
Optimization and validation of a multiresidue method for pesticide determination in maize using gas chromatography coupled to tandem mass spectrometry
J. F.
Facco
,
M. L.
Martins
,
G.
Bernardi
,
O. D.
Prestes
,
M. B.
Adaime
and
R.
Zanella
*
Federal University of Santa Maria, Chemistry Department, Lab. of Pesticide Residue Analysis (LARP), 97105-900 Santa Maria-RS, Brazil. E-mail: rzanella@base.ufsm.br; Fax: +55 55 32208011; Tel: +55 55 32208011
First published on 14th October 2014
Abstract
A modified QuEChERS method was optimized for pesticide multiresidue extraction from maize for further GC-MS/MS determination. Extraction was conducted using a simultaneous hydration/extraction process. The clean-up step was performed using low temperature precipitation of lipids followed by dispersive solid-phase extraction (d-SPE). Low temperature precipitation at −18 °C during 12 hours enabled a reduction of co-extractives. Factorial experimental designs allowed the determination of optimum extraction and clean-up conditions. The proposed method was evaluated through the following analytical parameters: linearity, limits of detection (LOD) and quantification (LOQ), accuracy and precision. Results for the instrumental LOD and LOQ were 1.5 and 5.0 μg L−1, respectively, while the practical method LOQ was 40 or 100 μg kg−1 depending on the compounds. Recoveries from spike levels 40, 100 and 200 μg kg−1 were from 70 to 120% with RSD ≤ 20% for most compounds. Evaluation of the matrix effect was performed relating the areas of analytes in pure solvents to areas obtained from organic extracts. The results found were lower than 16.7%, confirming the efficiency of the proposed extraction and clean-up method.
1. Introduction
Maize, wheat and rice are the most important cereal grains in the world, representing 65% of the animal feed production and 15% of food production for humans. Maize is also used as a basic material for fuel production.1 During maize production and storage, grains can be damaged by a wide range of pests, decreasing grain quality and causing economic losses. Thus, the application of pesticides is a common practice to preserve quality and prolong storage life. Based on the toxicity of these compounds it is very important to determine the pesticide residues in crops such as maize. Due to this concern laws have been established in most countries to set maximum residue levels (MRLs) for pesticides in food.2 However, sample preparation techniques to determine pesticides at trace levels in maize still remain a challenge due to the complexity of the matrix. Different from fruits and vegetables, which have high water contents, the maize composition includes high contents of lipids, carbohydrates, amino acids, carotenoids, vitamins and starch.3This complexity becomes evident as co-extractives affect the extraction efficiency and instrument performance.4,5 Several methods have been developed in the past decade for multiresidue determination of pesticides in fatty matrices.6,7 Norman et al.8 evaluated supercritical fluid extraction (SFE) and solid phase extraction (SPE) techniques for extraction and clean-up, respectively, of wheat and maize samples for determination of organophosphate insecticides. Furthermore, a liquid–liquid extraction (LLE) using acetone–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and gel permeation chromatography (GPC) was evaluated. The analyses were conducted by gas chromatography with a flame photometric detector (GC-FPD) and gas chromatography coupled to mass spectrometry (GC-MS). The evaluated techniques showed good recovery results and low values for LODs. However, SFE with a SPE procedure time was 2.5 h and LLE with a GPC procedure time was 9 h. In addition, the first procedure uses less solvent than LLE with GPC. Using SFE with SPE, the authors obtained good results for recovery, in the range of 70 to 120%, with RSD from 12.8 to 13.5% at the different levels studied. Introduced by Anastassiades et al.9 the QuEChERS (Quick, Easy, Cheap, Rugged and Safe) method is based on partitioning with acetonitrile followed by a dispersive SPE clean-up. This method was developed for fruits and vegetables using small quantities of sorbents in the clean-up step.10–15 However, over the years the QuEChERS method was modified (e.g. buffering step,16,17 other extraction solvents,18 new sorbents,19 low temperature precipitation clean-up20 and others) and extracted a large number of different classes of pesticides and also other compounds such as mycotoxins,21 veterinary drugs,22 polycyclic aromatic hydrocarbons,23 dyes24 from different food and environmental matrices.25 QuEChERS has been shown to minimize co-extractives in the extraction of fatty matrices due to the low solubility of the lipids in acetonitrile.26 However, when the fatty content of the matrix is too high an additional clean-up procedure is still necessary prior to injection.27 Most extraction methods for fatty matrices employ organic solvents such as hexane, ethyl acetate, acetone and dichloromethane in order to dissolve the lipids. In this case, a clean-up step is required since lipids are extracted together with the analytes but this becomes expensive and time consuming.28 In this sense, the use of acetonitrile is a good alternative with high recoveries for a wide range of pesticide polarities with less matrix interferences extracted but a clean-up is still necessary, mainly when the GC system is employed. In recent years pesticide determination in maize has been conducted using different extraction techniques. More recently, Marchis et al.29 employed the QuEChERS method for the determination of organophosphorus insecticides and pyrethroids in raw maize samples by GC-MS. The obtained recoveries were in the range of 60 to 105% except for malathion. This technique is classified as simple, high throughput, utilizes a low amount of organic solvent and has been applied in combination with different extractions methods for residue and contaminant determination in different samples such as cereals, milk, eggs, oil, fruits and soil with good results.30 Marchis et al.29 and Walorczyk et al.4 applied a freezing-out step as a pre-clean-up. Since both extraction and clean-up methods require optimization, many authors have chosen the use of experimental designs aiming to obtain the best sample preparation conditions, with advantages such as the time and reagent economy.31 Considering the few existing studies reporting the determination of pesticides in maize1,8,32 the aim of this work was to reduce co-extractives using a modified QuEChERS method with low temperature precipitation and d-SPE, optimize the extraction conditions applying factorial experimental designs and determine pesticide residues of different classes in maize using triple quadrupole GC-MS/MS with high selectivity and sensitivity.
