
Heparin-induced conformational changes of fibronectin within the extracellular matrix promote hMSC osteogenic differentiation†
Bojun
Li
,
Zhe
Lin
,
Maria
Mitsi
,
Yang
Zhang
and
Viola
Vogel
*
Department of Health Sciences and Technology, ETH Zurich, Ch-8093 Zurich, Switzerland. E-mail: viola.vogel@hest.ethz.ch; Fax: +41 44 632 10 73; Tel: +41 44 632 08 87
First published on 14th August 2014
Abstract
An increasing body of evidence suggests important roles of extracellular matrix (ECM) in regulating stem cell fate. This knowledge can be exploited in tissue engineering applications for the design of ECM scaffolds appropriate to direct stem cell differentiation. By probing the conformation of fibronectin (Fn) using fluorescence resonance energy transfer (FRET), we show here that heparin treatment of the fibroblast-derived ECM scaffolds resulted in more extended conformations of fibrillar Fn in ECM. Since heparin is a highly negatively charged molecule while fibronectin contains segments of positively charged modules, including FnIII13, electrostatic interactions between Fn and heparin might interfere with residual quaternary structure in relaxed fibronectin fibers thereby opening up buried sites. The conformation of modules FnIII12–14 in particular, which contain one of the heparin binding sites as well as binding sites for many growth factors, may be activated by heparin, resulting in alterations in growth factor binding to Fn. Indeed, upregulated osteogenic differentiation was observed when hMSCs were seeded on ECM scaffolds that had been treated with heparin and were subsequently chemically fixed. In contrast, either rigidifying relaxed fibers by fixation alone, or heparin treatment without fixation had no effect. We hypothesize that fibronectin's conformations within the ECM are activated by heparin such as to coordinate with other factors to upregulate hMSC osteogenic differentiation. Thus, the conformational changes of fibronectin within the ECM could serve as a ‘converter’ to tune hMSC differentiation in extracellular matrices. This knowledge could also be exploited to promote osteogenic stem cell differentiation on biomedical surfaces.
Introduction
Human mesenchymal stem cells (hMSCs) have become an attractive cell source for bone tissue-engineering applications,1 since a variety of signals can induce their osteogenic differentiation,2,3 and thereby promote bone healing and remodeling in both animal4 and human models.5 Adhesion of hMSCs to biomaterials is mediated by extracellular matrix (ECM) proteins and the nature of these adsorbed ECM proteins can have versatile effects on the differentiation pathways of hMSCs.6,7 Since collagen type I (Col-I) and glycosaminoglycans (GAG), including hyaluronan, are important components of the bone (ECM), it is not unexpected that the initial adhesion of hMSCs to collagen type I coated biomaterials promotes hMSC osteogenesis.8 Osteogenic differentiation of human mesenchymal stromal cells was also enhanced in sulfated hyaluronan containing collagen matrices.9,10Less attention has been given to investigating the role of Fn and its contributions to attracting hMSCs and to promoting bone healing. Fn is a major ECM protein and found to be crucial for the assembly and integrity of collagen matrix,11,12 whereby the latter comprises up to 90% of the total protein within the skeleton.13,14 In addition, Fn has been localized in the periosteum of rat calvaria15 and in the osteoid surrounding implants.16 These studies raise the question whether Fn could guide early stages of osteogenic differentiation. In vitro studies have revealed that the effects of Fn on hMSC physiology are complex: Fn-coated biomaterials promoted hMSC attachment17,18 and culture of hMSCs on Fn-coated surfaces promoted their migration, adhesion and proliferation, however, it did not affect their osteogenic differentiation.7 Several studies suggest that Fn's conformation is an important factor for regulating the osteogenic differentiation of osteoblast-like cells and hMSCs.19–23 Mechanical forces24–28 and interactions with heparin,29,30 both of which have been reported to influence Fn conformations might thus tune the osteogenic differentiation potential of hMSCs.
Heparin is a highly sulfated glycosaminoglycan belonging to the heparin sulfate family and it has high negative charge density.31–33 Heparin is naturally produced by mast cells where it serves as an inhibitor of the proteases contained within these cells, and is secreted upon mast cell stimulation.34,35 Heparins have traditionally been used in the clinic as anticoagulants, but over the years additional therapeutic and biological functions of heparins have emerged.36,37 An important aspect of heparin biology is the ability of the glycosaminoglycan chains to interact with numerous proteins including growth factors and molecules of the extracellular matrix,38 which suggests a role for heparin in tissue engineering applications.39–41 Fn contains at least two heparin binding sites,42–45 one of which serves also as a promiscuous binding site for many growth factors.46 Of particular interest for hMSCs differentiation is the ability of heparin to interact with numerous proteins that are associated with hMSC adhesion (e.g. Fn and vitronectin),47,48 proliferation (e.g. bFGF)49 and osteogenic differentiation (e.g. bone morphogenetic proteins (BMPs)).50 It has been reported that heparin-functionalized 2D or 3D hydrogels increase hMSC osteogenic differentiation,51,52 whereas continuous treatment of MSCs with heparin in the medium inhibits MSC osteogenesis.53 However, the mechanisms by which immobilization of heparin can regulate these effects on osteogenesis are not well understood.
