Heat-triggered drug release systems based on mesoporous silica nanoparticles filled with a maghemite core and phase-change molecules as gatekeepers†
Ji
Liu
ac,
Christophe
Detrembleur
a,
Marie-Claire
De Pauw-Gillet
b,
Stéphane
Mornet
c,
Luce Vander
Elst
d,
Sophie
Laurent
d,
Christine
Jérôme
*a and
Etienne
Duguet
*c
aCenter for Education and Research on Macromolecules (CERM), University of Liège, B6 Sart Tilman, B-4000 Liège, Belgium. E-mail: c.jerome@ulg.ac.be; Fax: +32-4-36663497; Tel: +32-4-3663565
bLaboratory of Mammalian Cell Culture (GIGA-R), University of Liège, B6 Sart Tilman, B-4000 Liege, Belgium
cCNRS, Univ. Bordeaux, ICMCB, UPR 9048, F-33600 Pessac, France. E-mail: duguet@icmcb.u-bordeaux1.fr; Fax: +33 540 002 761; Tel: +33 540 002 651
dDepartment of General, Organic & Biomedical Chemistry, NMR & Molecular Imaging Laboratory, University of Mons, Avenue Maistriau, 19, B-7000 Mons, Belgium
First published on 4th November 2013
Abstract
Core–shell nanoparticles made of a maghemite core and a mesoporous silica shell were developed as drug delivery systems (DDS). Doxorubicin® (DOX, DNA intercalating drug) was loaded within the mesoporous cavities, while phase-change molecules (PCMs), e.g. 1-tetradecanol (TD) with a melting temperature (Tm) of 39 °C, were introduced as gatekeepers to regulate the release behaviours. An overall loading amount of ca. 20 wt% (TD/DOX ca. 50/50 wt/wt) was confirmed. Heat-triggered release of DOX evidenced a “zero premature release” (<3% of the entire payload in 96 h release) under physiological conditions (37 °C), and however, a sustainable release (ca. 40% of the entire payload in 96 h) above Tm of TD (40 °C). It also demonstrated the possibility to deliver drug payloads in small portions (pulsatile release mode) via multiple heating on/off cycles, due to the reversible phase change of the PCMs. In vitro heat-triggered release of DOX within cell culture of the MEL-5 melanoma cell line was also tested. It was found that DOX molecules were trapped efficiently within the mesopores even after internalization within the cytoplasm of MEL-5 cells at 37 °C, with the potential toxicity of DOX strongly quenched (>95% viability after 72 h incubation). However, continuous cell apoptosis was detected at cell culture temperature above Tm of TD, due to the heat-triggered release of DOX (<50% viability after 72 h incubation at 40 °C). Moreover, due to the presence of a maghemite core within the DDS, T2-weighted magnetic resonance imaging performance was also confirmed. These as-designed core–shell nanoparticles are envisaged to become promising DDS for “on-demand” heat-triggered release.
1. Introduction
Mesoporous nanomaterials, e.g. mesoporous silica nanoparticles (MSNPs), have received considerable attention in the biomedical field in recent years, especially in drug delivery systems (DDS), due to their specific properties, such as high surface area, large pore volume, interconnected pore structure, facile and discriminatory external and internal surface derivatization, etc.1–4 Indeed, it is widely proposed that MSNPs could be used as DDS for guest molecules, such as drugs or bio-molecules.5–10 However, inevitable premature release occurs during the delivery routine, due to the open porosity structure and weak physical interactions between the guest molecules and internal surface of MSNPs.To overcome these drawbacks, a great variety of chemical ingredients has been attempted as gatekeepers to regulate the storage and release behaviours of cargo payloads.11,12 The loaded cargoes were reported to be well-trapped within the cavities. Upon the introduction of different triggers, the cavities could be opened and triggered release was accomplished. This promising type of DDS manifested themselves with a “zero premature release” character and would be particularly useful when the drugs to be delivered are extremely toxic or possessing some serious side-effects. Moreover, achieving an “on-demand” control over the release behaviours should definitely bring a breakthrough to the drug release application.1,13 As far as we know, nanoparticles,14–16 small organic molecules17–21 and supramolecular assemblies22–26 have already been attempted as gating functionality in the MSNP-based DDS, while triggered release was activated by reductive agents, enzymes, light, pH, glucose or temperature, etc. However, even if these concepts were validated, those preliminary gating systems might still suffer from some drawbacks, such as complicate design needed to introduce those gating components, unknown biocompatibility, difficulty in “on-demand” triggered release, irreversibility of the gating mechanism, incapability in manipulation for a pulsatile release, etc. Nevertheless, another promising molecule, 1-tetradecanol (TD), was recently reported in another DDS based on gold nanocages,20,21 and most of those drawbacks could be well addressed. As temperature-induced phase-change materials (PCMs), fatty alcohols and fatty acids are indeed promising candidates as gatekeepers due to their hydrophobic essence and good biocompatibility. Taking TD as an example, it is in a solid state at human physiological temperature, whereas melts into a fluid and therefore permeable state above its melting temperature (Tm = 39 °C). Thus, after the introduction of TD as a gatekeeper, the cavities are expected to remain closed during the circulation; however, they might be opened after exposure to a heating source, thus triggered release of the cargoes would be accomplished.
In this study, we combined for the very first time the complementary advantages of MSNPs as drug reservoirs and PCMs as gatekeepers to design a new DDS (Scheme 1). We also introduced a maghemite core within each MSNP for extra functionalities, e.g. MRI-tracking of biodistribution and heat-triggered release induced by alternating magnetic fields,3 even if the full evaluation of these potential benefits is not the main aim of this paper but will be addressed in a forthcoming study. Core–shell γ-Fe2O3@MSNPs were prepared from 7.5 nm γ-Fe2O3 seeds via the alkaline hydrolysis of tetraethyl orthosilicate (TEOS), while cetyltrimethylammonium bromide (CTAB) acting as both stabilizers for the seeds and soft templates for the mesopores. γ-Fe2O3@MSNPs were then PEGylated in order to improve their colloidal stability and stealthiness with regard to the complement system activation and mononuclear phagocyte system (MPS). Doxorubicin® (DOX), known as a DNA intercalating anticancer agent, was loaded within the mesoporous cavities, while TD was selected as a gatekeeper for its ideal melting temperature. Thus, when using such a DDS accompanying the cell culture, it was expected to open the cavities by a slight increase of the temperature of the cell culture (<42 °C) in order to trigger the drug release, but avoid severe heat-induced cell apoptosis. The human melanoma MEL-5 cell line was chosen as a model cancer cell line, and the cellular uptake and the efficiency of the heat-triggered DOX release within the MEL-5 cell culture were studied.
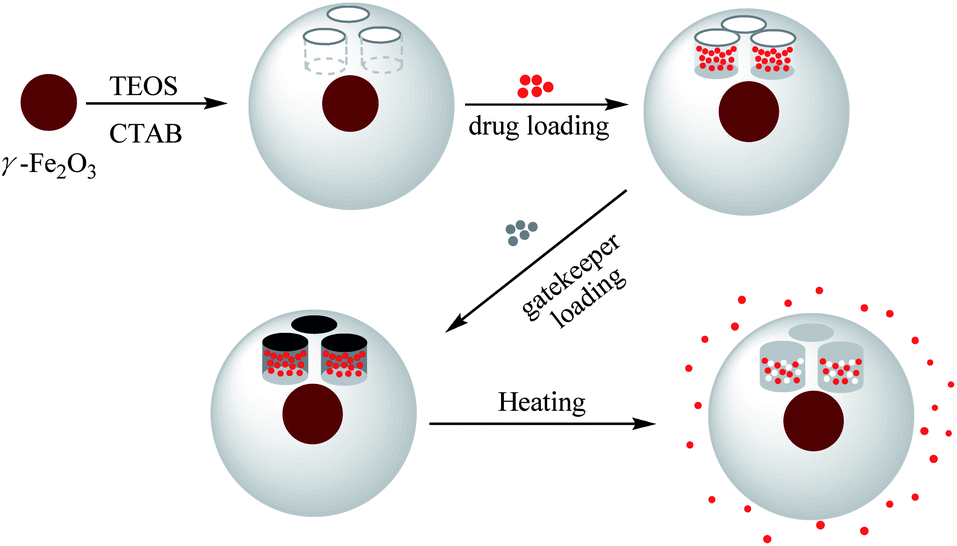 | ||
| Scheme 1 Preparation of γ-Fe2O3@MSNPs, drug loading of Doxorubicin®, immobilization with 1-tetradecanol as a gatekeeper, and triggered drug release by conventional external heating. | ||
2. Experimental
2.1. Materials
(3-Aminopropyl)triethoxysilane (APTES, 97%), tetraethyl orthosilicate (Si(OC2H5)4, 99%), sodium hydroxide (NaOH, 98.5%), cetyltrimethylammonium bromide (CTAB, 97%), succinic acid (SA, 99%), poly(ethylene glycol) methyl ether (mPEG1000-OH, Mn 1000 g mol−1), 4-(dimethylamino)pyridine (DMAP, 99%), ammonium hydroxide solution (NH3·H2O, 30 wt%), iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), iron(II) chloride tetrahydrate (FeCl2·4H2O, 98%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 98%), hydrochloric acid (HCl, 37 wt%), hydrofluoric acid (HF, 48 wt%), 1-tetradecanol (TD, 98%), fluorescein isothiocyanate isomer I (FITC, 98%), 4,6-diamidino-2-phenylindole (DAPI, 98%) and Doxorubicin® (DOX, 97%) were purchased from Aldrich and used without further purification. DMEM (low glucose, without sodium pyruvate), L-glutamine, PBS buffer solution (without Ca2+ and Mg2+), foetal bovine serum (FBS) and trypsin were obtained from Biowhittaker (Walkersville, MD). PBS buffer solution (with Ca2+ and Mg2+), penicillin G and streptomycin were purchased from GIBCO BRL (Gaithersburg, MD). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was purchased from Promega (Madison, USA).2.2. Preparation of γ-Fe2O3@MSNPs
γ-Fe2O3 NPs were prepared via the alkaline co-precipitation of iron(II) and iron(III) precursors with ammonium hydroxide solution according to Massart's method.27 A final 7.5 nm γ-Fe2O3 NP aqueous suspension was obtained with a concentration of 61.5 g mL−1, which was confirmed via titration.Preparation of γ-Fe2O3@MSNPs was carried out according to a protocol previously reported28 with minor modifications. Typically, 0.24 mL of the γ-Fe2O3 NP aqueous suspension was dispersed in 200 mL of CTAB solution (0.01 M) at 50 °C, and the mixture was stirred vigorously for 30 min. After the pH was tuned to 10 with KOH solution (0.1 M), 2 mL of TEOS and 5 mL of ethyl acetate were added sequentially. After 10 min, 0.2 mL of APTES was injected and the mixture was stirred overnight. The as-synthesized γ-Fe2O3@MSNPs were centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min) to remove the un-reacted species and dispersed in 50 mL of anhydrous THF under sonication (10 min). Then, 0.1 mL of APTES was added and the mixture was stirred under reflux for 3 h, and amino-functionalized γ-Fe2O3@MSNPs were obtained after centrifugation (6000 rpm, 15 min). To extract the CTAB and obtain the mesoporous nanostructures, the as-prepared γ-Fe2O3@MSNPs were dispersed in 50 mL of ethanol–water (50/50 vol/vol) mixture. After addition of 10 μL of HCl solution (37%), the mixture was stirred for 12 h under reflux, and then purified via three centrifugation–rinsing cycles (8000 rpm, 10 min). This refluxing–purification cycle was repeated another two times, before the amino-functionalized γ-Fe2O3@MSNPs were dried via freeze-drying.
