
An adaptable stage perfusion incubator for the controlled cultivation of C2C12 myoblasts†
Felix
Kurth
a,
Alfredo
Franco-Obregón
b,
Christoph A.
Bärtschi
a and
Petra. S.
Dittrich
*a
aETH Zurich, Department of Chemistry and Applied Biosciences, Department of Biosystems Science and Engineering, Vladimir-Prelog-Weg 3, 8093 Zürich, Switzerland. E-mail: petra.dittrich@bsse.ethz.ch; Fax: +41 44 632 1292; Tel: +41 44 633 6893
bDepartment of Surgery, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
First published on 24th October 2014
Abstract
Here we present a stage perfusion incubation system that allows for the cultivation of mammalian cells within PDMS microfluidic devices for long-term microscopic examination and analysis. The custom-built stage perfusion incubator is adaptable to any x–y microscope stage and is enabled for temperature, gas and humidity control as well as equipped with chip and tubing holder. The applied double-layered microfluidic chip allows the predetermined positioning and concentration of cells while the gas permeable PDMS material facilitates pH control via CO2 levels throughout the chip. We demonstrate the functionality of this system by culturing C2C12 murine myoblasts in buffer free medium within its confines for up to 26 hours. We moreover demonstrated the system's compatibility with various chip configurations, other cells lines (HEK-293 cells) and for longer-term culturing. The cost-efficient system are applicable for any type of PDMS-based cell culture system. Detailed technical drawings and specification to reproduce this perfusion incubation system is provided in the ESI.
Introduction
Since the first development of microfluidic platforms for cell culturing and analyses in the mid 90s, technological innovations have greatly improved their utility in basic research and drug discovery.1,2 Microfluidic platforms are conducive for precise cell manipulation and positioning3,4 as well as allow for an unprecedented high level of control over the cellular environment with respect to chemical gradients and biomechanical conditions.5,6 As much as the elaboration of more complicated designs will lay the foundation for advances in in vitro cell studies, so will the more precise control over basic biological parameters such as temperature, pH and humidity.7Several strategies are available to tackle these challenges. The simplest solution is to directly place the entire microfluidic device inside a conventional cell culture incubator maintaining a constant humidified environment at 37 °C and equilibrated with CO2.8,9 The drawback of this strategy, however, is that live cell imaging is significantly hampered. Whereas temperature control is relatively straight forward to achieve and various solutions are available,7 pH regulation commonly relies on the use of buffers with implicit biochemical limitations, such as HEPES.10 Moreover, for the monitoring of certain biological processes, such as transmembrane channel dynamics, the use of particular buffers should be restricted as they may modulate the response.11,12 Furthermore, HEPES possesses the serious drawback of generating reactive oxygen species when exposed to light.13 To overcome these limitations, custom-made concepts have been developed for the specific control of pH that inherent to this requirement often restricts the device design and needs to be technically adapted for each new task.14,15 CO2-independent medium is available for the growing of cells, but is limited in the amount of time that cells can remain viable. A few commercially available systems also provide well-established incubation chambers for microscopes. They provide stable conditions for which to set basic parameters, allow live cell imaging and can occasionally be modified to the user's specific needs. Yet, these systems are expensive, bulky and restricted in use with only one type of microscope setup. More flexible commercially available stage incubation chambers have been developed within the last years that are comparable in performance to the much larger microscope-housing models as well as compatible with standard multi-well plates or cell culture dishes. Only a few of them, however, are amenable for use with common microfluidic devices including their necessity for multiple-port tubing access.16 In addition, a small and microscope-independent incubation system would be required in applications where the microchip requires transportation for use in the field.17 The recent appearance of publications featuring stand-alone stage incubation chambers for microfluidic devices reflect the perceived requirement by the scientific community for such systems.18
Herein we report on a stage perfusion incubation chamber that can be used in conjunction with common microfluidic devices for the cultivation of mammalian cells (Fig. 1). The system is adaptable to any x–y stage of a conventional inverted microscope and regulates temperature and medium pH. It allows long-term observation of cultures on chip and provides gas tight tubing access for media perfusion as well as pressure lines used for the actuation of chip incorporated valves and other features that are based on flexible membranes.19–21 The pH regulation is achieved by the introduction of a CO2 containing gas mixture compatible with buffer free medium. The circular shaped chamber is composed of an aluminium alloy for effective heat transfer originating from incorporated resistance heaters for temperature control. Closable cutouts in the lid and the bottom of the chamber are amenable for bright field and fluorescence microscopy. Tubing ports are supplied along the sidewall of the chamber, granting access for multiple perfusion or pressure lines. The temperature is regulated by a closed loop control system and gas-tight sealing is achieved using either o-rings or flat gaskets between the lids and the chip. A gas stream is introduced via a high accuracy pressure regulator, supplying a constant CO2 feed at low flow rates. The diffusion of CO2 gas into cultivation sites on the chip was assured with the use of the elastomer, PDMS, in chip fabrication. PDMS is frequently used in microfluidic technology for its biocompatibility and gas permeability.22 The material costs for the portable incubation chamber summed to approximately 500 Euro (in 2014).