:1, v/v) and gel permeation chromatography (GPC) was evaluated. The analyses were conducted by gas chromatography with a flame photometric detector (GC-FPD) and gas chromatography coupled to mass spectrometry (GC-MS). The evaluated techniques showed good recovery results and low values for LODs. However, SFE with a SPE procedure time was 2.5 h and LLE with a GPC procedure time was 9 h. In addition, the first procedure uses less solvent than LLE with GPC. Using SFE with SPE, the authors obtained good results for recovery, in the range of 70 to 120%, with RSD from 12.8 to 13.5% at the different levels studied. Introduced by Anastassiades et al.9 the QuEChERS (Quick, Easy, Cheap, Rugged and Safe) method is based on partitioning with acetonitrile followed by a dispersive SPE clean-up. This method was developed for fruits and vegetables using small quantities of sorbents in the clean-up step.10–15 However, over the years the QuEChERS method was modified (e.g. buffering step,16,17 other extraction solvents,18 new sorbents,19 low temperature precipitation clean-up20 and others) and extracted a large number of different classes of pesticides and also other compounds such as mycotoxins,21 veterinary drugs,22 polycyclic aromatic hydrocarbons,23 dyes24 from different food and environmental matrices.25 QuEChERS has been shown to minimize co-extractives in the extraction of fatty matrices due to the low solubility of the lipids in acetonitrile.26 However, when the fatty content of the matrix is too high an additional clean-up procedure is still necessary prior to injection.27 Most extraction methods for fatty matrices employ organic solvents such as hexane, ethyl acetate, acetone and dichloromethane in order to dissolve the lipids. In this case, a clean-up step is required since lipids are extracted together with the analytes but this becomes expensive and time consuming.28 In this sense, the use of acetonitrile is a good alternative with high recoveries for a wide range of pesticide polarities with less matrix interferences extracted but a clean-up is still necessary, mainly when the GC system is employed. In recent years pesticide determination in maize has been conducted using different extraction techniques. More recently, Marchis et al.29 employed the QuEChERS method for the determination of organophosphorus insecticides and pyrethroids in raw maize samples by GC-MS. The obtained recoveries were in the range of 60 to 105% except for malathion. This technique is classified as simple, high throughput, utilizes a low amount of organic solvent and has been applied in combination with different extractions methods for residue and contaminant determination in different samples such as cereals, milk, eggs, oil, fruits and soil with good results.30 Marchis et al.29 and Walorczyk et al.4 applied a freezing-out step as a pre-clean-up. Since both extraction and clean-up methods require optimization, many authors have chosen the use of experimental designs aiming to obtain the best sample preparation conditions, with advantages such as the time and reagent economy.31 Considering the few existing studies reporting the determination of pesticides in maize1,8,32 the aim of this work was to reduce co-extractives using a modified QuEChERS method with low temperature precipitation and d-SPE, optimize the extraction conditions applying factorial experimental designs and determine pesticide residues of different classes in maize using triple quadrupole GC-MS/MS with high selectivity and sensitivity.
2. Materials and methods
2.1. Reagents, apparatus and samples