000 rpm, 10 min) to remove the un-reacted species and dispersed in 50 mL of anhydrous THF under sonication (10 min). Then, 0.1 mL of APTES was added and the mixture was stirred under reflux for 3 h, and amino-functionalized γ-Fe2O3@MSNPs were obtained after centrifugation (6000 rpm, 15 min). To extract the CTAB and obtain the mesoporous nanostructures, the as-prepared γ-Fe2O3@MSNPs were dispersed in 50 mL of ethanol–water (50/50 vol/vol) mixture. After addition of 10 μL of HCl solution (37%), the mixture was stirred for 12 h under reflux, and then purified via three centrifugation–rinsing cycles (8000 rpm, 10 min). This refluxing–purification cycle was repeated another two times, before the amino-functionalized γ-Fe2O3@MSNPs were dried via freeze-drying.
FITC-labelled γ-Fe2O3@MSNPs were prepared via the same protocol as described above, while during the growth of the silica shell via basic hydrolysis, APTES was replaced with the APTES/APTES-FITC (19/1 mol/mol) mixture, with the same overall feeding amount of APTES. APTES-FITC was prepared via the reaction between FITC (38.9 mg) and APTES (0.47 mL) (FITC:APTES = 1:20 mol/mol) in 5 mL of ethanol in the dark overnight, then stored at 4 °C for further use.
PEGylation of γ-Fe2O3@MSNPs: one gram of poly(ethylene glycol) methyl ether (mPEG1000-OH, 10 mmol –OH group) was reacted with succinic anhydride (12 mmol) in 20 mL of anhydrous THF at 60 °C overnight with DMAP (12 mmol) as a catalyst. The macromolecules were precipitated with cold diethyl ether and dried in a vacuum to obtain mPEG1000-COOH. Compared to mPEG1000-OH, the FTIR spectrum of mPEG1000-COOH showed an extra band at 1735 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), due to the presence of carboxyl groups (see ESI, Fig. S1a†). Moreover, the 1H-NMR spectrum exhibited a new peak at 2.5 ppm, which is attributed to methylene protons from the succinic acid segments (see ESI, Fig. S1b†). Amino-functionalized γ-Fe2O3@MSNPs (0.2 g) were first dispersed in 40 mL of anhydrous THF under sonication for 15 min, then mPEG1000-COOH (0.1 mmol –COOH group) and DMAP (0.1 mmol) in 10 mL of anhydrous THF were added. The mixture was stirred at 40 °C overnight. The non-reacted macromolecules were removed through three centrifugation–rinsing cycles (6000 rpm, 10 min), and then PEGylated γ-Fe2O3@MSNPs were lyophilized. Without specific notes, γ-Fe2O3@MSNPs in the following context refer to the γ-Fe2O3@MSNPs after PEGylation.
O), due to the presence of carboxyl groups (see ESI, Fig. S1a†). Moreover, the 1H-NMR spectrum exhibited a new peak at 2.5 ppm, which is attributed to methylene protons from the succinic acid segments (see ESI, Fig. S1b†). Amino-functionalized γ-Fe2O3@MSNPs (0.2 g) were first dispersed in 40 mL of anhydrous THF under sonication for 15 min, then mPEG1000-COOH (0.1 mmol –COOH group) and DMAP (0.1 mmol) in 10 mL of anhydrous THF were added. The mixture was stirred at 40 °C overnight. The non-reacted macromolecules were removed through three centrifugation–rinsing cycles (6000 rpm, 10 min), and then PEGylated γ-Fe2O3@MSNPs were lyophilized. Without specific notes, γ-Fe2O3@MSNPs in the following context refer to the γ-Fe2O3@MSNPs after PEGylation.
2.3. Drug loading and triggered release of γ-Fe2O3@MSNPs
Loading of DOX and TD into γ-Fe2O3@MSNPs: typically, 20 mg of γ-Fe2O3@MSNPs were dispersed in 5 mL of ethanol under sonication, and then DOX (5 mg in 1 mL of ethanol) was added. The mixture was stirred at 80 °C for 5 h, and at the end of which ethanol was supposed to be totally evaporated, then 10 mg of TD were added. After stirring for another 1 h, 5 mL of hot water (80 °C) was added, and phase-separation was observed. The upper oil moiety was the TD–DOX mixture, while the lower TD/DOX-loaded γ-Fe2O3@MSNPs and free DOX in water, due to the insolubility of TD in water and hydrophilic essence of γ-Fe2O3@MSNPs. Finally, the TD/DOX-loaded γ-Fe2O3@MSNPs were purified by centrifugation–rinsing (with cold water) cycles for at least six times to remove all free DOX molecules, and then dispersed in ice-cold PBS buffer under sonication (30 s). The amount of loaded DOX was determined spectrophotometrically by dispersing 5 mg of dry TD/DOX-loaded γ-Fe2O3@MSNPs into 1 mL of acetone under 10 min sonication in order to extract and transfer both DOX and TD into acetone. After centrifugation, the supernatant was collected, and the UV/vis absorbance at 495 nm was utilized to estimate the amount of loaded DOX, with the help of a pre-determined calibration curve. The drug loading capacity (DLC) and drug loading efficiency (DLE) were used to evaluate the loading capacity of the γ-Fe2O3@MSNPs: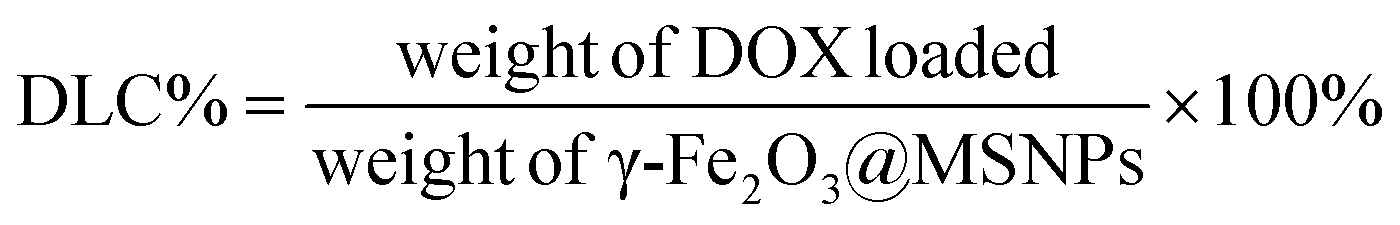 | (1) |
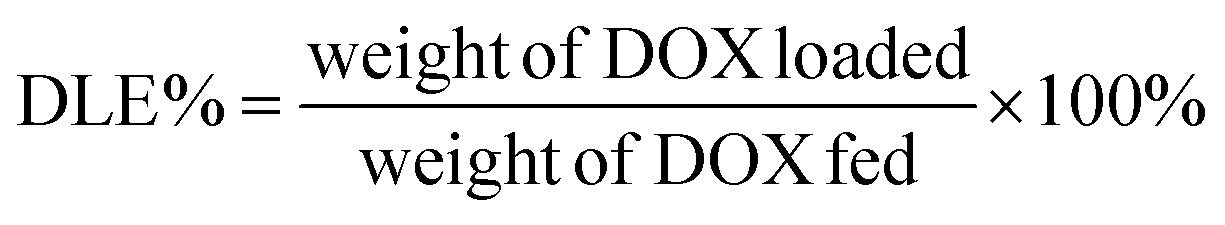 | (2) |
The heat-triggered release behaviours by external heating were studied via the conventional dialysis strategy, while an external water bath circulation was used to tune the temperature of the release system. Typically, 5 mL of TD/DOX-loaded γ-Fe2O3@MSNP suspension (5 g L−1) in PBS buffer solution was sealed in a dialysis bag (cut-off: 3500 g mol−1), and then dialyzed against 10 mL of PBS buffer solution (10 mM, pH 7.4).
The external heating was introduced either in a continuous mode or in an intermittent mode for which 37/40 °C water-circulation bathes were alternatively introduced every 12 h; while for the continuous mode, the water bath was kept at a constant temperature. At each pre-determined interval of time, 0.5 mL of the release medium was withdrawn for UV/vis absorbance measurement (495 nm) to quantify the amount of released DOX with a pre-determined calibration curve; at the same time 0.5 mL of fresh PBS buffer solution was added to keep a fixed volume.
2.4. Preparation of 50 nm MCM-41 MNPs
Fifty nm MCM-41 NPs were prepared according to a well-established protocol previously reported.29 Typically, 1 mL of KOH solution (1.5 M) was added to 100 mL of CTAB solution (6 mM) at 80 °C. After stirring for 20 min, 1.34 mL of TEOS (6 mM) was added and stirred for another 2 h at 80 °C. The as-synthesized MSNPs were centrifuged (10000 rpm, 15 min) to remove the un-reacted species. The following surface PEGylation was also done via the above-mentioned protocol for the γ-Fe2O3@MSNPs, and an average size of ca. 50 nm was confirmed by TEM. This MCM-41 MNP aqueous suspension was used as a reference for the T2-weighted MR image of the γ-Fe2O3@MSNP suspension with the same SiO2 concentration.