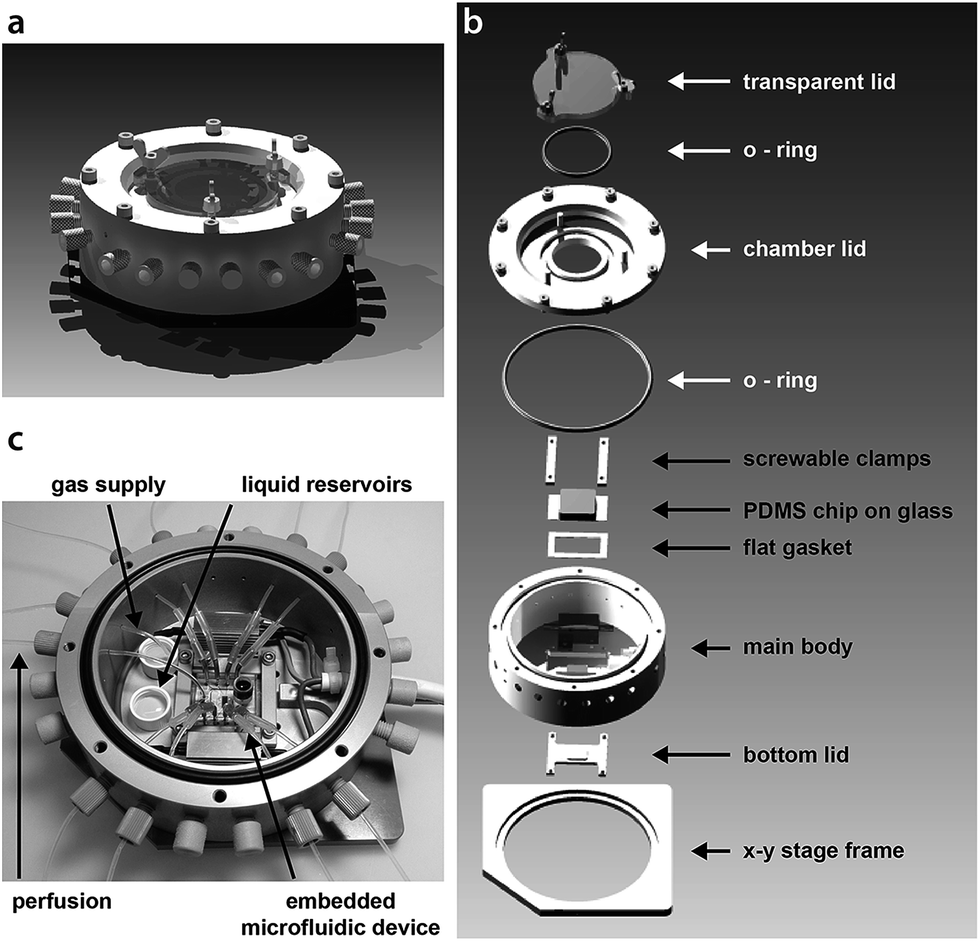 | ||
| Fig. 1 (a) A schematic of the assembled stage perfusion incubation system. (b) An exploded view of the incubation chamber showing all essential parts. (c) Photograph of the open chamber including the embedded chip with tubing connections shown in the middle. Specifically marked are the liquid reservoirs that account for the humidified atmosphere inside the chamber and the gas supply tubing. Tubing access ports that are not to be used will be sealed by blind caps. The outer diameter of the main body is 11.9 cm. Detailed technical drawings (Fig. S1 and S2†) and CAD files are available in the ESI.† | ||
Experimental
Working principle of the stage perfusion incubation system
Two feedback regulated resistance heaters (Arcol, HS25 Aluminium Housed Resistor, Switzerland) control the temperature of the incubator; for this purpose a PT100 sensor is adapted into the main body frame.A defined gas mixture (7% CO2, 93% synthetic air, Pangas, Switzerland) is applied for the control of the pH of the cell culture medium. The gas flow into the incubator is controlled by a high accuracy single stage diaphragm pressure regulator (Beswick, Greenland, NH) equipped with fluorinated ethylene propylene (FEP) tubing of specified length, accurately regulating the gas supply to a predefined pressure of 25 mbar. The gas mixture is sterile filtered (0.22 μm) prior to entering the incubation chamber through a FEP tubing of 200 mm length and 0.25 mm ID. A final gas mixture flow rate of 4.23 ml min−1 is achieved into the incubator chamber based on the following equation:
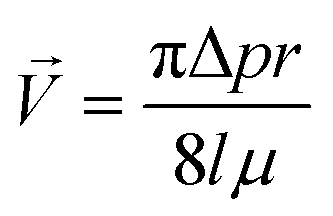 | (1) |
![[V with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0056_20d1.gif) as the volumetric flow rate, p the gas pressure, r the tubing ID, l the tubing length and μ the gas viscosity.
as the volumetric flow rate, p the gas pressure, r the tubing ID, l the tubing length and μ the gas viscosity.
To compensate for liquid evaporation from the open chip reservoir as well as to prevent the formation of air bubbles inside the microfluidic device, the incoming gas is first introduced into a reservoir containing 1 ml ddH2O that also serves to generate a humidified atmosphere inside the chamber. A stable gas flow through the chamber is enabled by opening one tubing port in the sidewall of the main body, allowing the heated air mixture to pass through the chamber while also preventing the diffusion of colder atmospheric air into the system, which would lead to reduction of CO2 levels.
A thorough description of the stage perfusion incubation system is given in the (ESI†) as well as detailed technical drawings (ESI, Fig. S1 and S2†). Furthermore, all individual components of the incubation system are listed in table S1 in the ESI† and separate Inventor CAD files are available for download.