Analytical standards are listed in Table 1 and the internal standard (IS) triphenylphosphate was acquired from Dr Ehrenstorfer (Augsburg, Germany) with purity between 94.0 and 99.5%. As a surrogate standard (SS), isotopically modfied trifluralin-d14 (99.1%), purchased from C/N/D Isotopes (Quebec, Canada) was used. Acetonitrile HPLC grade was acquired from Mallinckrodt (Phillipsburg, USA). Anhydrous magnesium sulfate (MgSO4) and sodium chloride p.a. (NaCl) were from J. T. Baker (Phillipsburg, USA). Sorbents primary secondary amine (PSA) and octadecylsilane (C18), with 40 μm of particle size, were purchased from Agilent Technologies (Santa Clara, USA). Nylon filters of 13 mm and 0.2 μm of porosity were from Agilent Technologies (Santa Clara, USA). Ultrapurified water was obtained with a Milli-Q Direct UV3 system from Millipore (Billerica, USA). Vortex mixer model QL-901 Microtécnica (Curitiba, Brazil), analytical balances AUW-220D and UX-420H from Shimadzu (Kyoto, Japan), refrigerated centrifuge NT 825 Novatécnica (Piracicaba, Brazil), centrifuge Centribio, (Curitiba, Brazil), reciprocal shaker TE 240/I and mill TE633 Tecnal (Piracicaba, Brazil) were used. Measurements were carried out on a gas chromatograph CP 3800 coupled to a triple quadrupole mass spectrometer MS 1200 from Varian (Walnut Creek, USA). The system was equipped with an autosampler CP 8400; injector 1079 with 3.4 mm i.d. liner packing with carbofrit® plug Restek (Bellefonte, USA) and data acquisition software MS Workstation 6.9.2. Maize blank samples (1 kg) were obtained from organic production, processed in the mill and then stored at −20 °C. Samples (1 kg) were processed in the mill and then stored in 200 g polypropylene containers before extraction.| Compounds | MRL (mg kg−1) | 1st transition (quantification) | 2nd transition (confirmation) | ||
|---|---|---|---|---|---|
| EU | Brazil | ||||
| a SS: surrogate standard; IS: internal standard; *: collision energy, V. | |||||
| 1 | Trifluralin d-14 (SS) | — | — | 315 → 267 (8)* | 315 → 209 (10) |
| 2 | Trifluralin | 0.05 | 0.05 | 306 → 264 (10) | 306 → 206 (15) |
| 3 | Quintozene | 0.02 | — | 295 → 237 (10) | 295 → 265 (10) |
| 4 | Alachlor | 0.01 | 0.2 | 188 → 130 (10) | 188 → 160 (10) |
| 5 | Pirimiphos-methyl | 5.0 | 10.0 | 290 → 151 (15) | 290 → 180 (10) |
| 6 | Fenitrothion | 0.05 | 0.2 | 277 → 109 (25) | 277 → 260 (10) |
| 7 | Malathion | 8.0 | 8.0 | 173 → 99 (15) | 173 → 127 (10) |
| 8 | Chlorpyrifos-ethyl | 0.05 | 0.1 | 314 → 258 (15) | 314 → 286 (15) |
| 9 | Trifloxystrobin | 0.02 | 0.05 | 222 → 162 (10) | 222 → 190 (5) |
| 10 | Triphenylphosphate (IS) | — | — | 325 → 169 (18) | 325 → 226 (18) |
| 11 | Bifenthrin | 0.05 | 0.02 | 181 → 165 (20) | 181 → 166 (10) |
| 12 | Tetradifon | 0.01 | — | 229 → 201 (15) | 229 → 199 (15) |
| 13 | Esfenvalerate | 0.02 | 1.0 | 225 → 91 (25) | 225 → 147 (10) |
| 14 | Deltamethrin | 2.0 | 1.0 | 253 → 174 (10) | 253 → 172 (5) |
2.2. GC-MS/MS conditions
Separation was achieved on a CP-Sil 8 CB Low bleed/MS capillary column (30 m × 0.25 mm i.d. and 0.25 μm of film thickness). Aliquots of 2 μL of the final clean extract were injected in splitless mode with the injector at 280 °C. Helium with a purity of 99.9999% (Air Products, São Paulo, Brazil) was used as the carrier gas. The oven temperature program was initially 45 °C (1.0 min) and then was increased at 30 °C min−1 to 280 °C and maintained for 4.1 min. Finally, the temperature was increased at 20 °C min−1 to 300 °C, resulting in a final run time of 17 min. The transfer line temperature was set at 250 °C and the ion source was electron impact ionization set at 70 eV and 210 °C. The collision gas for MS/MS experiments was argon (99.9992%) (Air Products, São Paulo, Brazil). The MS/MS system was operated in Selected Reaction Monitoring (SRM) mode using one transition for quantification and another for confirmation as shown in Table 1.2.3. Sample preparation procedure