2.5. Cell culture and cytotoxicity assessment
The mouse fibroblast-like L929 cells (ATCC CCL-1) were grown at 37 °C under humidified air (5 vol% of CO2) in Dulbecco's modified Eagle medium (DMEM), which was supplemented with 5 vol% of FBS, 1 vol% of glutamax, 1 vol% of penicillin/streptomycin (10000 units of penicillin and 10000 units of streptomycin per mL) (DMEM complete medium). The human melanoma MEL-5 cells (originated from a non-pigmented clone 32, gift from Dr G. Degiovanni, University of Liège) were cultured in DMEM complete medium. After rinsing with PBS (Ca2+/Mg2+ free) buffer solution, the cells were detached with trypsin (0.2 vol%)/PBS (Ca2+/Mg2+ free) buffer solution.
Cytotoxicity assessment was carried out with the L929 and MEL-5 cell lines. The cells were first seeded in 96-well plates at a density of 5 × 103 cells per well and grown in DMEM complete medium for 24 h. Cells were then treated with γ-Fe2O3@MSNPs in DMEM complete medium with different concentrations (1, 10, 100 and 1000 μg mL−1) for 24 and 48 h, respectively. For each concentration, 5 parallel measurements were carried out at the same time. After each incubation period, cells were rinsed with PBS (with Ca2+/Mg2+) buffer solutions and cell viabilities were assessed via the MTS assay. 20 μL of MTS and 100 μL of PBS (with Ca2+/Mg2+) buffer solution were added to each well, and then the plates were incubated for another 30 min at 37 °C. The absorbance at 490 nm was measured by using a Power wave X (Biotek instrument Inc.) micro-plate reader. Percentage viabilities were determined relative to the untreated cells, which were taken as a control (100% viability).
2.6. Cellular uptake within melanoma MEL-5 cells
Quantitative studies on cellular uptake with a cytofluorometer: MEL-5 cells (3.8 × 105) were seeded in a 12-well plate with 2 mL of DMEM complete medium. After 24 h, the medium was replaced with 2 mL of fresh DMEM complete medium (blank) or FITC-labelled γ-Fe2O3@MSNPs in DMEM complete medium (20, 50, 100 or 200 μg mL−1), respectively. After incubating for a predetermined period (3, 6, 15 or 24 h), the culture medium was removed and the cells were rinsed twice with PBS buffer (Ca2+/Mg2+ free) to eliminate the free γ-Fe2O3@MSNPs. After detaching with trypsin (0.2 vol%)/PBS (Ca2+/Mg2+ free) buffer solution, the treated cells were centrifuged (1300g, 5 min), and then re-dispersed in fresh PBS (Ca2+/Mg2+ free) buffer solution. Studies on cellular uptake of FITC-labelled γ-Fe2O3@MSNPs into MEL-5 cells were conducted with a FACScan fluorescence-activated cell sorter (FACS, Becton-Dickinson). The fluorescence intensities and percentage of cell-associated fluorescence were determined using the CellQuest software. The ratio of fluorescence intensity of 10000 treated MEL-5 cells to that of 10000 untreated cells was expressed as the mean fluorescence intensity (MFI). The bar graphs in the figures represent mean values (±standard deviation) from three independent experiments.
Qualitative studies on cellular uptake using confocal laser scanning microscopy (CLSM) were carried out with MEL-5 cells internalized with FITC-labelled γ-Fe2O3@MSNPs, which were obtained according to the above-mentioned protocol. After removing the free FITC-labelled γ-Fe2O3@MSNPs, the cells were treated with paraformaldehyde (4 vol%)/DAPI (1 vol%)/PBS buffer solution (with Ca2+/Mg2+) at room temperature for 15 min in the dark. After rinsing with PBS buffer solution (with Ca2+/Mg2+) for another two times, 2 mL of PBS buffer solution (with Ca2+/Mg2+) were added. Analysis of the treated cells was performed with a confocal laser scanning microscope (Nikon, A1R hybrid resonant).
Qualitative studies on cellular uptake using transmission electron microscopy (TEM) were carried out with the treated MEL-5 cells, which were also obtained by the above-mentioned protocol. After detachment with trypsin (10 vol %)/PBS (Ca2+/Mg2+ free) buffer solution and centrifugation (1300 rpm, 5 min), the treated cell pellets were fixed with glutaraldehyde (4 wt%)/PBS (Ca2+/Mg2+ free) buffer solution. After 24 h, the pellets were rinsed with PBS (Ca2+/Mg2+ free) buffer solution again to remove the free fixatives, dehydrated in alcohol series, embedded in Epon, and then sliced (ca. 70 nm in thickness) for TEM observation with a Philips CM-100 microscope.
Quantitative studies on cellular uptake via ICP analysis: MEL-5 cells internalized with the γ-Fe2O3@MSNPs were obtained according to the above-mentioned protocol. After the treated cells were detached with trypsin (0.2 wt%)/PBS buffer solution (Ca2+/Mg2+ free) and centrifuged (1300 rpm, 5 min), 0.1 mL of HCl (37 wt%) and 0.1 mL of HF (48 wt%) were added, in order to dissolve the internalized γ-Fe2O3@MSNPs. After neutralization with 1 M NaOH solution, the solution was diluted to 20 mL, and the supernatant solution after centrifugation (1300 rpm, 5 min) was used for elemental analysis with an inductively coupled plasma optical emission spectrometer (ICP/OES 720ES Varian), while untreated cells with the same number were taken as a control.
Statistical analysis: cell culture experiments were performed in triplicate. Results are presented as mean value ± standard deviation. Statistical analyses of the data were performed using unpaired and two-tailed Student's t-test. Statistical significance was determined at p < 0.05.
2.7. Characterization
Dynamic light scattering (DLS) measurements were carried out with a Malvern Instrument Nano-ZS, which was equipped with a He–Ne laser (λ = 663 nm) and a scattering angle of 90° at 25 °C. The correlation function was analysed via the CONTIN method, and the hydrodynamic diameter (Dh) was determined using the Stokes–Einstein equation. Standard deviation was used to evaluate the size distribution (PDI). The electrophoretic mobility of the γ-Fe2O3@MSNPs was checked at 25 °C and zeta potential (ζ) was obtained with the Smoluchowski approximation. The average Dh and ζ were obtained by averaging data from three different runs.Transmission electron microscopy (TEM) was performed with a Philips CM-100 microscope. A drop of the nanoparticle aqueous suspension was placed onto a copper grid and left to dry under air.
Fourier transform infrared spectra (FTIR) of the PEG1000-OH and PEG1000-COOH macromolecules, and γ-Fe2O3@MSNPs were recorded with a PerkinElmer FTIR instrument. Samples were mixed and ground with potassium bromide, and then compressed for IR analysis.
1H nuclear magnetic resonance (NMR) spectra of PEG1000-OH and PEG1000-COOH macromolecules were measured with a 250 MHz Bruker spectrometer at room temperature in THF-d4.
Thermogravimetric analysis (TGA) of the γ-Fe2O3@MSNPs and TD/DOX-loaded γ-Fe2O3@MSNPs was performed from 20 to 600 °C at a heating rate of 20 °C min−1 under air with a TA Q500 Instrument.
Differential scanning calorimetry (DSC) of TD and TD/DOX-loaded γ-Fe2O3@MSNPs after freeze-drying was performed from 0 to 80 °C at a heating rate of 10 °C min−1 in nitrogen with a TA Q100 Instrument.
Isothermal absorption–desorption profiles of the γ-Fe2O3@MSNPs before and after drug loading were recorded at a liquid nitrogen temperature (77 K) with a Micromeritics ASAP 2010 instrument via the BET (Brunauer–Emmett–Teller) model. Specific surface areas were calculated from the adsorption data in the low pressure range, while pore size was determined following the BJH (Barrett–Joyner–Halenda) method.
Proton relaxometry measurement of the γ-Fe2O3@MSNPs was performed with a Stelar Fast Field-Cycling Spectrometer FFC-2000 equipped with a permanent magnet for the relaxation measurements in the range of 0.01–40 MHz at 37 °C. Additional data were obtained at 60 MHz on a Minispec mq-60 (Bruker).