Incubation system characterisation
A thermocouple type K (chromel-alumel, 1/16′′ OD, custom-built) was implemented for the determination of the chamber air and chip temperatures, Tair and Tchip, respectively. The thermocouple was introduced into the chamber via a tubing port in the sidewall of the chamber and placed in the centre of the chamber for Tair determination. Tchip was determined by introducing the thermocouple into a 1.5 mm hole punched in the middle of a test chip (ESI, Fig. S3†).Fluorescent measurements were taken using 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS) as a pH indicator at 1 μM in phosphate buffered saline (PBS; pH 7.4, Life Technologies, Switzerland) and cell culture growth medium (GM). The HPTS stock solution was prepared in ddH2O at 10 mM. Measurements were taken at 37 °C in a humidified environment under non-regulated (no CO2 supply, PBS, GM) and regulated conditions (7% CO2, GM). For regulated measurements, the chip device was additionally incubated in a standard cell culture incubator over night in a humidified atmosphere at 37 °C and 7% CO2 prior to the experiments. Images were taken with an EMCCD camera (iXon, Andor Technologies, Ireland) and a 20× objective. Data readout was analysed using imageJ.23
Peripheral systems
The incubation system setup was mounted onto an inverse microscope (Olympus IX70 and IX71 series) equipped with a mechanical x–y stage and right handle (IX-MVR). Condenser types were either IX-LWUCD or IX-ULWCD. Fluid control was mediated by a high accuracy syringe pump system (Nemesys, Cetoni, Germany) using a gas tight glass syringe (1 ml or 250 μl, Agilent Technologies, Switzerland) connected to FEP tubing (0.25 mm ID) by a PEEK Luer lock system (10–32 threaded female to female Luer and a finger-tight PEEK nut; Ercatech AG, Switzerland). Nitrogen supply for the control layer was regulated by a pressure system from fluigent (Fluigent, France).Chip design
Our design for the culture of C2C12 cells is a multi-layered microfluidic chip device made of poly-(dimethylsiloxane) (PDMS; Sylgard 184, Dow Corning), consisting of a fluid layer and an overlying control layer, separated by a thin and flexible PDMS membrane.21,24 The layers are fabricated separately and subsequently aligned and fixed for final implementation.The inlet channel of the fluid layer branches stepwise into eight cultivation chambers of 400 μm width and 2 mm length. The channel height is 40 μm. The control layer is comprised of eight separate channels used for physical manipulation of the cells and cell capture. Six lines of horseshoe-shaped pillar structures account for the localised collection and deposition of cells within the fluid channel. Upon pressurisation of the control layer the deformable PDMS membrane intervening between the two layers is depressed into the fluid layer allowing cell collection (Fig. 3a). After sufficient time has been allowed for the collected cells to attach to the fluid layer substrate, the control membrane is withdrawn into its original non-deflected state; cells are then free to migrate onto the channel surface (Fig. 3b). In this manner cell migration studies can be conducted within the device. Two control channels located at the first fluid channel branch point enable regulated cell supply to either the upper or the lower 4 cultivation chambers (ESI, Fig. S4†). We further analysed the performance of the pressure control layer in relation to the induced pressure. Details are provided in the ESI,† the results are depicted in Fig. S5 (ESI†).
For the culture of HEK-293 cells a single-layered chip design with a channel height of 100 μm was used. The chip design is similar to the one used for the culture of C2C12 cells except that the longer culture chambers in the middle of the chip were replaced by two shorter culture chambers in series. A micrograph of this design is depicted in Fig. S6 (ESI†).
All details on the wafer and chip device fabrication are provided in the ESI.†
Chip preparation
All channels of the chip were filled with DMEM without supplements (Life Technologies, Switzerland) by centrifugation (800 × g, 5 min) immediately after plasma bonding. As the liquid was pulled through the chip during the experiments, the inlet hole was equipped with a custom-built poly(methyl methacrylate) (PMMA) reservoir for easy fluid exchange (Fig. S7†). The chip was then equilibrated under a humidified atmosphere at 37 °C and 7% CO2 over night. The following day, the chip was assembled into the perfusion incubation system and the channel surface was coated with fibronectin (FN; Life Technologies, Switzerland) by flushing the channels with a 0.1 mg ml−1 FN in PBS at −0.2 μl min−1 for at least 30 min. The fluid was exchanged to culture medium prior to cell seeding.Cell culture
C2C12 mouse skeletal myoblasts were obtained from the American Type Culture Collection (ATCC; LGC Standards, France). Culturing was conducted in growth medium (GM) containing Dulbecco's Modified Eagle Medium (DMEM; Life Technologies, Switzerland) including 4.5% glucose, 1 mM sodium pyruvate and 2 mM L-glutamine completed with 20% foetal bovine serum (FBS; Life Technologies, Switzerland) in a humidified atmosphere at 37 °C and 7% CO2. Cells were passaged every 1.5 d, seeded into 75 cm2 flasks (TPP, Switzerland) and kept at low confluences (20% to 40 %) to prevent differentiation. For on-chip experiments, cells were seeded in 25 cm2 flasks (TPP, Switzerland) and grown until 40% confluence was reached. The cells were trypsinised, re-suspended in 5 ml GM and centrifuged at 200 × g for 5 min. Supernatant was discarded, cells were re-suspended in 1 ml GM and subsequently filtered via a 20 μm pore diameter cell filter (CellTrics, Partec, Germany) for cell suspension supply of single cells to the chip.Human embryonic kidney cells (HEK-293) were obtained from the American Type Culture Collection (ATCC; LGC Standards, France). Cells were grown in DMEM (Life Technologies, Switzerland) containing 1.0% glucose, 1 mM sodium pyruvate, 2 mM L-glutamine, 1% non-essential amino acids (PAA, Austria), 1% penicillin–streptomycin (Life Technologies, Switzerland) and supplemented with 10% FBS (Life Technologies, Switzerland). Cells were passaged twice a week into 25 cm2 culture flasks (TPP, Switzerland) prior full confluence was reached. For passaging, cells were trypsinised, centrifuged at 700 × g for 5 min and seeded at an initial confluence of 20% after re-suspension in fresh medium.