Recent studies have shown that heparin induces a conformational change of surface-adsorbed Fn, from a compactly folded quaternary structure to a more extended configuration.29 In the more extended conformation, Fn exposes cryptic binding sites for vascular endothelial growth factor (VEGF)54 that remained available even after the removal of heparin.30 Since it has been reported that VEGF has positive effects on hMSC osteogenesis,55 and the exposed binding site can interact with additional growth factors involved in osteogenesis (Fig. 1),46 we asked here whether heparin-induced conformational changes of Fn within the fibrillar ECM could have an effect on hMSCs differentiation. Since a cell-derived fibrillar ECM represents a 3D environment that is closer to physiological,56 human foreskin fibroblast (HFF) derived ECM scaffolds were used here to test the effect of heparin induced Fn conformational changes on hMSC differentiation. We compared the differentiation of hMSCs, either seeded on Fn-coated surfaces or reseeded into fibroblast-derived ECM scaffolds after pretreating with or without heparin. The conformation of Fn in ECM was directly probed by fluorescence resonance energy transfer (FRET).27,57
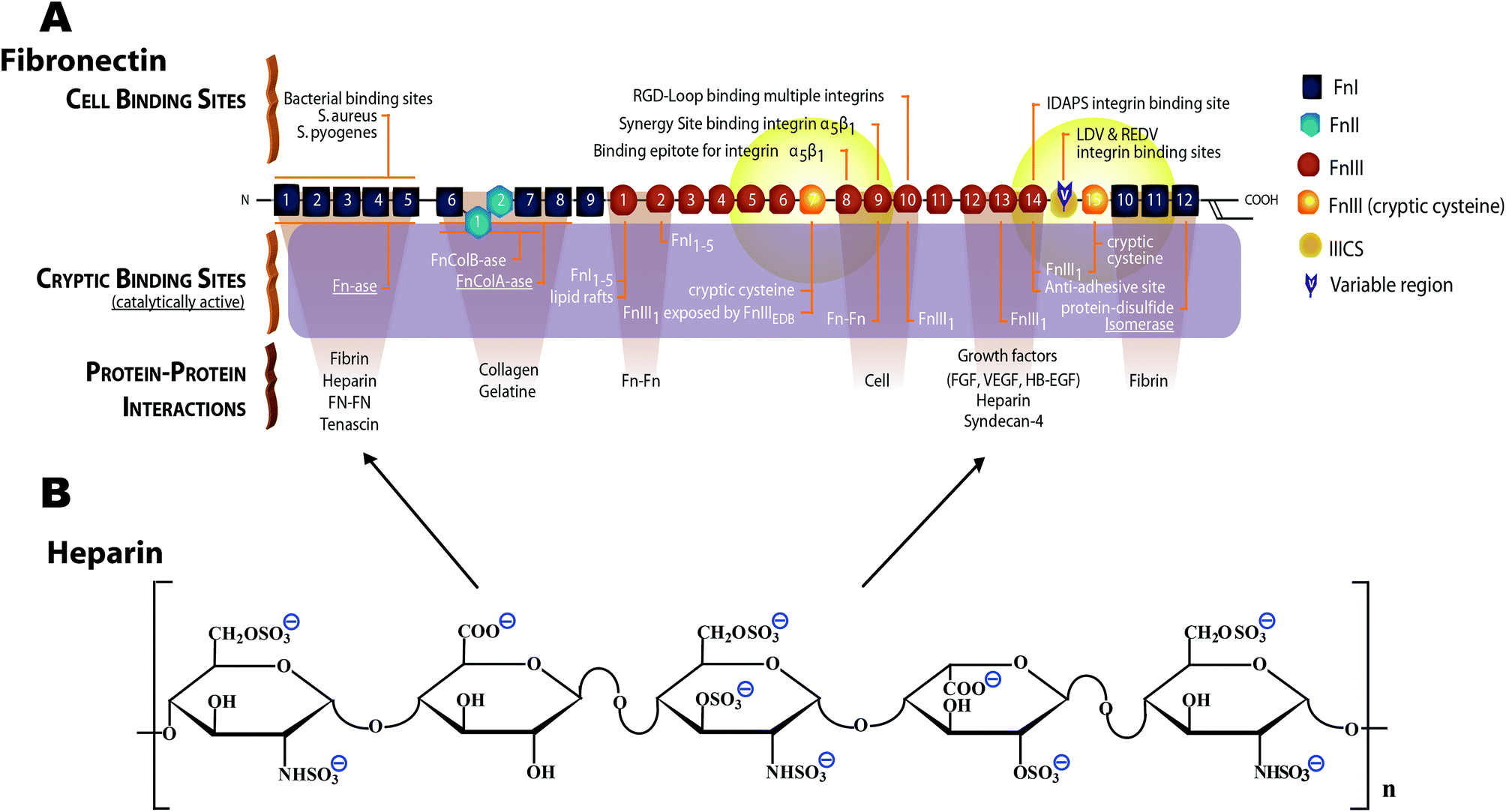 | ||
| Fig. 1 Schematic structure of monomeric plasma fibronectin and heparin. (A) Fn contains a large number of cell binding and protein–protein interaction sites, including the cell binding site RGD88 on FnIII10 and the synergy site PHSRN on FnIII9.89,90 Fn contains at least two heparin binding sites,42–45 one of which (FnIII12–14) also serves as promiscuous binding site for various growth factors.46 Two cryptic, non-disulfide bonded cysteines in FnIII7 and FnIII15 (shown with orange color) were used in our studies for FRET-labeling using Alexa 546 as acceptor, and about 3.5 amines per monomeric Fn were randomly labeled with Alexa 488 as donor. The Förster radius of this fluorophore pair is ∼6 nm (from Invitrogen). Hence the energy transfer is limited to within 12 nm of FnIII7 and FnIII15 (yellow fading spheres). Adapted from ref. 28. (B) Heparin is a highly sulfated glycosaminoglycan. The heparin polymeric chain is composed of repeating disaccharide unit of D-glucosamine and uronic acid linked by interglycosidic bond. The uronic acid residue could be either D-glucuronic acid or L-iduronic acid.32 The key structural unit of heparin is a unique pentasaccharide sequence. This sequence consists of three D-glucosamine and two uronic acid residues. | ||