3. Results and discussion
3.1. Preparation of the γ-Fe2O3@MSNPs
Usually, in vivo circulation half-life of nanoparticles is highly decided by both particle size and surface properties. A prolonged retention of nanoparticles within the vasculature and attenuated physiological renal clearance can be obtained via an optimal size control.30 As reported, for particles of size in the micrometre scale, they could be easily metabolized via the active phagocytosis process of the mononuclear phagocyte system (MPS). While for those small-sized particles or molecules (<5 nm), they are capable of passing through the cell membrane rapidly due to the small size, however, quickly eliminated via the physiological renal clearance.31 Nanoparticles of size of a few tens of nm up to 100 nm might remain in the blood circulation somewhat longer than the former ones.32,33 Here, γ-Fe2O3@MSNPs were prepared by using CTAB as both stabilizer and soft-template according to a previous report,28 and the thickness of the mesoporous silica shell could be controlled by optimizing the CTAB concentration and TEOS feeding amount. Morphological and structural features of the γ-Fe2O3@MSNPs were studied by TEM (Fig. 1a), and core–shell structured nanoparticles, with an average size of ca. 59 nm, were observed. In the partially magnified image, the porous structure was also detected. The amount of maghemite in the γ-Fe2O3@MSNPs was estimated to be ca. 2.7 wt% by titration.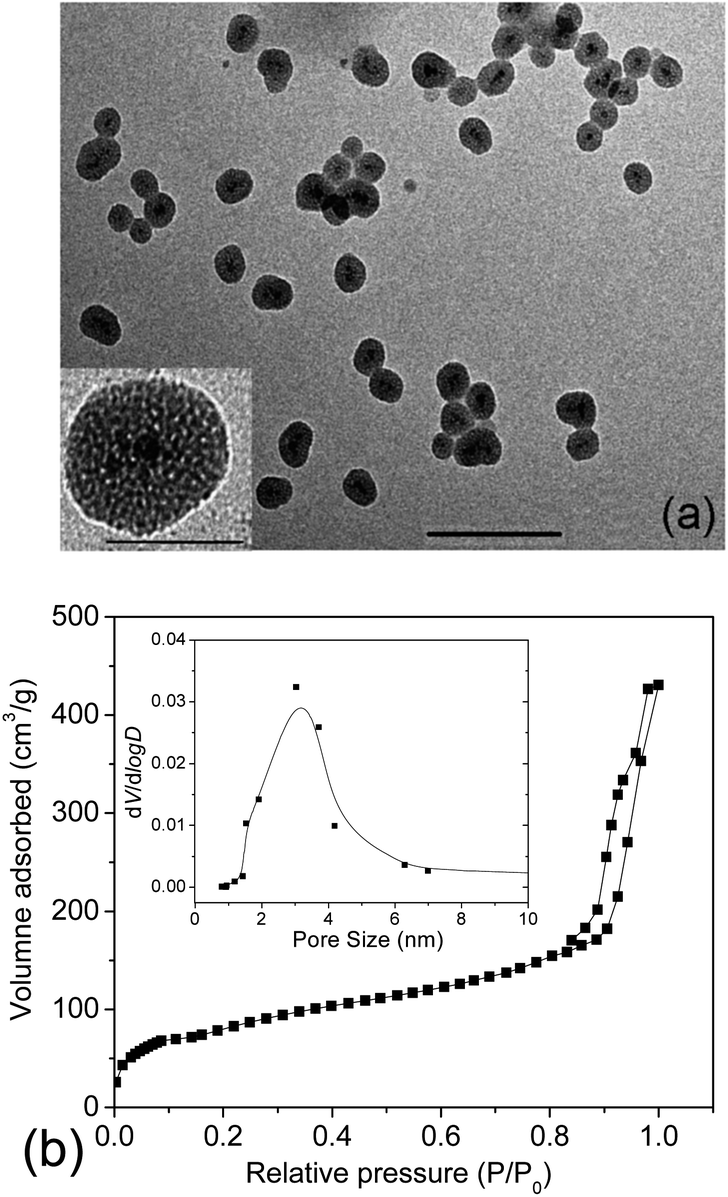 | ||
| Fig. 1 Representative TEM image of the γ-Fe2O3@MSNPs (scale bar: 100 nm) and inset: partially magnified image (scale bar: 50 nm) (a), and N2 adsorption–desorption isotherm profiles of the γ-Fe2O3@MSNPs (inset: pore size distribution derived from the adsorption branch) (b). | ||
Besides the size control, surface properties might also post influence on the toxic potential and intracellular fate of the nanoparticles.32,34,35 PEGylation is widely accepted as an effective method to improve the compatibility and long-term colloidal stability of nanoparticles under hydrophilic or lipophilic conditions.36 Furthermore, PEGylation of inorganic nanoparticles was also reported to increase the blood circulation half-life, and inhibit both uptake by kidneys31 and macrophages in the MPS.37 Here, mPEG1000-COOH was introduced via the amidation reaction with (3-aminopropyl)triethoxysilane (APTES) grafted on the silica surface. The PEGylation process was confirmed by FTIR spectroscopy with the characteristic band at a ν(CO) stretching vibration of 1750 cm−1 (see ESI, Fig. S2a†). Moreover, the surface PEGylation was also evidenced by the bands at 2947, 2882 and 1344 cm−1, which are assigned to νas(C–H), νs(C–H) and νas(C–O–C) vibrations, respectively. Surface PEGylation was also confirmed from DLS analysis (see ESI, Fig. S2b†) by the increase in Dh from 120 nm (PDI 0.13) to 210 nm (PDI 0.19) after PEGylation, and change in ζ from +33 mV to −2 mV, with a neutral surface obtained. The large deviation in size from DLS and TEM analyses could be attributed to the differences in polymer chain conformation between TEM observation (dehydrated state) and DLS measurement (hydrated state), other than particle aggregation, as confirmed by TEM images (ESI, Fig. S2c and d†). Actually, PEGylation is well-known for greatly improving the colloidal stability in aqueous medium and to increase readily their hydrodynamic radius. Then the soft template of CTAB was removed via the extraction strategy, the efficiency of which has already been confirmed in the work of Zhao38 and Lin5via infrared analysis. It is deserved to note that we could still observe the peak in the range of 2800–3000 cm−1, and this might be more readily attributed to the presence of alkyl groups of the APTES moieties. We will see in Section 3.3 that the complete extraction of CTAB, known to be highly cytotoxic,39,40 was indirectly confirmed.
To check the pore volume as well as the pore size of the mesoporous silica cavities, N2 adsorption–desorption isotherm profiles of the γ-Fe2O3@MSNPs were recorded with a porosimeter (Fig. 1b). A specific surface area of 475 m2 g−1 and an overall pore volume of 0.34 cm3 g−1 were obtained. It also showed that the γ-Fe2O3@MSNPs contained small pores of ca. 3 nm, consistent with other similar studies.6,28,41
3.2. Drug loading and heat-triggered release behaviours of the γ-Fe2O3@MSNPs as DDS
As discussed before, the main drawback of using DDS with open pores, such as mesoporous silica, is their incapacity to avoid premature drug release during the storage and drug delivery routine.12,13 In order to circumvent this limitation, herein, 1-tetradecanol molecules were co-loaded as gatekeepers to stably entrap the DOX payloads within the mesoporous silica cavities. A stable dispersion of the TD/DOX-loaded γ-Fe2O3@MSNP aqueous suspension was obtained, suggesting that those hydrophobic molecules were efficiently entrapped within the silica mesopores. The drug loading capacity (DLC) and drug loading efficiency (DLE) of the TD/DOX-loaded γ-Fe2O3@MSNPs were estimated spectrophotometrically to be ca. 9.3 wt% and 37.3%, respectively. Compared to the previous reports on the DLC of ca. 18.1 wt% of Ibuprofen within Fe3O4@MSNP DDS (0.41 cm3 g−1) by Lin,5 the relatively lower DLC reached in the present work could be due to the lower overall pore volume and the physico-chemical differences between DOX and Ibuprofen. At this stage, we did not try to optimize/maximize this value, because (i) a fraction of the pore volume had to be saved for accommodating later TD molecules and (ii) a lower DLC value is not detrimental to our strategy which should allow us to safely deliver much more efficient drugs whose dose could be, hence, lower. The differential scanning calorimetry (DSC) curve of the TD/DOX-loaded γ-Fe2O3@MSNP powders (after freeze-drying) exhibited a melting peak at ca. 39 °C, suggesting the presence of TD, and a proportion of ca. 8.9 wt% was estimated from the melting enthalpy of both TD/DOX-loaded γ-Fe2O3@MSNPs and pure TD (see ESI, Fig. S3a†). According to the TGA measurement (see ESI, Fig. S3b†), the loading percentage of TD/DOX together was confirmed to be ca. 18.8 wt%. After subtracting the DOX percentage of 9.3 wt%, the proportion of TD was estimated to be 9.5 wt%, close to the value of 8.9 wt% from DSC analysis.Triggered release of DOX by external heating was first investigated via dialysis against PBS buffer solutions at different temperatures (Fig. 2a). Due to the presence of TD as a gatekeeper, as well as the Tm of ca. 39 °C, it was conjectured that there should be no release below this temperature. As expected, negligible release (<4%) was observed at 37 °C after 96 h. However, when the release medium was heated up to 40 °C, TD molecules were melted into fluids, but still remained within the cavities due to their hydrophobic essence. In this case, the DOX molecules diffused through the TD fluid into the aqueous release medium, similar to a dialysis process. Thus, an incremental release could be expected after longer heating treatment, and an overall release of ca. 37% of the entire payload was detected after 96 h at 40 °C. In contrast, without TD as a gatekeeper, ca. 25% of the loaded DOX released after 60 h at 37 °C, while a little bit faster release was detected at 40 °C due to the higher diffusion coefficient (see ESI, Fig. S4†). The incomplete release might be attributed to the equilibrated electrostatic interaction between the mesoporous silica inner cavities and the DOX molecules, similar to other previous reports.42–44 All these results corroborated the role of TD as a gatekeeper and also the resulting “zero premature release”. Moreover, the triggered release can be manipulated by tuning the heating temperature over Tm of the gatekeepers.