On-chip cell culture
For the on-chip cell seeding of C2C12 cells, the cell suspension was added to the chip reservoir and next drawn through the chip by suction. Cells were collected by pressurising the pressure pad lines with 900 mbar N2 pressure starting from the last pad in flow direction and sequentially pressurising in a forward direction; cells were fed into the chip at a flow rate of −2.5 μl min−1. After cell capture, the flow was stopped and the N2 pressure was released from the control layer. The cell suspension in the chip reservoir was exchanged for fresh GM (equilibrated to 37 °C and 7% CO2) and flushed into the chip at −0.2 μl min−1 for 10 min to sustain the cells.For perfusion of C2C12 cultures on chip, the open bottom area of the perfusion chamber was closed with an aluminium lid. Fresh growth medium was supplied every 0.5 h at 0.01 μl min−1 for 5 min, regulated by an automated syringe pump script.
For cell counting images of growing cultures were taken every 2 h by a CCD camera (UK-1117, EHD, Germany) via a 10× objective. The bottom lid of the perfusion chamber was removed for imaging only. Cell number at each time point was normalised to the original seeding density.
For long-term culturing of HEK-293 cells, the cell suspension was added into the reservoir and flushed into the chip at 1.2 μl min−1 until 20 to 30 % confluence was reached. Medium was renewed twice a day at 0.333 μl min−1 for 1 h. The flow speed was chosen to keep the induced shear force at a minimal level, i.e. ≤0.1 dyn cm−2, a rate that is considered unproblematic for most perfusion cultures.7
Cell viability assay
To evaluate the cell viability trypan blue (Sigma, Switzerland) was used to identify dead cells. Cell culture flasks were rinsed once with phosphate buffered saline (PBS; pH 7.4, Life Technologies, Switzerland) and replaced with a trypan blue buffer solution consisting of a trypan blue to PBS ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. For on-chip cultures the reservoir solution was exchanged directly with the same trypan blue buffer solution and flushed through the chip for 5 min at 0.333 μl min−1 prior to imaging using an EMCCD camera (iXon Ultra, Andor Technologies, Ireland) at 20× magnification. Cell counting was done using Image J.23
:3. For on-chip cultures the reservoir solution was exchanged directly with the same trypan blue buffer solution and flushed through the chip for 5 min at 0.333 μl min−1 prior to imaging using an EMCCD camera (iXon Ultra, Andor Technologies, Ireland) at 20× magnification. Cell counting was done using Image J.23
Stress assay
The stress assay was conducted as previously described by Crocetti et al. in 25 cm2 culture flasks.25 Briefly, as a positive control for cell viability under steady state culturing conditions HEK-293 cells were stressed with 1 mM hydrogen peroxide (Merck, Germany) overnight on the fourth day of culturing. The following day (day 5) dead cells were stained with trypan blue as described previously.Results and discussion
Temperature and pH control
To evaluate the performance of the temperature control system the inner air temperature (Tair) and the chip temperature (Tchip) were recorded for a preset temperature (Tset) using a thermocouple (Fig. 2a) and a gas inlet feed of 4.23 ml min−1 (the gas is not pre-heated). For Tset the feedback loop control guarantees an accuracy of ±0.1 °C for all tested ranges above room temperature (RT). At a preset temperature of Tset = 37 °C, Tair was measured at 36.63 °C ± 0.25 °C, i.e. 1% aberration. During long-term microscopic observation, for which the bottom lid had to remain open, Tchip was recorded with 36.5 °C ± 0.1 °C at Tset = 37 °C, i.e. 1.35% offset, which could be easily compensated for with only minor Tset adjustments. When the bottom lid was closed, Tchip was coincident with Tset, due to the minimal heat drain through the sealed chip base. Offsets at lower temperatures for Tair and Tchip are affected by room temperature. Therefore, in addition to mammalian cultures (37 °C) our system is also amenable to bacterial and yeast cultures (≤37 °C).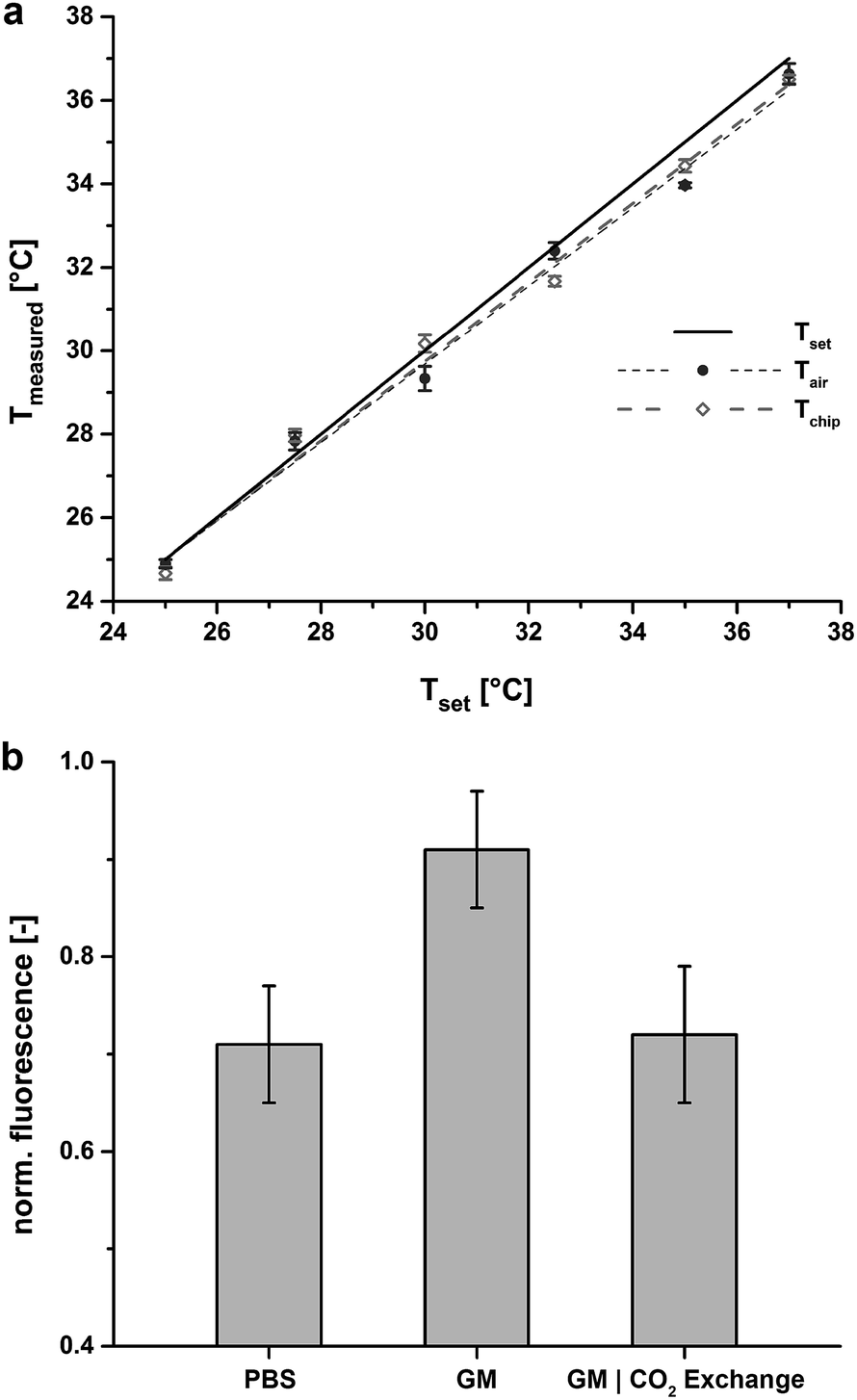 | ||