Extraction of pesticides from maize samples was conducted using a modified QuEChERS method based on Mastovska et al.33 An aliquot (2.5 g) of the maize sample was submitted to simultaneous hydration/extraction process. In order to evaluate the accuracy of the method, blank samples were spiked at 40, 100 and 200 μg kg−1 using a stock solution containing all the studied pesticides. For the extraction step, 10 mL of purified water were added to the sample in a 50 mL polypropylene tube. After 1 min of agitation using a vortex, 10 mL of acetonitrile were added to the tube that was agitated in a reciprocal shaker (200 rpm) for 1 h. Addition of water makes sample pores more accessible to the extraction solvent promoting the swelling of the matrix and allow better recoveries of the analytes.33,34 After this, NaCl (1 g) and MgSO4 (6 g) were added to the tube that was shaken in a vortex for 1 min and then centrifuged (3400 rpm) for 8 min at 10 °C. At this point, the low-temperature clean-up step was performed prior to dispersive-SPE. For this, 4 mL of the supernatant was transferred into a 15 mL polypropylene tube and stored for at least 12 h in a freezer (−10 to −20 °C). Co-extractives, mainly fatty acids, were then separated from the extract by simple decantation, since during these steps the fatty acid portion present in the matrix settles to the bottom of the tube. After that, an aliquot of 1 mL of the supernatant (organic phase) was transferred to a polypropylene tube of 15 mL, containing 150 mg of MgSO4, 50 mg of PSA and 50 mg of C18 for the clean-up step by d-SPE. Tubes were centrifuged for 8 min at 3400 rpm and an aliquot of the supernatant was filtered (nylon filter 0.2 μm) and transferred to an auto sampler vial for GC-MS/MS analysis. To monitor errors during sample preparation, a surrogate standard (trifluralin-d14) was added before extraction to all samples at 100 μg kg−1. Fig. 1 shows the proposed sample preparation procedure.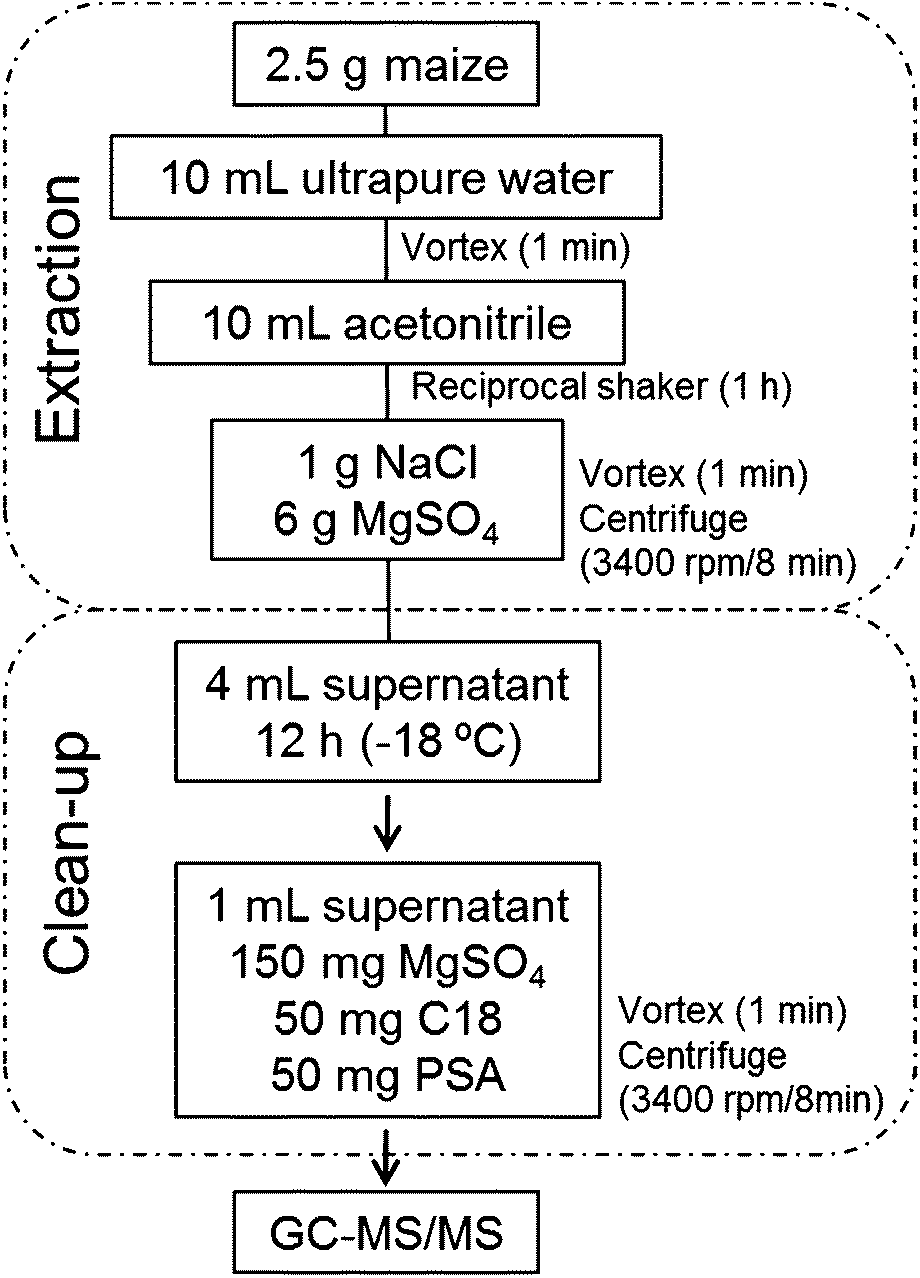 | ||
| Fig. 1 Flowchart of the validated procedure, showing the steps involved in the extraction of the maize samples. | ||
2.4. Experimental design
| Test | mg C18 (coefficients) | mg PSA (coefficients) |
|---|---|---|
| 1 | 26 (−1) | 26 (−1) |
| 2 | 150 (+1) | 26 (−1) |
| 3 | 26 (−1) | 150 (+1) |
| 4 | 150 (+1) | 150 (+1) |
| 5 | 88 (0) | 88 (0) |
| 6 | 88 (0) | 88 (0) |
| 7 | 88 (0) | 88 (0) |
| 8 | 0 (−α = √2) | 88 (0) |
| 9 | 88 (0) | 176 (+α = √2) |
| 10 | 176 (+α = √2) | 88 (0) |
| 11 | 88 (0) | 0 (−α = √2) |
The choice of the quantities was made to use only PSA or C18 in the experiments 8 and 11, respectively, so that the mathematical model represented by the response surface can show with more evidence the effect of one or another sorbent. The number of compounds with recovery between 70 and 120% was the response of the model.
2.5. Method performance
The modified QuEChERS method was validated in order to evaluate the performance of the method for sample preparation and quantification by GC-MS/MS. Linearity was evaluated using calibration curves in concentrations of 5, 10, 25, 50, 100 and 200 μg L−1, in acetonitrile and in matrix blank extract, and method accuracy and precision at the spike levels of 40, 100 and 200 μg kg−1, with seven replicates each (n = 7). To evaluate matrix effects, the slopes obtained in the calibration with matrix-matched standards were compared with those obtained with solvent-based standards, calculating matrix/solvent slope ratios for each pesticide.36 The instrumental limits, LODi and LOQi, were estimated using the signal-to-noise ratio considering the concentration in matrix-matched calibration solutions injected into GC-MS/MS that resulted in ratios of 3 and 10, respectively, for LODi and LOQi. The limits of detection and quantification of the method (LODm and LOQm) were established experimentally. The value of LOQm, in μg kg−1, was established as the lowest concentration of the spiked blank matrix that provided recoveries between 70 and 120% with RSD% ≤ 20%. The LODm was calculated dividing the LOQm by 3.3. Accuracy was expressed as recovery (%) and precision in terms of repeatability (RSDr) and due to the complexity of the studied matrix, the same experiment with three spiked levels was repeated on another day in order to estimate the intermediate precision (RSDip).372.6. Method applicability