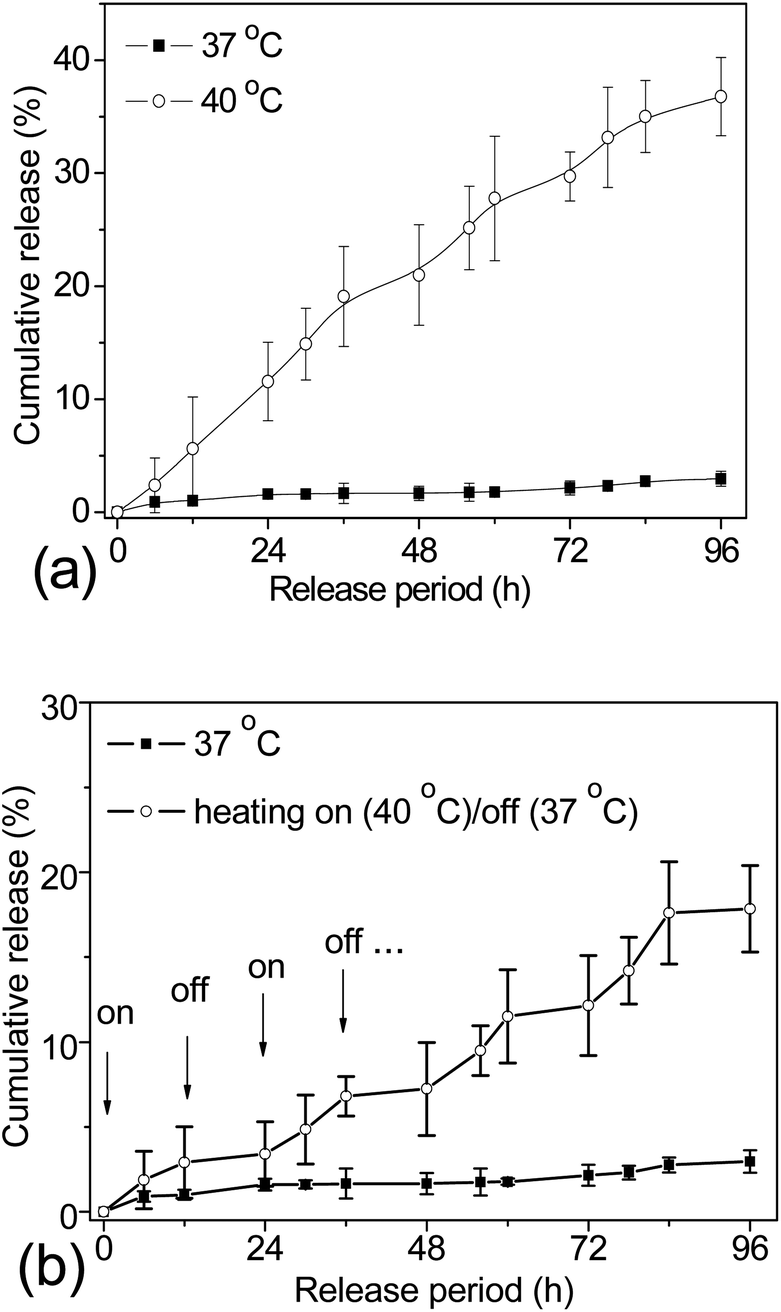 | ||
| Fig. 2 Cumulative release of DOX from the TD/DOX-loaded γ-Fe2O3@MSNP aqueous suspension (5 g L−1) into PBS buffer solutions at different temperatures (a); cumulative release of DOX from the TD/DOX-loaded γ-Fe2O3@MSNP aqueous suspension (5 g L−1) into PBS buffer solutions under multiple heating on (40 °C)/off (37 °C) cycles with an interval of 12 h, while a 96 h cumulative release profile at 37 °C external heating was taken for comparison (b). The cumulative release was presented as mean value ± standard deviation (n = 3), and the solid lines just serve to guide the eyes. | ||
For an ideal DDS, it is not only important to achieve a “zero premature release” during the delivery as required, but also for some special purposes it might be necessary to release the loaded cargos in small portions, which is called “pulsatile release”. To fulfil this requirement, the DDS should be able to open/close the gates on-demand. Bearing in mind that the phase change of TD is reversible, studies directed toward the possibility of releasing drug payloads in small portions were also carried out. The TD/DOX-loaded γ-Fe2O3@MSNP suspension was dialyzed against PBS buffer solutions under multiple heating on (40 °C)/off (37 °C) cycles for a time interval of 12 h, and cumulative release is summarized in Fig. 2b. The starting temperature was set at 40 °C, where the release of DOX was activated (marked with arrows, on); whereas upon suddenly cooling down to 37 °C, the release was quenched. As expected, on-demand “switch on/off” release was successfully manipulated via multiple heating on/off cycles due to the reversible phase-change of TD gatekeepers, and increasing release could be obtained with more heating on/off cycles, evidencing the accomplishment of on-demand pulsatile release.
3.3. Cytotoxicity, cellular uptake and in vitro heat-triggered release of DOX within culture of melanoma MEL-5 cells
To evaluate the possibility of γ-Fe2O3@MSNPs being promoted as effective DDS, cytotoxicity against the human melanoma MEL-5 cell line (Fig. 3a) and also mouse fibroblast-like L929 cell line (Fig. 3b) was first evaluated via the MTS assay. As shown, the γ-Fe2O3@MSNPs exhibited very low cytotoxicity against these two cell lines even at a high concentration (1000 μg mL−1) after 96 h incubation, which might also evidence the role of PEGylation in improving the biocompatibility.31,36,37 Since CTAB has been reported to be highly toxic against commonly used cell lines,39,40 we also tested the cytotoxicity of the γ-Fe2O3@MSNPs before CTAB extraction. As expected, significant loss in cell viability was detected, especially for those with high incubation concentration and long period (see ESI, Fig. S5†). Moreover, this might also indirectly confirm the efficiency of the procedure of the CTAB extraction from our γ-Fe2O3@MSNPs.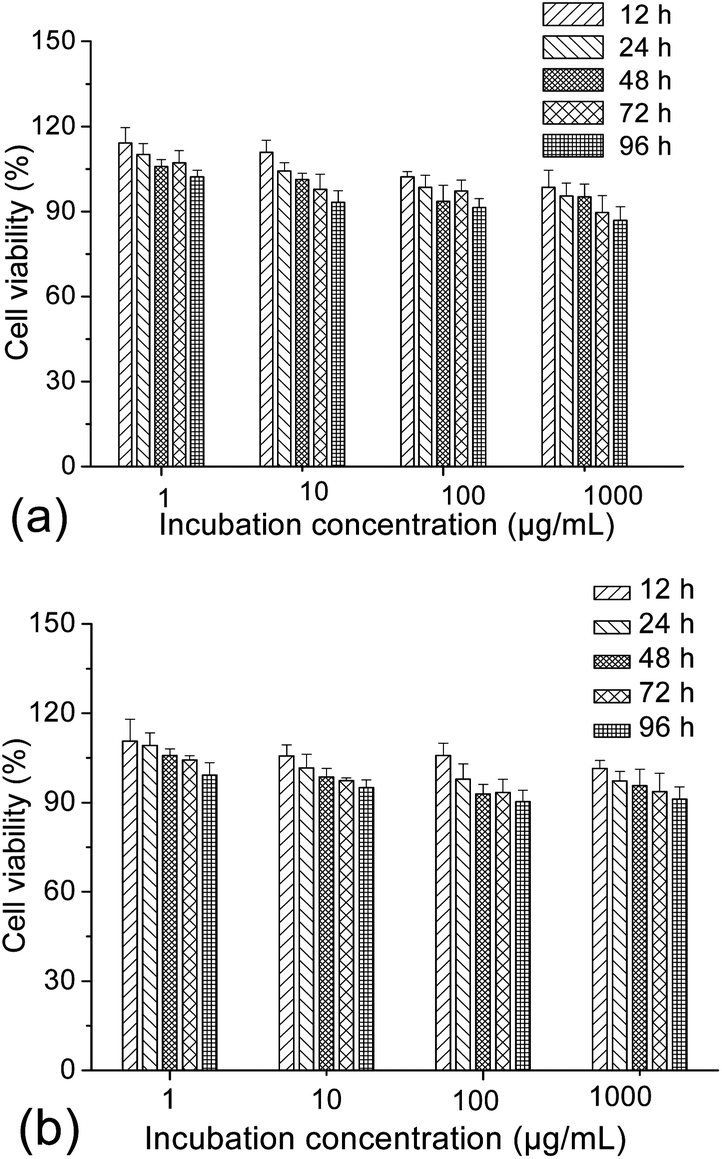 | ||
| Fig. 3 Cytotoxicity profiles of the γ-Fe2O3@MSNPs against the melanoma MEL-5 (a) and fibroblast-like L929 (b) cell lines via the MTS assay, with different incubation concentrations for different periods at 37 °C. Untreated cells were taken as a control (100% viability), and percentage cell viabilities were all expressed relative to the control. Results were all presented as mean value ± standard deviation (n = 5). | ||
Besides the in vitro compatibility of the nanoparticles, cellular uptake efficiency is another key issue for those aimed at DDS applications. To locate the γ-Fe2O3@MSNPs intracellularly, they were first labelled with fluorescein isothiocyanate (FITC), by replacing APTES with an APTES/APTES-FITC (19/1 mol/mol) mixture during the preparation, as depicted in the Experimental part. A confocal laser scanning microscope (CLSM) was used to trace the internalized FITC-labelled γ-Fe2O3@MSNPs by examining the green fluorescence originating from the FITC groups. As depicted in Fig. 4a, DAPI-stained nuclei were observed by exciting the cells with a UV laser at a wavelength of 350 nm. Green fluorescence was observed as small spots upon excitation at 490 nm (Fig. 4b), which strongly indicates the presence of γ-Fe2O3@MSNPs. The contrast field pattern of the treated MEL-5 cells (Fig. 4c) and also the merged image (Fig. 4d) confirmed the accumulation of the γ-Fe2O3@MSNPs within the cytoplasm around the nucleus.
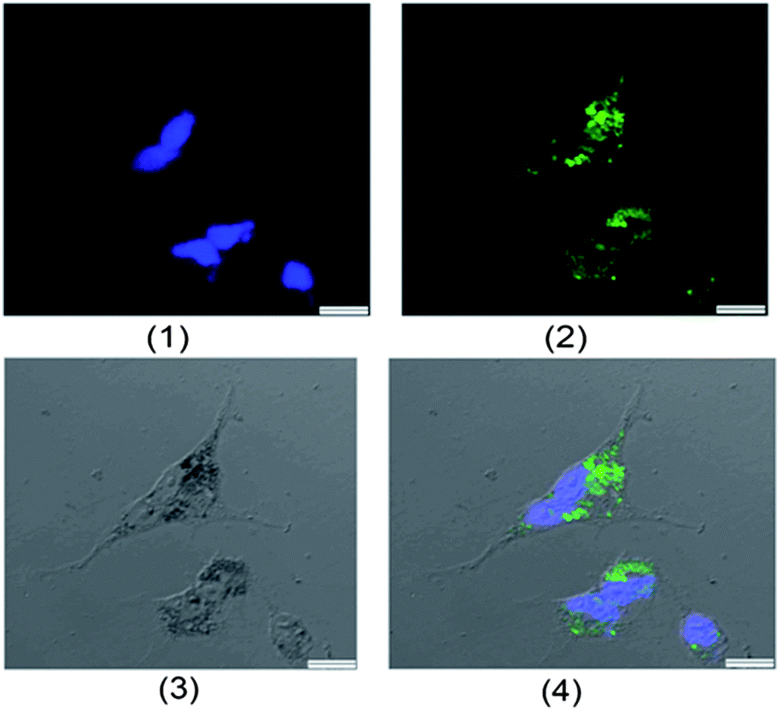 | ||
| Fig. 4 Confocal laser scanning microscopic (CLSM) images of the treated MEL-5 cells after 12 h incubation with the FITC-labelled γ-Fe2O3@MSNPs (100 μg mL−1) at 37 °C (scale bar: 1 μm): (1) nuclei stained with DAPI (blue), (2) FITC (green), (3) contrast field pattern, and (4) merged images of (1), (2) and (3). | ||
Unlike CLSM, TEM could reveal the precise location and spatial distribution of individual nanoparticles within the cells. Here, we performed TEM to further characterize the treated MEL-5 cells. After removal of the free nanoparticles and fixation of the cells, the cells were then embedded in Epon and sliced for TEM observation. As shown in ESI Fig. S6,† TEM images of the treated cells highlight the presence of γ-Fe2O3@MSNPs within the cells, which appear as dark dots (marked with arrows). Furthermore, we also observed that γ-Fe2O3@MSNPs did not enter into the nucleus but only clustered inside the cytoplasm, and the presence of Fe2O3@MSNPs within vehicles inside the cells was also reported on the cellular uptake of 43 nm Fe3O4 by Feng45 and 10 nm silica-coated Fe3O4 NPs by Kiessling.46 It is particularly remarkable that the cells still retained their viability and continued to proliferate even after the internalization of γ-Fe2O3@MSNPs (Fig. 3) indicating non-cytotoxicity.