| Fig. 2 (a) Temperature validation of Tair (solid circle) and Tchip (open diamond) of the incubation system including linear fits. Tset (black line) indicates a temperature accuracy of 0.1 °C for all preset values above room temperature. At 37 °C Tair shows an offset of 1% from Tset and Tchip shows an offset of 0.5 °C with the bottom lid open after 1.5 minutes. When the bottom lid is closed Tchip resembles Tset for all measured values. All data points are mean values of three independent measurements. (b) pH control inside the perfusion incubation chamber with and without a constant feed of a defined gas mixture (7% CO2, synthetic air). HPTS is used as a fluorescent pH indicator and PBS (pH = 7.4) as a pH reference solution. At uncontrolled conditions (no CO2 feed) growth medium (GM) clearly depicts a more basic pH (pH = 7.98, measured with regular pH meter). Under controlled atmospheric conditions GM is regulated to physiological pH. All data points include data from three independent measurements that have been normalized to the maximal measured value. | ||
Next, a defined gas mixture is fed into the chamber to establish a constant pH of the medium inside the open chip reservoir before being fed into the chip. Due to the gas permeability of PDMS, the pH of the medium inside the chip is controlled, allowing on-chip cell cultivation over longer time periods. We verified the constancy of the pH inside the chip with the pH-sensitive dye 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS) (Fig. 2b). Comparing uncontrolled conditions (no CO2 flow) to CO2-equilibrated conditions demonstrates that pH regulation of the cell culture medium within physiological range is clearly possible with this approach.
Double-layered chip performance
Here we describe a double-layered PDMS chip, whereby an upper control layer descends a pillar for cell trapping (Fig. 3a and b and ESI, Fig. S4†). Such horizontally movable cell traps were used to achieve selective cell seeding at predefined positions within the cultivation chambers. The actuation of the cell traps is achieved by stepwise pressurisation of the control layer from 0 mbar up to 900 mbar thereby regulating the distance between the descending pillar and the channel base (ESI, Fig. S5†). In addition, by pressurising the more distal cell trap first, followed in order (distal to proximal) by the other rows of pillars, a homogeneous distribution of seeded cells along the microchannel is achieved (Fig. 3a and b). When a straight chamber is utilised without traps, cells are seeded from high density to low density, front to rear, respectively (ESI, Fig. S8†). For instance, individualised C2C12 cells can be successfully trapped with a pressure of minimally 700 mbar applied to the trap. Once the cells have securely adhered to the chamber substrate (approximately 10 minutes) the traps can be released enabling free migration and division without any obstacles that would have originated from permanent cell hurdles, etc.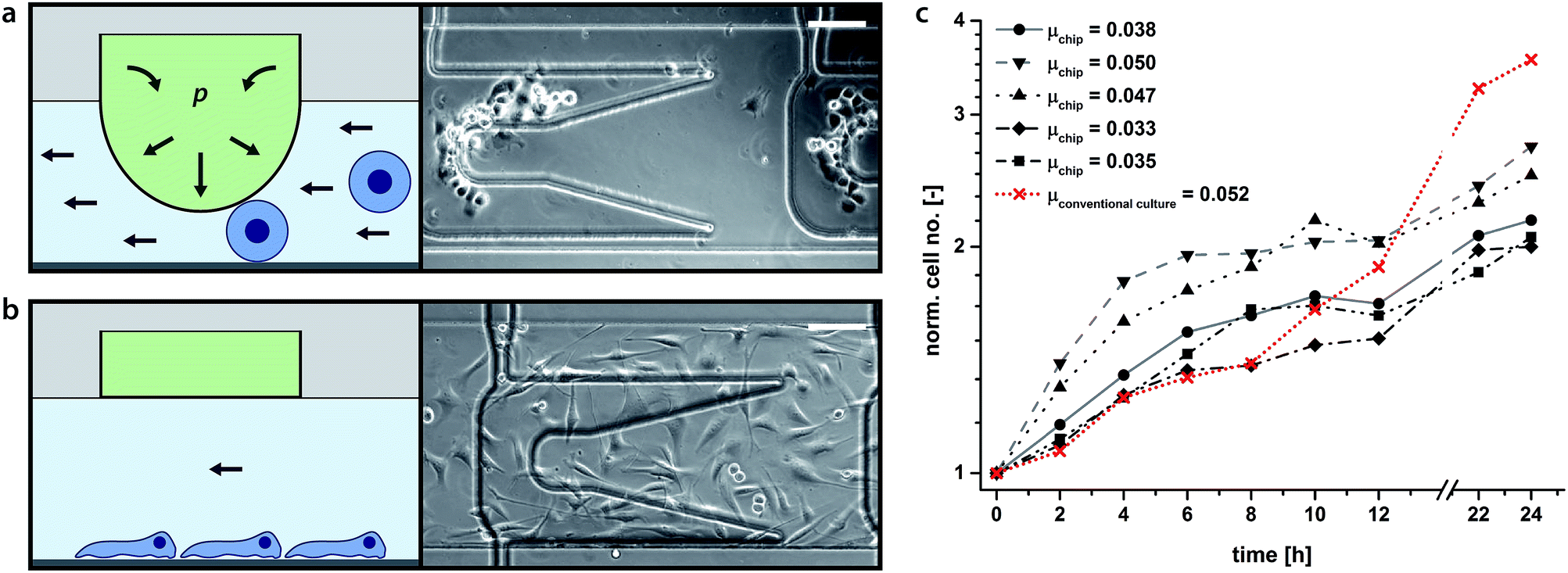 | ||
| Fig. 3 (a and b) Schematics (left) and micrographs (right) of newly captured (above) and adhered cells (below). Upon depression of the membrane cells are trapped between the substrate and the descending pillar (a). Upon adhesion to the substrate, the membrane pillar is retracted back to its original position and the cells can freely migrate and divide over the smooth surface without geometric obstacles (b). Image (b) was taken 14 h after cultivation start. Scale bars: 100 μm. Movies of cell capturing and cell migration and division are available in the ESI.† (c) C2C12 proliferation rates on chip and in a conventional cell culture flask. Example of representative growth curves obtained from 5 on-chip C2C12 cultures (grey scale). Cell growth in a conventional 75 cm2 cell culture flask is shown in red (mean values of 4 counts). Growth rates were independently calculated for 24 hour periods. For the on-chip cultures all doubling times are briefer than 24 h, consistent with cell culture results cited in the literature.27,28 The mean doubling time for the presented on-chip populations was 16.9 h ± 3.0 h (n = 5), correspondent to a specific growth rate of μ = 0.0409 ± 0.007. The presented conventional cell culture doubling time is 13.3 h ± 2.1 h with a specific growth rate of μ = 0.0523 ± 0.008. | ||