To evaluate the applicability of the proposed method, a total of 11 commercial samples were obtained from markets and rural supply stores in the city of Santa Maria-RS (Brazil) and were analyzed applying the proposed method.3. Results and discussion
3.1. Chromatographic determination
The optimized GC conditions allowed the multiresidue determination of the 14 analytes in a single run of 17 min. Fig. 2 shows the GC-MS/MS TIC chromatogram obtained with the conditions described in the Section 2.2 with a blank sample spiked at 100 μg kg−1.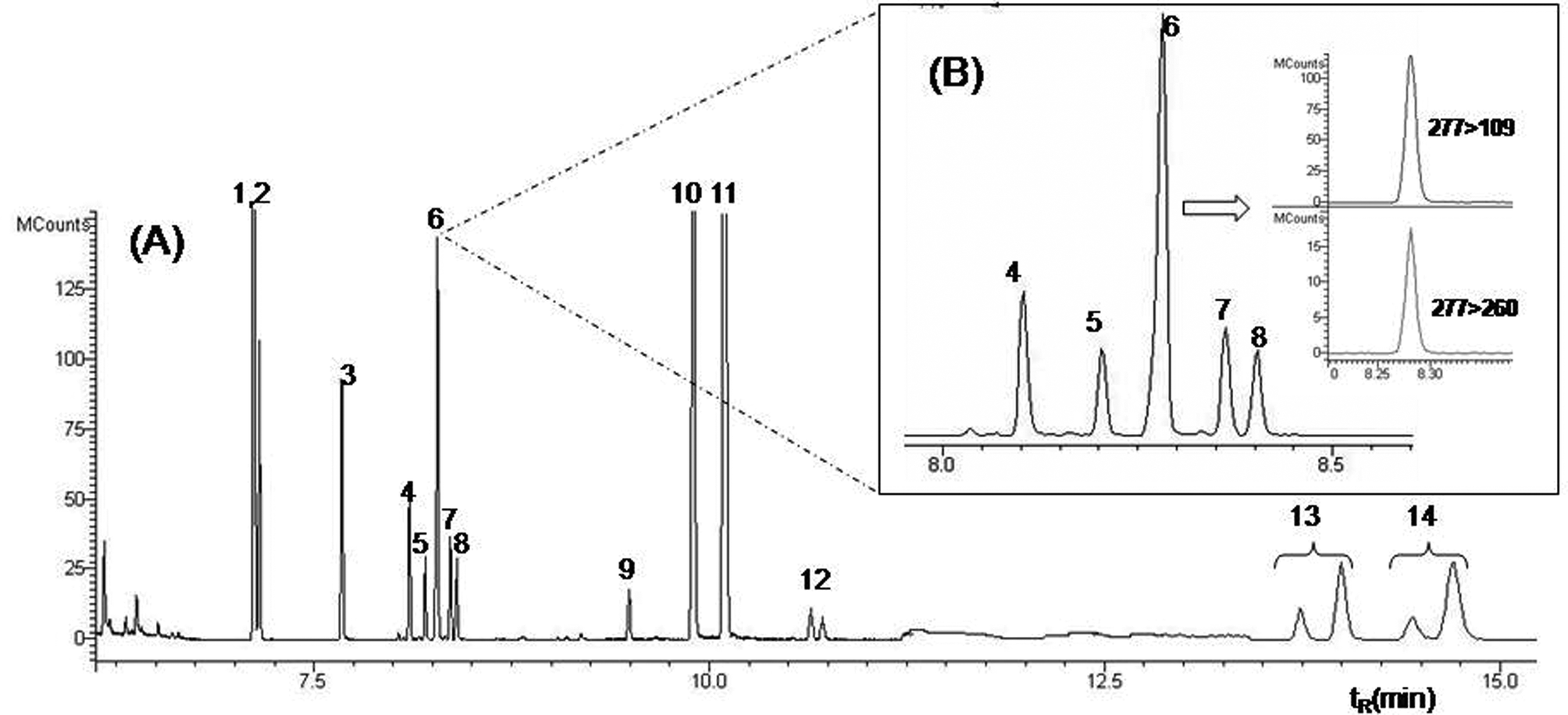 | ||
| Fig. 2 GC-MS/MS chromatogram of a blank sample spiked at 100 μg kg−1, highlighting the quantification and confirmation ions for the pesticide fenitrothion. The numbers of identification of each peak is presented in Table 1 | ||
3.2. Optimization of the extraction and clean-up procedure
As shown in Fig. 3, changing factor 1 (agitation time) from 15 to 60 min, factor 3 (cooling time) from 6 to 12 h and factor 4 (temperature) from −10 to −20 °C to −20 to −30 °C had no significant effect (p = 0.05) on the number of compounds with acceptable recovery. However, two-way interactions, 1 by 3 and 1 by 4, presented positive effects, indicating some relationships between agitation times during extraction and time/temperature of the freezing-out step. A strong negative effect was observed for factor 2, by changing from C18 to PSA. The sorbent C18 removes long chain fatty compounds, sterols and other nonpolar interferences and PSA removes sugars, fatty acids, organic acids, lipids and pigments.26 The high lipidic content in maize can explain this result. When PSA is used in combination with C18, additional lipids and sterols can be removed, therefore the use of both sorbents was investigated in method development. The two-way interaction 2 by 4 (PSA as a sorbent in the temperature range of −20 to −30 °C) also presented a strong negative effect. As both factors are associated with clean-up, this negative effect can be explained by excessive removal of analytes during the clean-up step. The remaining interactions had no significant effect (p = 0.05). Thus, the final conditions adopted were: agitation time of 1 h (better extraction), cooling time of 12 h (improved recoveries associated with 1 h of agitation) at the temperature range of −10 to −20 °C. Since the cooling step mainly promotes the removal of lipids but cannot provide sufficient removal of other co-extractives, an additional clean-up experimental design, focused on co-extractives arising from maize blank samples, was carried out to optimize the amounts of C18 and PSA sorbents. | ||
| Fig. 3 Pareto chart for the effect of single variables and two way interactions for the 24 full factorial design, where the column bars represent the effect (y) caused by one change in factors 1, 2, 3 and 4 or by the two way interactions between them (x by y). | ||
The interactions among variables studied by CCD are shown in Fig. 4, where the number of compounds with recovery between 70 and 120% is related to the sorbent amounts used. Two regions with maximum responses are presented in the response surface, one for high amounts of sorbents (near to 200 mg for C18 and PSA) and another region near the minimum amount of sorbents. The high response with maximum sorbent amounts agrees with the temperature range selected in the previous step to avoid excessive clean-up. The adopted procedure allowed optimization of sorbent amounts for the best clean-up conditions, since the low temperature range associated with high sorbent amounts enables removal of analytes from extracts. Lower quantities or the absence of sorbents could lead to more effective recovery, but may also affect the robustness of the method because of the excess of co-extractives remaining in sample extracts, leading to instrumental variability during routine applications.