Due to the limitation of CLSM and TEM techniques in quantitative studies on the cellular uptake efficiency, fluorescence-activated cell sorting (FACS) analyses were then carried out with the treated cells, while untreated cells were taken as a blank. Fig. 5a shows the histogram of the FACS analysis. The untreated cells exhibited weak green background; however, after internalization with the FITC-labelled γ-Fe2O3@MSNPs, the fluorescence intensity of the treated MEL-5 cells increased dramatically, and a totally different cell population was detected. To quantify the cellular uptake efficiency of γ-Fe2O3@MSNPs within the MEL-5 cells, the mean fluorescence intensity (MFI), which is denoted as the ratio of fluorescence intensity per 10000 treated cells to that of 10000 un-treated cells, was measured. This approach has been reported to study the uptake efficiency of a drug47 or siRNA delivery system.48 After incubation with γ-Fe2O3@MSNPs of different concentrations for different periods, the change in MFI is summarized in Fig. 5b. We can observe both incubation period-dependent (Fig. 5b-i) and also concentration-dependent (Fig. 5b-ii) increase in MFI in the period range of 3–24 h and concentration range of 20–200 μg mL−1. Another quantitative analysis of those treated MEL-5 cells was carried out via ICP/OES measurement, and an averaged cellular uptake amount of 9.96 ± 0.52 pg per cell was confirmed for the MEL-5 cells after 12 h incubation with γ-Fe2O3@MSNPs (100 μg mL−1). Even though no direct data on the cellular amount of magnetic mesoporous silica reported, this uptake amount is comparable to the uptake amount of 30–40 pg per cell for 10 nm silica-coated Fe3O4 nanoparticles by Kiessling,46 0.5 pg per cell for 500 nm Fe3O4@SiO2 hollow mesoporous nanospheres by Zhu49 and 10–20 pg per cell for those iron oxide nanoparticles reported by Chen.50
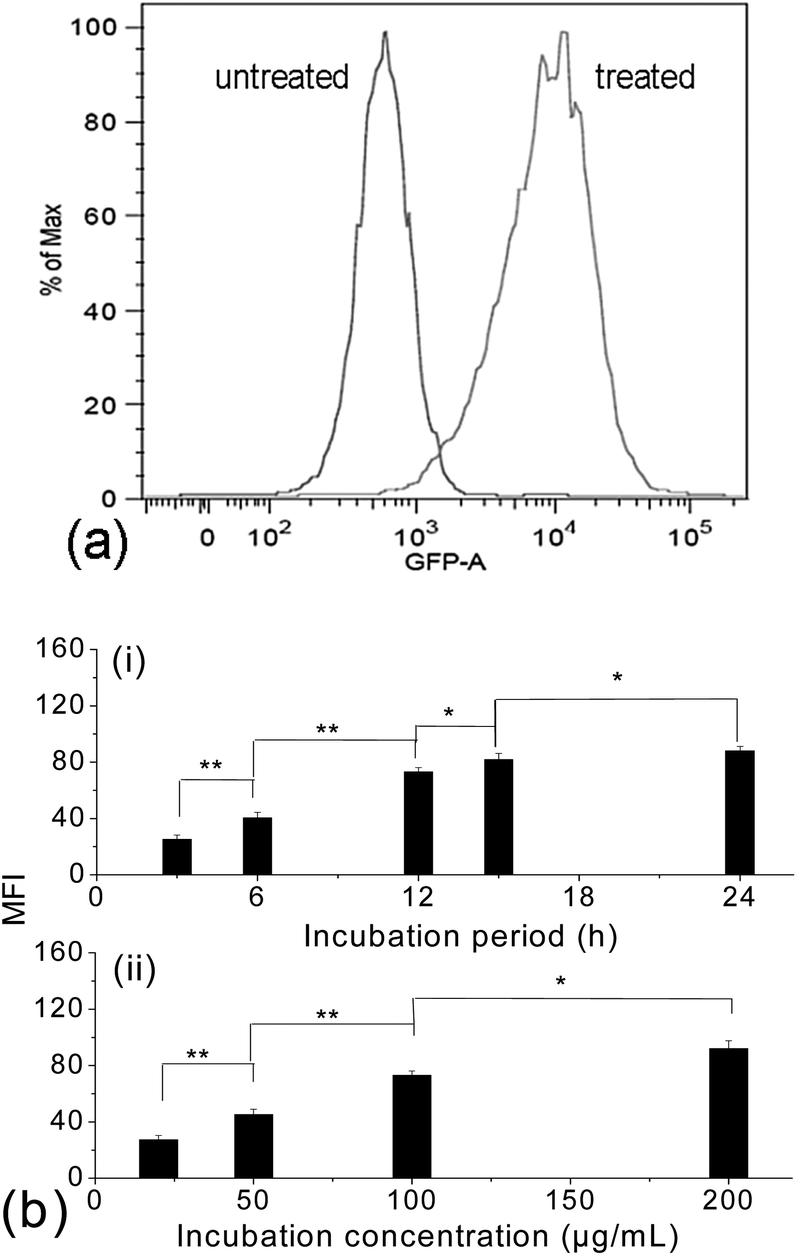 | ||
| Fig. 5 Fluorescence-activated cell sorting (FACS) histograms of the untreated MEL-5 cells and treated cells after 12 h incubation with the FITC-labelled γ-Fe2O3@MSNPs (100 μg mL−1), with the log of FITC intensity (GFP-A on the x-axis) plotted against the number of cells (counts on the y-axis) (a); dependence of the mean fluorescence intensity (MFI) of the treated MEL-5 cells on the incubation period (3, 6, 15 and 24 h) with a fixed incubation concentration of 50 μg mL−1 (b-i) and dependence of the MFI of the treated MEL-5 cells on the incubation concentration (20, 50, 100 and 200 μg mL−1) with a fixed incubation period of 6 h (b-ii). (* = P < 0.05, ** = P < 0.01 from Student's t-test.) | ||
To further verify the above-mentioned hypothesis that TD molecules could act as gatekeepers to control the DOX release within the cavities during the drug delivery, studies on cell viability were also carried out to evaluate the in vitro triggered release of DOX within MEL-5 cell culture. Prior to the in vitro heat-triggered DOX release, the cytotoxicity of DOX against the MEL-5 cell line was evaluated via the MTS assay. As shown in Fig. S7a and b,† significant cell proliferation inhibition was observed, when the MEL-5 cells were treated with DOX/DMEM medium solutions at both 37 and 40 °C, respectively. A manipulative toxic range of DOX from 1 to 10 μg mL−1 was confirmed. The concentration of TD/DOX-loaded γ-Fe2O3@MSNP suspension in DMEM complete medium was fixed at 100 μg mL−1 to ensure the DOX concentration (9.3 μg mL−1) in the manipulative toxic range.
From 41 °C, tumour cells are known to begin to show signs of apoptosis, while retaining their viability below 41 °C. Therefore we envisaged to increase the cell culture temperature from 37 to 40 °C, above Tm of TD, in order to open the gates and trigger the release of drug payloads, while avoiding severe cell apoptosis from overheating. To confirm this hypothesis, the cell viability at 40 °C was first examined, and a cell viability of ca. 91% was detected after 72 h incubation of the MEL-5 cells with TD-loaded γ-Fe2O3@MSNPs (without DOX loaded) at 40 °C (see ESI, Fig. S8a†), confirming the possibility of heating the cell culture medium to 40 °C without cell apoptosis. Furthermore, high cell viabilities were also observed after multiple heating off (37 °C)/on (40 °C) cycles (see ESI, Fig. S8b†). All these results corroborated our strategies to increase the cell culture temperature to 40 °C in order to melt the TD gatekeepers and trigger the drug release. So if the cell apoptosis occurs, it could only result from the activation of chemotherapeutic agents, rather than from thermal effects due to overheating.
The potential cytotoxicity of DOX-loaded γ-Fe2O3@MSNPs and TD/DOX-loaded γ-Fe2O3@MSNPs against MEL-5 cells was evaluated at various incubation concentrations, periods as well as temperatures. As shown in Fig. 6a, incubation concentration-dependent and also period-dependent cytotoxicity was detected for the DOX-loaded γ-Fe2O3@MSNPs at 37 °C, in the absence of TD gatekeepers. Moreover, similar phenomena were also observed at 40 °C in Fig. 6b, however, with even lower cell viability. The higher cytotoxicity at 40 °C could be explained by the competent cellular uptake rate and intracellular distribution of DOX, in agreement with the cytotoxicity profiles of bare DOX in Fig. S7a† (37 °C) and S7b† (40 °C). More importantly, it is interesting to note that the cytotoxicity of the DOX-loaded γ-Fe2O3@MSNPs (DLC of 11.6 wt%) was very similar to that of bare DOX. Taking the 60 h incubation with 50 μg mL−1 of DOX-loaded γ-Fe2O3@MSNPs as an example, the DOX concentration in the culture medium reached ca. 1.3 μg mL−1 after 60 h incubation (release of ca. 23% obtained from Fig. 2a), and cell viability was estimated to be ca. 55% (data drawn from Fig. S7a†), close to the value of ca. 46% in Fig. 6a. However, in the presence of TD, as expected, negligible cell apoptosis was observed for both high (100 μg mL−1) and low (50 μg mL−1) incubation concentrations after 72 h incubation at 37 °C (Fig. 6a). The cytotoxic potential of loaded DOX was obviously quenched, suggesting a stable storage of DOX within the mesoporous cavities with TD gatekeepers. When the cell culture temperature was increased to 40 °C, an obvious decrease in cell viability was detected for TD/DOX-loaded γ-Fe2O3@MSNPs in Fig. 6b, and also incubation concentration-dependent as well as period-dependent cell apoptosis was observed, due to the time-dependent drug release at 40 °C as detected in Fig. 2a.