It should be noted that the presented approach of removable traps could also be employed for reversible capture or sorting purposes for the selection of suspension cells or particles based on size. Additionally, reversible trap motifs have been previously used for the patterning of cells20 as well as could potentially be applied to the building of multi-layered cell sheets for tissue engineering purposes.26
Cell growth
The mechanobiological factors regulating the proliferation of C2C12 murine myoblasts have been studied.27,28 C2C12 cell growth rate was taken as a measure for cell viability within our culturing paradigm. The normal doubling time of C2C12 myoblasts is between 12 and 24 hours (μ > 0.029, μ: specific growth rate) depending on culture conditions and the existence of appropriate mechanical cues.27,28 Repetitive perfusion with medium every 30 min at low flow rates (i.e. 0.02 dyn cm−2) was found to provide the optimal mechanical stimulus for cell growth and viability. Growth rates declined with longer time intervals interspersed between media feeds (data not shown).Fig. 3c illustrates C2C12 growth rate in our microfluidic device compared to cell growth rate in a conventional cell culture flask. Representative growth curves obtained from five independent C2C12 cultures grown on-chip are depicted (grey scale) along with their respective calculated values of growth rate. For comparison, an example of the growth rate observed in a conventional tissue culture flask (75 cm2) is shown in red. C2C12 myoblasts cultured on-chip doubled once during the 24 hour observation period.
The mean calculated growth rate was 16.9 h ± 3.0 h (i.e. μ = 0.0409 ± 0.007), only slightly longer than that cited for cells under conventional tissue culturing conditions, i.e. 14 h, yet significantly faster than that for cells grown while mechanically-unloaded, i.e. 30 h (simulated microgravity).27 In general, rapid cell growth rate at the onset of plating was observed, followed by a slowing in growth rate upon the establishment of cell–cell contact, a process known as contact inhibition and associated with the down-regulation of key components of the cellular mechanotransduction apparatus.28,29 Mechanical stimulation of myoblasts increases their proliferation,30 whereas mechanical unloading stalls their proliferation.27 A subset of the myoblast on-chip cultures exhibited relatively rapid growth rates during the first few hours of plating likely due to the mechanical stimulation afforded by intermittent fluid flow (feeding) that was not available in the tissue culture flask (cf. Su et al. 2013).31 An abrupt reduction in proliferation, however, could also signify that myoblasts on-chip reach confluence earlier and withdraw from the cell cycle sooner than the myoblasts maintained in the tissue culture flasks. In support of this interpretation it was apparent that cells on-chip underwent a deceleration of proliferation before myoblasts grown in standard tissue culture plastic. C2C12 cultures could be cultured on-chip for up to 26 h before significant contact inhibition was apparent. Although plating myoblasts at lower density would prolong how long they would maintain proliferative on-chip before undergoing contact inhibition, they would also run the risk of going quiescent due to low cell density. By necessity, robust proliferation must segue into differentiation, otherwise tissue regeneration would be short-circuited. Longer culturing periods allowing for the fusion of myoblasts into differentiated myotubes were not conducted here, but it is anticipated that cell viability and differentiation would not be compromised as long as media exchange was provided for up to one week, a feature that as we have shown is achievable with this system as the incubation chamber provides stable cell culturing conditions as long as media exchange is maintained.