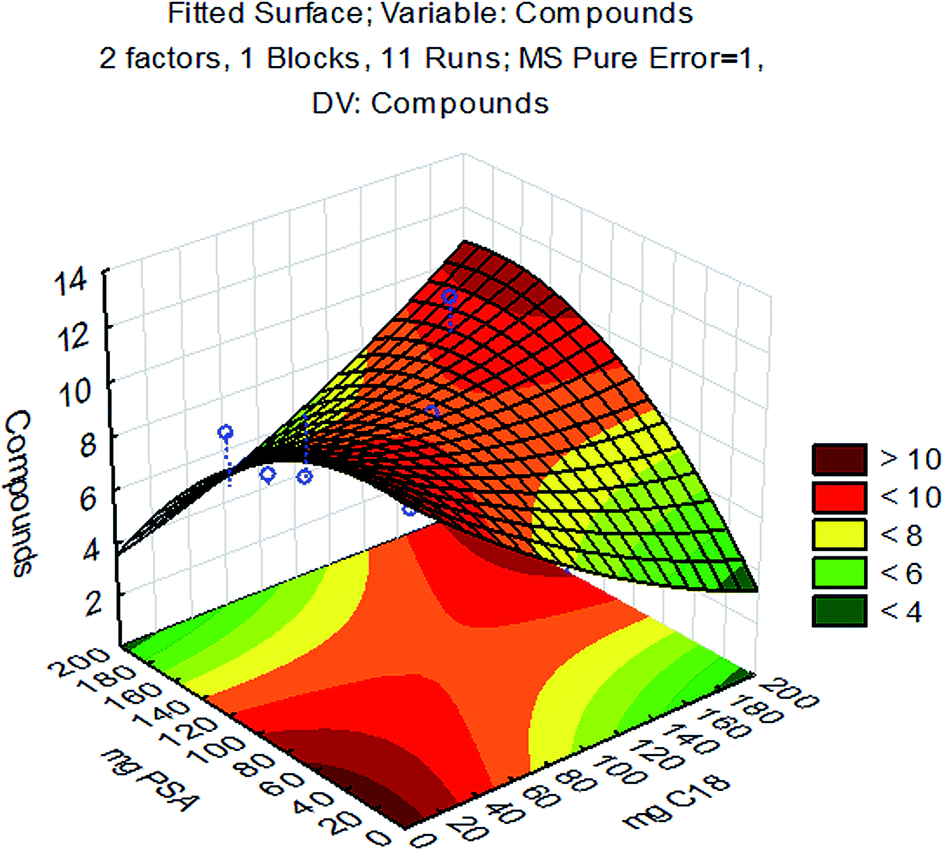 | ||
| Fig. 4 Response surface for PSA and C18 obtained by CCD. X and Y: the amount of sorbents and Z: the number of compounds recovered from 70 to 120%. | ||
Thus, some amount of sorbents and consequently more clean-up are recommended. However, high amounts of sorbents represent high analytical costs and being that for complex matrices some clean-up is desirable, 50 mg of PSA and 50 mg of C18 were selected for method validation. The need for a clean-up step is clear, but particularly, the need for an appropriate ratio of sorbents used, since this will define the method efficiency.
3.3. Method validation
Validation parameters were evaluated and selectivity was confirmed since no interferences were observed in the blank extract when compared with a spiked maize sample. Analytical curves were obtained for the range 5 to 200 μg L−1 and good linearity was observed with r2 higher than 0.992 for all the studied compounds. The LODi and LOQi values were established in both pure solvent solution and in the matrix extract. The values obtained for LODi and LOQi were 1.5 and 5.0 μg L−1, respectively. As presented in Table 3, for the compounds alachlor, esfenvalerate, malathion, pirimiphos-methyl and trifloxystrobin, the LODm and LOQm were 12 and 40 μg kg−1, respectively. These compounds presented recoveries in the range of 70 to 120%, with RSD < 20%. LODm and LOQm values for bifenthrin, chlorpyrifos-ethyl, deltamethrin, fenitrothion, quintozene, tetradifon and trifluralin were 30 and 100 μg kg−1, respectively. Therefore, the LODm and LOQm values for most of pesticides reached the MRL established in Brazil, except for bifenthrin and trifluralin. The LOQ values for these compounds may be further reduced by the concentration of the final extract. Moreover, it is important to confirm if this step will not compromise the analytical response due to noise increase. The results of accuracy, evaluated through recovery tests, and of precision in intraday and interday assays are shown in Table 3. Recovery values for the three concentration levels ranged between 55.3 and 114.4%. Good precision values were observed for the pesticides with RSD below 20%.| Compounds | Spike levels, μg kg−1 | LOQm μg kg−1 | LODm μg kg−1 | ME % | |||
|---|---|---|---|---|---|---|---|
| 40 | 100 | 200 | 100 | ||||
| Rec ± RSD (%), intraday | Rec ± RSD (%), interday | ||||||
| a n = 7; Rec = recovery; ME = matrix effect. | |||||||
| Alachlor | 94.7 ± 17.8 | 114.4 ± 16.0 | 99.5 ± 16.8 | 101.4 ± 7.1 | 40 | 12 | −6.1 |
| Bifenthrin | 68.1 ± 5.9 | 88.0 ± 12.4 | 75.0 ± 5.1 | 75.6 ± 8.7 | 100 | 30 | 4.2 |
| Chlorpyrifos-ethyl | 62.4 ± 5.9 | 90.1 ± 13.7 | 77.5 ± 10.6 | 89.3 ± 10.3 | 100 | 30 | −6.7 |
| Deltamethrin | 59.9 ± 8.0 | 99.1 ± 11.9 | 94.4 ± 18.1 | 89.5 ± 8.3 | 100 | 30 | 16.7 |
| Esfenvalerate | 71.3 ± 8.8 | 104.0 ± 13.2 | 94.0 ± 16.7 | 79.7 ± 6.2 | 40 | 12 | 1.7 |
| Fenitrothion | 61.6 ± 9.4 | 107.3 ± 16.8 | 93.1 ± 9.1 | 97.8 ± 20.0 | 100 | 30 | −8.5 |
| Malathion | 74.9 ± 9.0 | 106.8 ± 12.8 | 97.2 ± 9.4 | 96.0 ± 5.9 | 40 | 12 | −9.6 |
| Pirimiphos-methyl | 91.9 ± 13.8 | 106.1 ± 16.9 | 95.9 ± 7.0 | 78.7 ± 13.7 | 40 | 12 | −11.4 |
| Quintozene | 55.3 ± 6.0 | 87.0 ± 12.5 | 90.1 ± 18.3 | 77.5 ± 4.0 | 100 | 30 | −9.6 |
| Tetradifon | 61.1 ± 7.5 | 92.3 ± 15.6 | 75.1 ± 6.3 | 73.6 ± 5.5 | 100 | 30 | −8.3 |
| Trifloxystrobin | 93.9 ± 18.0 | 110.1 ± 14.1 | 90.2 ± 6.4 | 98.7 ± 11.2 | 40 | 12 | −7.8 |
| Trifluralin | 65.8 ± 5.8 | 102.5 ± 13.0 | 102.3 ± 17.3 | 91.0 ± 5.2 | 100 | 30 | −14 |