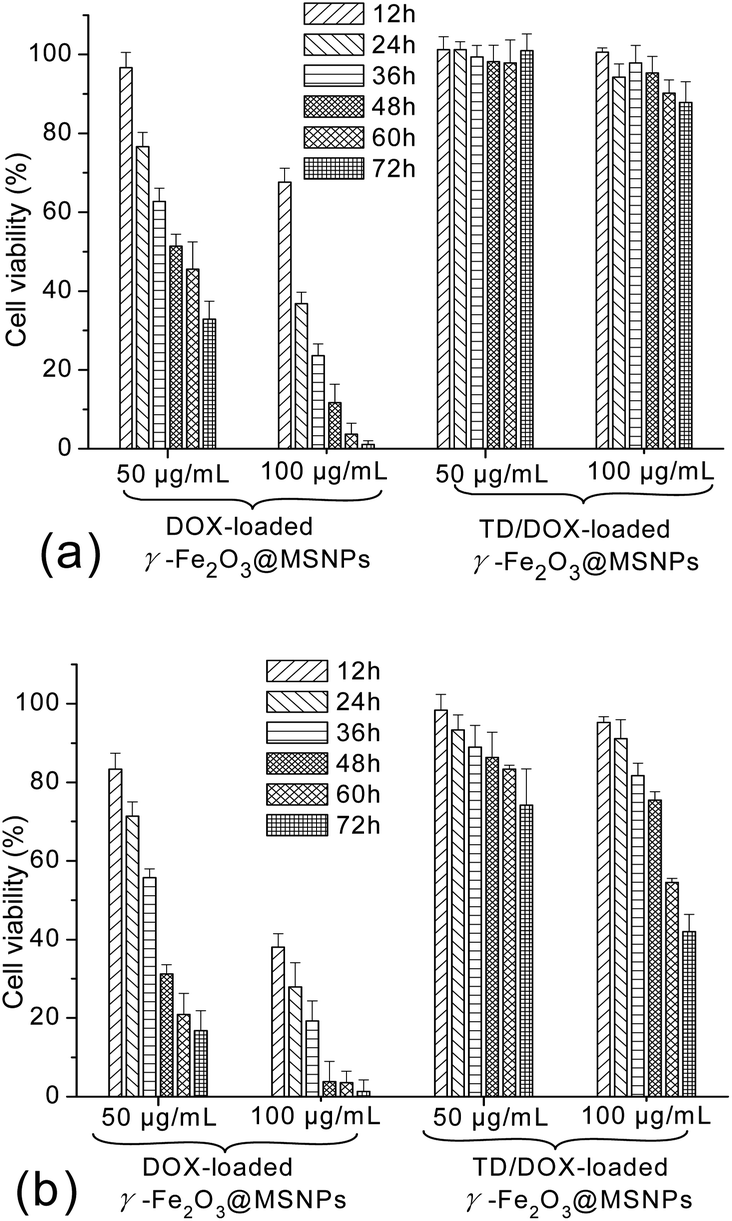 | ||
| Fig. 6 Cytotoxicity profiles of the DOX-loaded γ-Fe2O3@MSNPs and TD/DOX-loaded γ-Fe2O3@MSNPs against the melanoma MEL-5 cells with different incubation concentrations at 37 °C (a) and 40 °C (b), respectively; percentage cell viabilities of the treated MEL-5 cells were expressed relative to the untreated cells (control, 100% viability), results were all presented as mean value ± standard deviation (n = 5). | ||
CLSM was used again to visualize the in vitro heat-triggered drug release from the TD/DOX-loaded γ-Fe2O3@MSNPs. Here, γ-Fe2O3@MSNPs were labelled with an FITC group again to trace the bio-distribution of the nanoparticles. Moreover, it was very easy to visualize DOX directly since DOX itself is also a fluorescent molecule, showing red fluorescence (excitation: 480 nm and emission: 590 nm). The fluorescence characteristics of DOX and FITC were utilized to distinguish the localizations of the drug and γ-Fe2O3@MSNPs, respectively. As shown in Fig. 7a, after 24 h incubation of the TD/DOX-loaded FITC-labelled γ-Fe2O3@MSNPs (100 μg mL−1) at 37 °C, the treated MEL-5 cells emitted blue fluorescence (Fig. 7a1, DAPI) from the nuclei, green fluorescence (Fig. 7a2, FITC) from the γ-Fe2O3@MSNPs, while red fluorescence (Fig. 7a3, TRED) to the DOX molecules. Interestingly, after merging Fig. 7a1–a3 into Fig. 7a4, we found that most of the green (γ-Fe2O3@MSNPs) and red (DOX) fluorescent areas co-localized at the same spots. This phenomenon strongly indicates a stable storage of DOX within the mesoporous cavities with TD as a gatekeeper during the delivery routine, even after internalization. Furthermore, in agreement with the above-mentioned discussion, all those γ-Fe2O3@MSNPs accumulated within the cytoplasm around the nuclei. The internalization of DOX-loaded γ-Fe2O3@MSNPs (without TD gatekeepers) was also studied for comparison. After 24 h incubation, red fluorescence originating from DOX was also observed in the cytoplasm (see ESI, Fig. S9†), but without partitioning by the green fluorescence into several small dots as shown in Fig. 7a, indicating the diffusion of DOX into the cytoplasm rather than being trapped inside the γ-Fe2O3@MSNPs.
 | ||
| Fig. 7 CLSM images of the treated MEL-5 cells after 24 h incubation with the FITC-labelled TD/DOX-loaded FITC-labelled γ-Fe2O3@MSNPs (100 μg mL−1) at 37 °C (a), treated MEL-5 cells after 36 h (b) and 72 h (c) incubation with the FITC-labelled TD/DOX-loaded γ-Fe2O3@MSNPs (100 μg mL−1) at 40 °C: (1) nuclei stained with DAPI (blue), (2) FITC (green), (3) DOX (red), and (4) merged images of (1), (2) and (3) (scale bar: 100 μm). | ||
Furthermore, to confirm the possibility of opening the gates via external heating, the MEL-5 cells were incubated with the TD/DOX-loaded FITC-labelled γ-Fe2O3@MSNPs at 40 °C for different periods, and CLSM images were taken after 36 h (Fig. 7b) and 72 h incubation (Fig. 7c). In contrast to the incubation at 37 °C, the DOX molecules (red) did not co-localize in a few spots any more, but diffused within the cellular fluid (Fig. 7b3 and c3). By comparing the merged microscope images (Fig. 7b4 and c4), it is obvious that higher fluorescence intensity could be observed after longer incubation at 40 °C. These results strongly support the concept of in vitro heat-triggered drug release by increasing the cell culture temperature over the Tm of the TD gatekeepers.
To explore the possibility to release the drug payloads in a pulsatile mode with the MEL-5 cell culture, we followed the change in cell viability upon incubation with TD-loaded γ-Fe2O3@MSNPs (control, see ESI, Fig. S7b†) or TD/DOX-loaded γ-Fe2O3@MSNPs (see ESI, Fig. S10†) under multiple heating off (37 °C)/on (40 °C) cycles with an interval of 12 h. As expected, nearly no overheating-induced cell apoptosis after multiple heating off (37 °C)/on (40 °C) cycles due to the absence of DOX within the TD-loaded γ-Fe2O3@MSNPs (see ESI, Fig. S7b†). However, a decrease in cell viability was detected (ESI, Fig. S10†) once incubated at 40 °C, due to the release of DOX. The continuous decrease, even at the following 37 °C treatment, could be explained by the continuously lagged activation of DOX, which were released during the previous 40 °C treatment, suggesting that it is difficult to directly evidence the pulsatile release within cell culture by decrease in cell viability.
3.4. Potential as a contrast agent for magnetic resonance imaging (MRI)
The aim of the introduction of a maghemite core within each MSNP was not to compete with well-known MRI contrast agents,51–53 but to make the DDS trackable in order to easily monitor its biodistribution. Here, the relaxivity of the γ-Fe2O3@MSNPs was first checked by Nuclear Magnetic Resonance (NMR) relaxometry, and the longitudinal r1 relaxivity was recorded (Fig. 8a). For a better understanding, the NMRD profiles were fitted via the SPM theory developed by Muller and co-workers.54 The simulated curves fit relatively well with the experimental NMRD profiles (Fig. 8a). Similar to a previous report,55 a shift of the peak towards lower frequencies and a decrease in maximum relaxivity were also observed, due to the presence of the partially permeable mesoporous SiO2 shell, which highly constrained the mobility of inner water molecules and decreased the magnetization. Moreover, MRI images were collected at 7 T on Bruker Pharmascan 70:16. As shown in Fig. 8b, γ-Fe2O3@MSNPs (right side) give T2-weighted images as dark as (if not slightly darker than) the background, while no change for the maghemite-free MSNPs used as a blank. Therefore, these results indicate the possibility of in vivo tracking the γ-Fe2O3@MSNP DDS with the MRI technique and further establishing their final bio-distribution.