Long-term culture viability
To investigate operational stability and versatility with respect to other chip designs and cell strains experiments were conducted with HEK-293 cells in long-term experiments. To this end, HEK-293 cells were seeded in a modified single-layered chip device (ESI, Fig. S6a†) and cultured for 5 days. At the end of five days cells were tested for Trypan blue inclusion to ascertain viability, dead cells staining blue (ESI, Fig. S6b†). As further reference conditions for our stage incubation system, HEK-293 cells were seeded in an identical chip device as well as in a standard tissue culture flask, but maintained in a conventional cell culture incubator for identical durations. As a positive control another batch of cells were induced to undergo oxidative stress (1 mM hydrogen peroxide) prior to the viability check on day 5.Fig. 4a summarises the results of the long-term cell culture experiment. The on-chip cultures grown on stage demonstrated similar viability to that achieved in the tissue culture flask control grown within an incubator (∼98% to 99%), whereas the viability of cultures having undergone oxidative stress and maintained with the incubator was significantly decreased (∼85%). The viability of cells cultured on-chip, but maintained inside a conventional cell culture incubator was also similar to that from either the flask-incubator or chip-stage condition. Generally, cultures on-chip reached full confluence at around the same time as the cultures in flasks (Fig. 4b). This fact is also reflected by the microscopy images depicted in Fig. 4c here shown for day 2 and day 5 of the culture time.
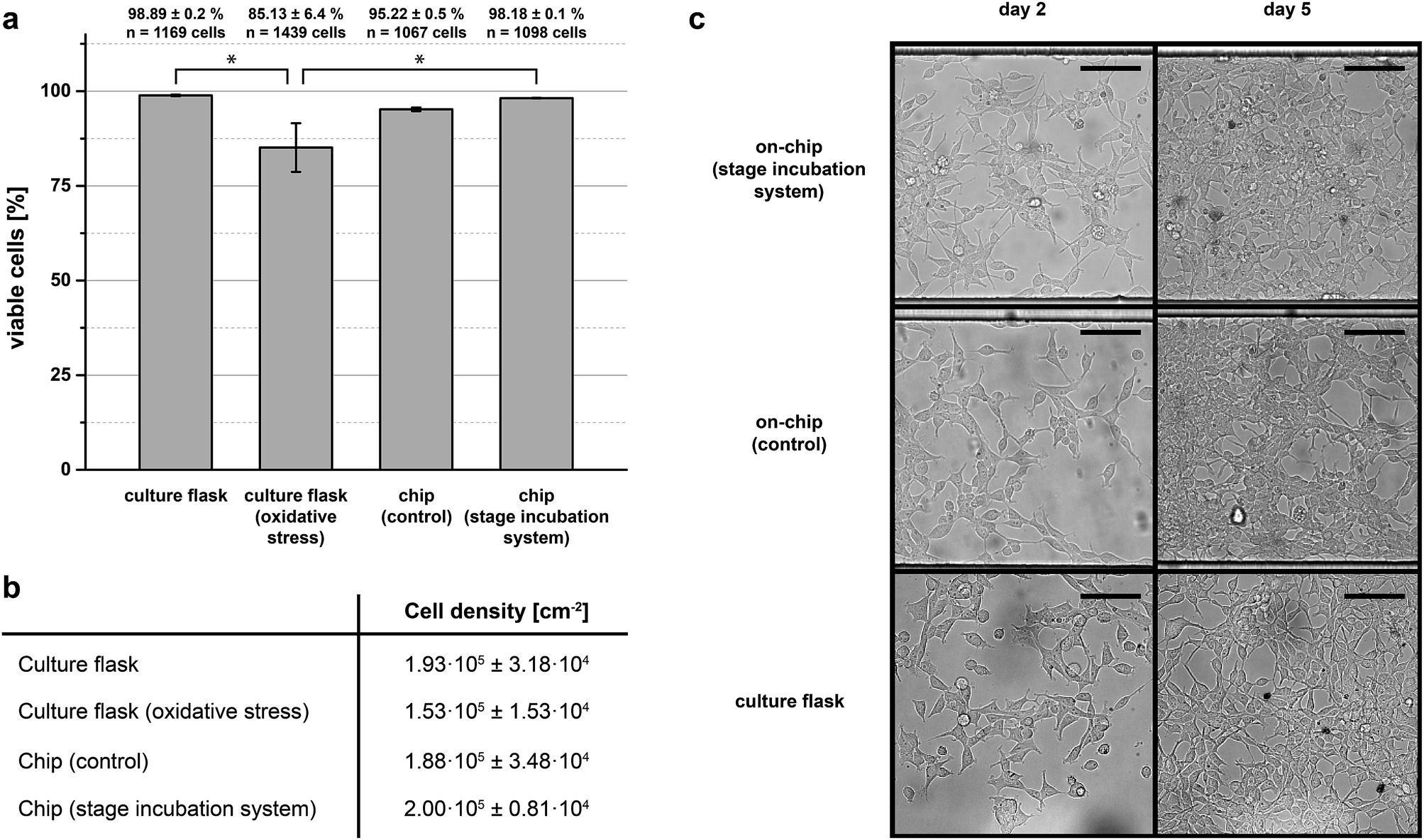 | ||
| Fig. 4 (a) HEK-293 viability after 5 days in culture for cells grown in conventional culture flasks and on-chip. Cells grown in conventional culture flasks or on-chip and maintained on stage exhibited similar values of viability, 98% and 99%, respectively. Cells having undergone oxidative stress show significantly reduced viability, 85% (*: p < 0.05). (b) Cell densities after 5 days in culture for all conditions demonstrate that cell growth is not limited in our on-stage incubation system. (c) Selected images of the cell cultures for on-chip and tissue flask cultures on day 2 and day 5 of the trials. Cell confluence is similar in all cases for each day. | ||
Conclusions
Here we present the design of an inexpensive perfusion incubation chamber for the cultivation of mammalian cells under regulated conditions to be combined with standard PDMS-based microfluidic devices. The incubation chamber is adaptable to any x–y stage of conventional inverted microscopes, but can also be used as a stand-alone cell incubation system; the system is portable as long as it is connected to a portable CO2 gas mixture flask. The presented concept is especially valuable for the observation of biological processes that are sensitive to buffer-containing media.Acknowledgements
Funding from the European Research Council under the 7th Framework Programme (ERC Starting Grant no. 203429 nμLIPIDS) is gratefully acknowledged. We thank H. Benz for construction of the electronic heating circuit, P. Kuhn for the fabrication of the master moulds and K. Eyer for fruitful discussions.References
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- D. G. Spiller, C. D. Wood, D. A. Rand and M. R. H. White, Nature, 2010, 465, 736–745 CrossRef CAS PubMed.