In the last few years, different methods have been published in the literature about determination of pesticide residues in food matrices. However, only a few approaches present the determination of these compounds in maize samples. For example, Norman & Panton8 developed an automated method using supercritical CO2 and clean-up by solid phase extraction employing graphitized carbon black for determination of 10 organophosphate pesticide residues in maize. Lower LODs (7.0 to 53 μg kg−1) were possible because the extraction procedure used 25 g of sample and more than 200 mL of organic solvents. Cunha & Fernandes1 for determination of 41 pesticide residues in maize, combined the QuEChERS method and dispersive liquid–liquid microextraction (DLLME) using carbon tetrachloride as the extractive solvent and the extract obtained by QuEChERS as the dispersive solvent. The DLLME procedure effectively provided an enrichment of the extract and low LOQs were obtained (12.5 to 25 μg kg−1), however the use of a chlorinated solvent is required. In our proposed method, an easy approach was developed. The extraction step uses 2.5 g sample and only 10 mL of acetonitrile, and the clean-up was performed by low temperature precipitation followed by d-SPE. All the steps were easy to perform and the procedure was also environmental friendly. These characteristics make the method feasible and applicable in the routine practices by any laboratory.
3.4. Matrix effects
The matrix effect is directly related to the concentration of the pesticide present in the extract, especially at lower concentrations if the matrix/analyte ratio is too high.38 In the present study, the matrix effect was not significant since the values were lower than ±20%.39 These results indicate the optimized extraction and clean-up procedures were satisfactory. However, even after optimization, the method may be subject to variations as shown in Table 3. Most compounds had a negative matrix effect characterizing a suppression of the chromatographic signal of the analytes based on the matrix complexity.4 In order to maintain the matrix effect controlled, curves in matrix extract were used during the optimization of the method as well as in the method applications.383.5. Real samples
The developed method was applied to the determination of pesticide residues in 11 maize commercial samples. Among the analyzed samples, two samples showed pirimiphos-methyl in a concentration <LOQm and one sample in a concentration of 80 μg kg−1. The results found were below the MRLs established by Brazilian (10 mg kg−1)40 and EU (5 mg kg−1)41 legislations. No residues of the studied compounds were found in the other samples.4. Conclusions
The optimized QuEChERS method employing a clean-up step at a low temperature followed by d-SPE proved to be adequate for the determination of pesticides in maize samples, since it reduced the co-extractives present in the extract and decreased the quantities of sorbents necessary for the clean-up step. Using GC-MS/MS in the SRM mode allowed a very efficient quantitative and qualitative analysis with high sensitivity and selectivity. The use of an experimental design allowed for the obtainment of optimum conditions for analyte extraction and matrix clean-up with fewer optimization assays, making this step faster and cheaper.Acknowledgements
The authors gratefully acknowledge the financial support and fellowship grants from CNPq, CAPES, FINEP/SIBRATEC, Brazil.References
- S. C. Cunha and J. O. Fernandes, J. Chromatogr. A, 2011, 1218, 7748–7757 CrossRef CAS PubMed.
- U. Koesukwiwat, S. J. Lehotay, K. Mastovska, K. J. Dorweiler and N. Leepipatpiboon, J. Agric. Food Chem., 2010, 58, 5950–5958 CrossRef CAS PubMed.
- D. P. Chaudhary, S. Kumar and O. P. Yadav, Nutritive Value of Maize: Improvements, Applications and Constraints, in Maize: Nutrition Dynamics and Novel Uses, ed. D. P. Chaudhary, S. Kumar and S. Singh, Springer India, New Delhi, 2014, pp. 3–17 Search PubMed.
- S. Walorczyk and D. Drożdżyński, J. Chromatogr. A, 2012, 1251, 219–231 CrossRef CAS PubMed.
- L. Zhang, S. Liu, X. Cui, C. Pan, A. Zhang and F. Chen, Cent. Eur. J. Chem., 2012, 10, 900–925 CrossRef CAS PubMed.
- S. J. Lehotay, K. Mastovská and S. J. Yun, J. AOAC Int., 2005, 88, 630–638 CAS.
- B. Gilbert-López, J. F. García-Reyes and A. Molina-Díaz, Talanta, 2009, 79, 109–128 CrossRef PubMed.
- K. N. T. Norman and S. H. W. Panton, J. Chromatogr. A, 2001, 907, 247–255 CrossRef CAS.
- M. Anastassiades, S. J. Lehotay, D. Štajnbaher and F. J. Schenck, J. AOAC Int., 2003, 86, 412–431 CAS.