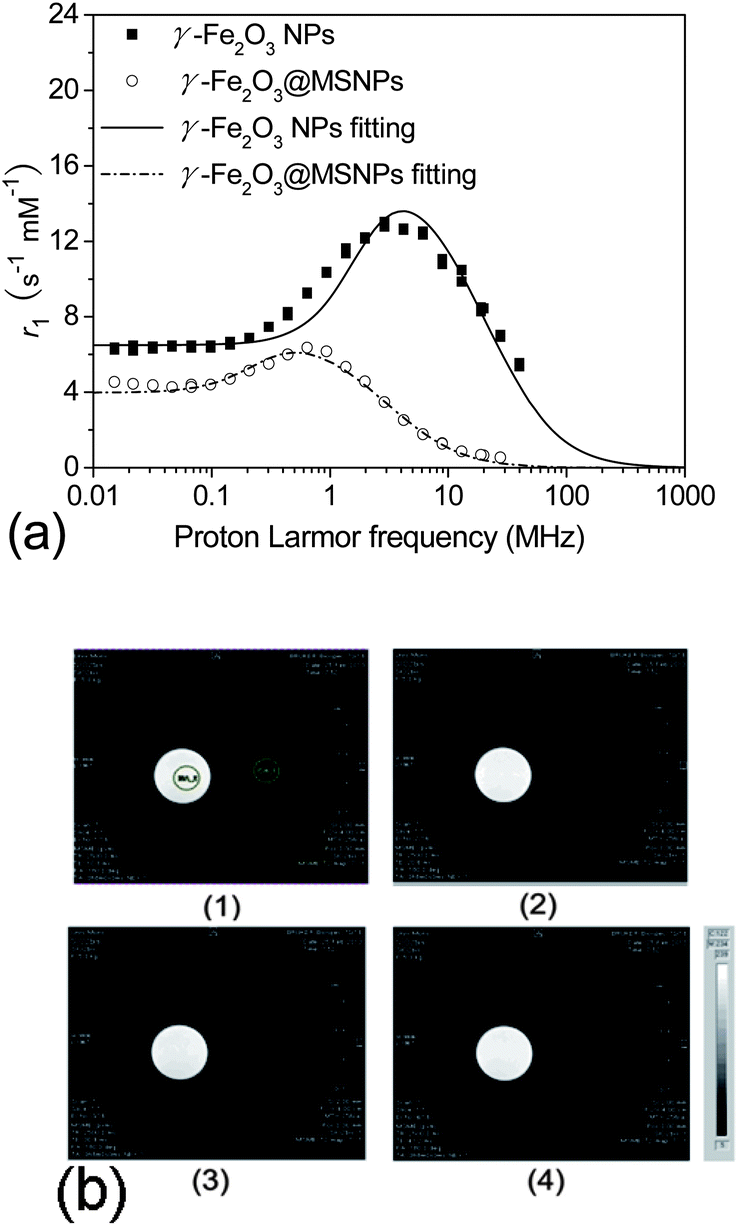 | ||
| Fig. 8 NMRD profiles of the γ-Fe2O3@MSNPs and the bare γ-Fe2O3, as well as their fitting via SPM theory (a); T2-weighted MR images of the γ-Fe2O3@MSNP aqueous suspension (right side of each images, [Fe] = 0.2 mM) and 50 nm MCM-41 NP aqueous suspension (left side of each images, 2.5 mg L−1) at echo time equal to 10.3 ms (b-1), 20.6 ms (b-2), 30.9 ms (b-3) and 41.2 ms (b-4), with TR = 2500 ms (b). | ||
4. Conclusions
In this study, a multifunctional drug delivery system γ-Fe2O3@MSNPs was successfully designed with mesoporous silica cavities as reservoirs for DOX and phase-change molecules (TD) as gatekeepers. The gating mechanism involves the reversible phase-change of TD under the heating on/off process. A “zero premature release” was achieved under physiological conditions (37 °C, PBS buffer); however, release was triggered in a controlled manner under external heating. Multiple heating on/off could contribute to a pulsatile release of drug payloads in small portions, which is essential for some specific treatments requiring precise dosage within a desired period. In vitro release of DOX within MEL-5 cell culture disclosed that toxic potential of DOX could be strongly quenched at 37 °C; however, cell apoptosis was observed when the culture temperature was increased to 40 °C, due to the opening of gates and the subsequent heat-triggered release of DOX. Hence, the concept of using PCMs as gatekeepers in MSNP-based DDS was successfully validated.For taking full advantage of these as-designed γ-Fe2O3@MSNPs for “on-demand”, i.e. remotely heat-triggered release, developments are in progress for making use of the ability of maghemite cores to heat in alternating magnetic fields (AMFs) and therefore making the DDS self-heating.
Acknowledgements
The authors thank the National Fund for Scientific Research (F.R.S.-FNRS), European Community and the Erasmus Mundus international doctoral school IDS-FunMat for their financial support. The authors are also grateful to Mrs Laetitia Etienne in ICMCB for ICP/OES analyses. The authors are also grateful to the GIGA-imaging and flow cytometry platform in University of Liège for their help with CLSM and FACS measurements.Notes and references
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- J. M. Rosenholm, C. Sahlgren and M. Linden, Nanoscale, 2010, 2, 1870–1883 RSC.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Acc. Chem. Res., 2011, 44, 893–902 CrossRef CAS PubMed.
- M. Manzano, M. Colilla and M. Vallet-Regi, Expert Opin. Drug Delivery, 2009, 6, 1383–1400 CrossRef CAS PubMed.
- P. P. Yang, Z. W. Quan, Z. Y. Hou, C. X. Li, X. J. Kang, Z. Y. Cheng and J. Lin, Biomaterials, 2009, 30, 4786–4795 CrossRef CAS PubMed.
- B. S. Chang, X. Y. Sha, J. Guo, Y. F. Jiao, C. C. Wang and W. L. Yang, J. Mater. Chem., 2011, 21, 9239–9247 RSC.
- L. Y. Xia, M. Q. Zhang, C. E. Yuan and M. Z. Rong, J. Mater. Chem., 2011, 21, 9020–9026 RSC.
- C. Tourne-Peteilh, D. A. Lerner, C. Charnay, L. Nicole, S. Begu and J. M. Devoisselle, ChemPhysChem, 2003, 4, 281–286 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- C. Coll, A. Bernardos, R. Martinez-Manez and F. Sancenon, Acc. Chem. Res., 2013, 46, 339–349 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- M. W. Ambrogio, C. R. Thomas, Y. L. Zhao, J. I. Zink and J. F. Stoddart, Acc. Chem. Res., 2011, 44, 903–913 CrossRef CAS PubMed.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, P. Amoros and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS PubMed.
- R. Liu, Y. Zhang, X. Zhao, A. Agarwal, L. J. Mueller and P. Y. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS PubMed.
- C. L. Zhu, C. H. Lu, X. Y. Song, H. H. Yang and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 1278–1281 CrossRef CAS PubMed.
- S. W. Choi, Y. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 7904–7908 CrossRef CAS PubMed.
- G. D. Moon, S. W. Choi, X. Cai, W. Li, E. C. Cho, U. Jeong, L. V. Wang and Y. Xia, J. Am. Chem. Soc., 2011, 133, 4762–4765 CrossRef CAS PubMed.
- L. Tian, N. Gandra and S. Singamaneni, ACS Nano, 2013, 7, 4252–4260 CrossRef CAS PubMed.
- N. Singh, A. Karambelkar, L. Gu, K. Lin, J. S. Miller, C. S. Chen, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2011, 133, 19582–19585 CrossRef CAS PubMed.
- A. Bernardos, E. Aznar, C. Coll, R. Martinez-Manez, J. M. Barat, M. D. Marcos, F. Sancenon, A. Benito and J. Soto, J. Controlled Release, 2008, 131, 181–189 CrossRef CAS PubMed.
- R. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- R. Casasus, M. D. Marcos, R. Martinez-Manez, J. V. Ros-Lis, J. Soto, L. A. Villaescusa, P. Amoros, D. Beltran, C. Guillem and J. Latorre, J. Am. Chem. Soc., 2004, 126, 8612–8613 CrossRef CAS PubMed.
- E. Ruiz-Hernandez, A. Baeza and M. Vallet-Regi, ACS Nano, 2011, 5, 1259–1266 CrossRef CAS PubMed.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- Q. Cai, Z. S. Luo, W. Q. Pan, Y. W. Fan, X. H. Chen and F. Z. Cui, Chem. Mater., 2001, 13, 258–263 CrossRef CAS.
- V. I. Shubayev, T. R. Pisanic and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–3166 CrossRef CAS.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- A. P. Leonov, J. Zheng, J. D. Clogston, S. T. Stern, A. K. Patri and A. Wei, ACS Nano, 2008, 2, 2481–2488 CrossRef CAS PubMed.
- I. Gorelikov and N. Matsuura, Nano Lett., 2008, 8, 369–373 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS PubMed.
- F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo and G. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS PubMed.
- Y. Zhao, L. N. Lin, Y. Lu, S. F. Chen, L. Dong and S. H. Yu, Adv. Mater., 2010, 22, 5255–5259 CrossRef CAS PubMed.
- Y. J. Yang, X. Tao, Q. Hou, Y. Ma, X. L. Chen and J. F. Chen, Acta Biomater., 2010, 6, 3092–3100 CrossRef CAS PubMed.
- M. T. Zhu, B. Wang, Y. Wang, L. Yuan, H. J. Wang, M. Wang, H. Ouyang, Z. F. Chai, W. Y. Feng and Y. L. Zhao, Toxicol. Lett., 2011, 203, 162–171 CrossRef CAS PubMed.
- C. Zhang, B. Wangler, B. Morgenstern, H. Zentgraf, M. Eisenhut, H. Untenecker, R. Kruger, R. Huss, C. Seliger, W. Semmler and F. Kiessling, Langmuir, 2007, 23, 1427–1434 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Linden, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- P. Vader, L. J. van der Aa, J. F. Engbersen, G. Storm and R. M. Schiffelers, J. Controlled Release, 2010, 148, 106–109 CrossRef CAS PubMed.
- Y. Zhu, Y. Fang and S. Kaskel, J. Phys. Chem. C, 2010, 114, 16382–16388 CAS.
- A. Bhirde, J. Xie, M. Swierczewska and X. Chen, Nanoscale, 2011, 3, 142–153 RSC.
- E. Duguet, S. Vasseur, S. Mornet and J. M. Devoisselle, Nanomedicine, 2006, 1, 157–168 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- F. Liu, S. Laurent, H. Fattahi, L. Vander Elst and R. N. Muller, Nanomedicine, 2011, 6, 519–528 CrossRef CAS PubMed.
- A. Roch, R. Muller and P. Gillis, J. Phys. Chem. C, 1999, 110, 5403–5411 CAS.
- F. Ye, S. Laurent, A. Fornara, L. Astolfi, J. Qin, A. Roch, A. Martini, M. S. Toprak, R. N. Muller and M. Muhammed, Contrast Media Mol. Imaging, 2012, 7, 460–468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR and 1H NMR spectra of PEG–COOH; FTIR spectra and DLS curve of the γ-Fe2O3@MSNPs; evolution of the temperature of the release medium; TEM image of the treated MEL-5 cells; cytotoxic profiles of DOX or TD-loaded γ-Fe2O3@MSNPs against MEL-5 cells at 37 and 40 °C. See DOI: 10.1039/c3tb21229g |
| This journal is © The Royal Society of Chemistry 2014 |