- A. Schmid, H. Kortmann, P. S. Dittrich and L. M. Blank, Curr. Opin. Biotechnol., 2010, 21, 12–20 CrossRef CAS PubMed.
- V. Lecault, A. K. White, A. Singhal and C. L. Hansen, Curr. Opin. Chem. Biol., 2012, 16, 381–390 CrossRef CAS PubMed.
- F. Kurth, K. Eyer, A. Franco-Obregón and P. S. Dittrich, Curr. Opin. Chem. Biol., 2012, 16, 400–408 CrossRef CAS PubMed.
- Y. Chen, P. Li, P.-H. Huang, Y. Xie, J. D. Mai, L. Wang, N.-T. Nguyen and T. J. Huang, Lab Chip, 2014, 14, 626–645 RSC.
- L. Kim, Y.-C. Toh, J. Voldman and H. Yu, Lab Chip, 2007, 7, 681–694 RSC.
- A. Tourovskaia, X. Figueroa-Masot and A. Folch, Lab Chip, 2005, 5, 14–19 RSC.
- E. Leclerc, B. David, L. Griscom, B. Lepioufle, T. Fujii, P. Layrolle and C. Legallaisa, Biomaterials, 2006, 27, 586–595 CrossRef CAS PubMed.
- M. Kirsch, E. E. Lomonosova, H.-G. Korth, R. Sustmann and H. de Groot, J. Biol. Chem., 1998, 273, 12716–12724 CrossRef CAS PubMed.
- J. Vriens, G. Appendino and B. Nilius, Mol. Pharmacol., 2009, 75, 1262–1279 CrossRef CAS PubMed.
- J. W. Hanrahan and J. A. Tabcharani, J. Membr. Biol., 1990, 116, 65–77 CrossRef CAS PubMed.
- J. S. Zigler Jr, J. L. Lepe-Zuniga, B. Vistica and I. Gery, In Vitro Cell. Dev. Biol., 1985, 21, 282–287 CAS.
- S. P. Forry and L. E. Locascio, Lab Chip, 2011, 11, 4041–4046 RSC.
- J. Vukasinovic, D. K. Cullen, M. C. LaPlaca and A. Glezer, Biomed. Microdevices, 2009, 11, 1155–1165 CrossRef PubMed.
- S. Giulitti, E. Magrofuoco, L. Prevedello and N. Elvassore, Lab Chip, 2013, 13, 4430–4441 RSC.
- D. F. Schaffhauser, O. Andrini, C. Ghezzi, I. C. Forster, A. Franco-Obregón, M. Egli and P. S. Dittrich, Lab Chip, 2011, 11, 3471–3478 RSC.
- B. Harink, S. Le Gac, D. Barata, C. van Blitterswijk and P. Habibovic, Lab Chip, 2014, 14, 1816–1820 RSC.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113 CrossRef CAS PubMed.
- W. Liu, L. Li, J.-C. Wang, Q. Tu, L. Ren, Y. Wang and J. Wang, Lab Chip, 2012, 12, 1702–1709 RSC.
- K. Eyer, P. Kuhn, C. Hanke and P. S. Dittrich, Lab Chip, 2012, 12, 765–772 RSC.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- P. Kuhn, J. Puigmarti-Luis, I. Imaz, D. Maspoch and P. S. Dittrich, Lab Chip, 2011, 11, 753–757 RSC.
- S. Crocetti, C. Beyer, G. Schade, M. Egli, J. Fröhlich and A. Franco-Obregón, PLoS One, 2013, 8, e72944 CAS.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- T. Benavides Damm, S. Richard, S. Tanner, F. Wyss, M. Egli and A. Franco-Obregón, FASEB J., 2013, 27, 2045–2054 CrossRef PubMed.
- S. Crocetti, C. Beyer, S. Unternährer, T. Benavides Damm, G. Schade-Kampmann, M. Hebeisen, M. Di Berardino, J. Fröhlich and A. Franco-Obregón, Cytometry, Part A, 2014, 85, 525–536 CrossRef PubMed.
- A. J. Wagers and I. M. Conboy, Cell, 2005, 122, 659–667 CrossRef CAS PubMed.
- A. Grossi, K. Yadav and M. A. Lawson, J. Biomech., 2007, 40, 3354–3362 CrossRef PubMed.
- X. Su, A. B. Theberge, C. T. January and D. J. Beebe, Anal. Chem., 2013, 85, 1562–1570 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed material and methods section including further referred to figures, CAD files of the incubation chamber and movie files of cells on-chip are available. See DOI: 10.1039/c4an01758g |
| This journal is © The Royal Society of Chemistry 2015 |