vallet-Regí, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef.
- P. Horcajada, A. Ramila, J. Perez-Pariente and M. Vallet-Regí, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS PubMed.
- B. Muńoz, A. Rámila, J. Pérez-Pariente, I. Díaz and M. Vallet-Regí, Chem. Mater., 2003, 15, 500–503 CrossRef.
- Y. Zhou, K. Li, J. Y. Yang, C. X. Guan, Y. Wang, C. J. Liu and J. H. Zhu, Small, 2012, 8, 1373–1383 CrossRef CAS PubMed.
- M. M. Wan, J. Y. Yang, Y. Qiu, Y. Zhou, Q. Hou, W. G. Lin and J. H. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 4113–4122 CAS.
- M. M. Wan, W. J. Qian, W. G. Lin, Y. Zhou and J. H. Zhu, J. Mater. Chem. B, 2013, 1, 3897–3905 RSC.
- V. B. Fenelonov, A. Y. Derevyankin, S. D. Kirik, L. A. Solovyov, A. N. Shmakov, J. L. Bonardet, A. Gedeon and V. N. Romannikov, Microporous Mesoporous Mater., 2001, 33, 44–45 Search PubMed.
- H. I. Lee, J. H. Kim, G. D. Stucky, Y. F. Shi, C. Pak and J. M. Kim, J. Mater. Chem., 2010, 20, 8483–8487 RSC.
- J. Fan, C. Z. Yu, L. M. Wang, B. Tu, D. Y. Zhao, Y. Sakamoto and O. Terasaki, J. Am. Chem. Soc., 2001, 123, 12113–12114 CrossRef CAS.
- Y. Zhou, W. J. Qian, J. Yang, S. L. Zhou, Y. J. Wang and J. H. Zhu, Chin. J. Chem., 2012, 30, 2073–2078 CrossRef CAS.
- J. Y. Yang, J. Yang, Y. Zhou, W. G. Lin, H. J. Wang and J. H. Zhu, J. Hazard. Mater., 2010, 176, 602–608 CrossRef CAS PubMed.
- Z. M. Wang, K. Hohsinoo, K. Shishibori, H. Kanoh and K. Ooi, Chem. Mater., 2003, 15, 2926–2935 CrossRef CAS.
- J. Yang, F. N. Gu, H. J. Wang, Y. Zhou, J. Y. Yang, Z. Y. Wu and J. H. Zhu, Catal. Today, 2009, 148, 88–96 CrossRef CAS PubMed.
- W. G. Lin, F. Wei, Q. Hou, T. Y. Zhang and J. H. Zhu, Microporous Mesoporous Mater., 2012, 156, 233–243 CrossRef CAS PubMed.
- C. M. Yang, Y. Q. Wang, B. Zibrowius and F. Schuth, Phys. Chem. Chem. Phys., 2004, 6, 2461–2467 RSC.
- S. W. Song, K. Hidajat and S. Kawi, Langmuir, 2005, 21, 9568–9575 CrossRef CAS PubMed.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- Y. L. Wei, Y. M. Wang, J. H. Zhu and Z. Y. Wu, Adv. Mater., 2003, 15, 1943–1945 CrossRef CAS.
- Y. Zhou, W. G. Lin, J. Yang, L. Gao, N. Lin, J. Y. Yang, Q. Hou, Y. Wang and J. H. Zhu, J. Colloid Interface Sci., 2011, 364, 594–604 CrossRef CAS PubMed.
- A. S. Maria Chong and X. S. Zhao, J. Phys. Chem. B, 2003, 107, 12650–12657 CrossRef.
- L. Gao, Y. Wang, J. Q. Wang, L. Huang, L. Y. Shi, X. X. Fan, Z. G. Zou, T. Yu, M. Zhu and Z. S. Li, Inorg. Chem., 2006, 45, 6844–6850 CrossRef CAS PubMed.
- F. Wei, J. Y. Yang, L. Gao, F. N. Gu and J. H. Zhu, J. Hazard. Mater., 2009, 172, 1482–1490 CrossRef CAS PubMed.
- M. Jaroniec and L. A. Solovyov, Langmuir, 2006, 22, 6757–6760 CrossRef CAS PubMed.
- M. S. Ahola, E. S. Sailynoja and M. H. Raitavuo, Biomaterials, 2001, 22, 2163–2170 CrossRef CAS.
- C. Y. Chen, S. Q. Xiao and M. E. Davis, Microporous Mater., 1995, 4, 1–20 CrossRef CAS.
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961–1968 CrossRef CAS.
- I. Izquierdo-Barba, E. Sousa, J. C. Doadrio, A. L. Doadrio, J. P. Pariente, A. Martínez, F. Babonneau and M. Vallet-Regí, J. Sol-Gel Sci. Technol., 2009, 50, 421–429 CrossRef CAS PubMed.
- O. P. Tkachenko, K. V. Klemetiev, E. Loffler, I. Ritzkopf, F. Schuth, M. Bandyopadhyay, S. Grabowski, H. Gies, V. Hagen, M. Muhler, L. Lu, R. A. Fischer and W. Grunert, Phys. Chem. Chem. Phys., 2003, 5, 4325–4334 RSC.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, 1982, pp. 111–218 Search PubMed.
- F. N. Gu, Y. Zhou, F. Wei, Y. Wang and J. H. Zhu, Microporous Mesoporous Mater., 2009, 126, 143–151 CrossRef CAS PubMed.
- L. X. Wen, Z. Z. Li, H. K. Zou, A. Q. Liu and J. F. Chen, Pest Manage. Sci., 2005, 61, 583–590 CrossRef CAS PubMed.
- N. Roveri, M. Morpurgo, B. Palazzo, B. Parma and L. Vivi, Anal. Bioanal. Chem., 2004, 381, 601–606 CrossRef PubMed.
- A. Karewicz, K. Zasada and K. Szxzubialka, Int. J. Pharm. Chem., 2010, 385, 163–169 CrossRef CAS PubMed.
- H. Ritter and D. J. Brühwiler, J. Phys. Chem. C, 2009, 113, 10667–10674 CAS.
- C. Xue, J. Wang, B. Tu and D. Zhao, Chem. Mater., 2010, 22, 494–503 CrossRef CAS.
- Y. M. Wang, Z. Y. Wu, L. Y. Shi and J. H. Zhu, Adv. Mater., 2005, 17, 323–327 CrossRef CAS.
- K. Park, G. Y. Lee, Y. S. Kim, M. Yu, R. W. Park, I. S. Kim, S. Y. Kim and Y. Byun, J. Controlled Release, 2006, 114, 300–306 CrossRef CAS PubMed.
- V. Hoffart, A. Lamprecht, P. Maincent, T. Lecompte, C. Vigneron and N. Ubrich, J. Controlled Release, 2006, 113, 38–42 CrossRef CAS PubMed.
- I. G. Shenderovich, G. Buntkowsky, A. Schreiber, E. Gedat, S. Sharif, J. Albrecht, N. S. Golubev, G. H. Findenegg and H. H. Limbach, J. Phys. Chem. B, 2003, 107, 11924–11939 CrossRef CAS.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–167 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tb21092h |
| This journal is © The Royal Society of Chemistry 2014 |