- W. Zhang, J. Xu, F. Dong, X. Liu, Y. Zhang, Y. Tao, X. Wuand and Y. Zheng, Anal. Methods, 2013, 5, 7102–7109 RSC.
- Y. Zhang, J. Xu, F. Dong, X. Liu, X. Li, Y. Li, X. Wu, X. Liang and Y. Zheng, Anal. Methods, 2013, 5, 1449–1455 RSC.
- L. Cherta, T. Portolés, J. Beltran, E. Pitarch, J. G. J. Mol and F. Hernández, J. Chromatogr. A, 2013, 1314, 224–240 CrossRef CAS PubMed.
- X. Wu, J. Xu, F. Dong, X. Liu, Y. Li and Y. Zheng, J. Chromatogr. A, 2014, 1329, 30–37 CrossRef CAS PubMed.
- A. R. Restrepo, A. F. G. Ortiz, D. E. H. Ossa and G. A. P. Mesa, Food Chem., 2014, 156, 153–161 CrossRef PubMed.
- M. D. H. Prodhan, E. N. Papadakis and E. Papadopoulou-Mourkidou, Food Anal. Methods, 2014 DOI:10.1007/s12161-014-9898-3.
- S. J. Lehotay, K. Mastovska and A. R. Lightfield, J. AOAC Int., 2005, 88, 615–629 CAS.
- M. Andrascikovaand and S. Hrouzkova, Anal. Methods, 2013, 5, 1374–1384 RSC.
- S. K. Sahoo, R. S. Battu and B. Singh, Am. J. Anal. Chem., 2011, 2, 26–31 CrossRef.
- M. B. R. Cerqueira, S. S. Caldas and E. G. Primel, J. Chromatogr. A, 2014, 1336, 10–22 CrossRef CAS PubMed.
- E. Sobahnzadeh, N. K. A. Bakar, M. R. B. Abas and K. Nemati, Environ. Monit. Assess., 2012, 184, 5821–5828 CrossRef PubMed.
- P. Yogendrarajah, C. V. Poucke, B. D. Meulenaer and S. D. Saeger, J. Chromatogr. A, 2013, 1297, 1–11 CrossRef CAS PubMed.
- J. Kang, C. L. Fan, Q. Y. Chang, M. N. Bu, Z. Y. Zhao, W. Wang and G. F. Pang, Anal. Methods, 2014, 6, 6285–6293 RSC.
- A. S. Rociek, M. Surma and E. Cieslik, Bull. Environ. Contam. Toxicol., 2013, 90, 508–513 CrossRef PubMed.
- W. Jia, X. Chu, Y. Ling, J. Huang, Y. Lin and J. Chang, J. Sep. Sci., 2014, 37, 782–791 CrossRef CAS PubMed.
- V. Fernandes, V. F. Domingues, N. Mateus and C. D. Matos, J. Sep. Sci., 2013, 36, 376–382 CrossRef CAS PubMed.
- S. J. Lehotay, K. Mastvoska and S. J. Yun, J. AOAC Int., 2005, 88, 630–638 CAS.
- H. R. Norli, A. Christiansen and E. Deribe, J. Chromatogr. A, 2011, 1218, 7234–7241 CrossRef CAS PubMed.
- S. J. Lehotay, A. Kok, M. Hiemstra and P. V. Bodegraven, J. AOAC Int., 2005, 88, 595–614 CAS.
- D. Marchis, G. L. Ferro, P. Brizio, S. Squadrone and M. C. Abete, Food Control, 2012, 25, 270–273 CrossRef CAS PubMed.
- P. Payá, M. Anastassiades, D. Mack, I. Sigalova, B. Tasdelen, J. Oliva and A. Barba, Anal. Bioanal. Chem., 2007, 389, 1697–1714 CrossRef PubMed.
- M. A. Bezerra, R. E. Santelli, E. P. Oliveira, L. S. Villar and L. A. Escaleira, Talanta, 2008, 76, 965–977 CrossRef CAS PubMed.
- A. M. Botero-Coy, M. Ibáñez, J. V. Sancho and F. Hernández, J. Chromatogr. A, 2013, 25, 157–165 CrossRef PubMed.
- K. Mastovska, K. J. Dorweiler, S. J. Lehotay, J. S. Wegscheid and K. A. Szpylka, J. Agr. Food Chem., 2010, 58, 5959–5972 CrossRef CAS PubMed.
- D. I. Kolberg, O. D. Prestes, M. B. Adaime and R. Zanella, Food Chem., 2011, 125, 1436–1442 CrossRef CAS PubMed.
- B. B. Neto, I. S. Scarminio and R. E. Bruns, Como fazer experimentos, Bookman, 4a edn, Porto Alegre, 2010 Search PubMed.
- I. R. Pizzutti, A. Kok, M. Hiemstra, C. Wickert and O. D. Prestes, J. Chromatogr. A, 2009, 1216, 4539–4552 CrossRef CAS PubMed.
- DG-SANCO, EUROPEAN COMISSION. Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. Document No. SANCO/12571/2013, 2013.
- G. P. Pinho, A. A. Neves, M. E. L. R. Queiroz and F. O. Silvério, Quím. Nova, 2009, 32, 987–995 CrossRef CAS PubMed.
- C. Ferrer, A. Lozano, A. A. Aguera, G. Jiménez and A. R. Fernández–Alba, J. Chromatogr. A, 2011, 1218, 7634–7639 CrossRef CAS PubMed.
- Agência Nacional de Vigilância Sanitária (ANVISA), Resolução RE No 2838 (2013) de 06/08/13, Diário Oficial da União, 157, 37.
- European Council (2005). Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Off. J. Eur. Union L70/1, p. 1–141.
| This journal is © The Royal Society of Chemistry 2015 |