Results
Heparin treatment induces a more extended conformation of fibrillar fibronectin within cell-derived ECM scaffolds
Fn-rich ECM was produced by culturing HFFs for 4 days in the presence of unlabeled Fn and trace amounts of Fn-FRET, which got incorporated into the assembled ECM. Afterwards, the ECM was decellularized and either treated with 100 µg ml−1 heparin for 12 hours or used without treatment (native). The Fn-FRET is used to monitor Fn's conformational changes. Briefly, the four free cryptic cysteines located on FnIII7 and FnIII15 of dimeric plasma Fn were labeled with Alexa 546 as acceptor fluorophores (A), while amines of Fn were randomly labeled with Alexa 488 as donor fluorophores (D) at an approximate ratio of 3.5 acceptors to 7 donors per Fn dimer. The average distance between our multiple donor and acceptor fluorophores, which is directly related to the measured FRET ratios, is sensitive to Fn conformational changes. When Fn changes to more extended conformations, the FRET ratio decreases due to the separation between acceptor and donor fluorophores.57 Therefore, the relative Fn conformations in different ECM can be roughly compared by measuring the relative emission intensities of these two fluorophore populations.27 Finally, low seeding densities (3000 cells cm−2) were used throughout our experiments to prevent cell–cell interactions which have been shown to strongly affect MSC differentiation.58Immunostaining of Fn ECM scaffolds as assembled by HFFs (which did not contain FRET labeled Fn) showed that the ECM scaffolds kept intact after decellularization and after heparin treatment (Fig. 2A and 2B). No obvious morphological differences were observed between heparin-treated and untreated ECM scaffolds. However, upon heparin treatment the Fn FRET IA/ID ratios were shifted to lower values (blue curve in Fig. 2E), suggesting that the Fn within heparin-treated ECM became more extended compared to untreated ECM. This change was also reflected on the average FRET IA/ID ratios of 10 independent samples (Fig. 2F). Indeed, the average FRET ratio of native ECM (mean IA/ID = 0.63 ± 0.03) is slightly higher than heparin-treated ECM (mean IA/ID = 0.59 ± 0.02). Additionally, measurement of FRET ratio of 3D ECM on individual Z-slices showed that heparin treatment slightly decreased FRET ratios of HFF derived ECM throughout the whole volume of measured ECM (Fig. S1†).
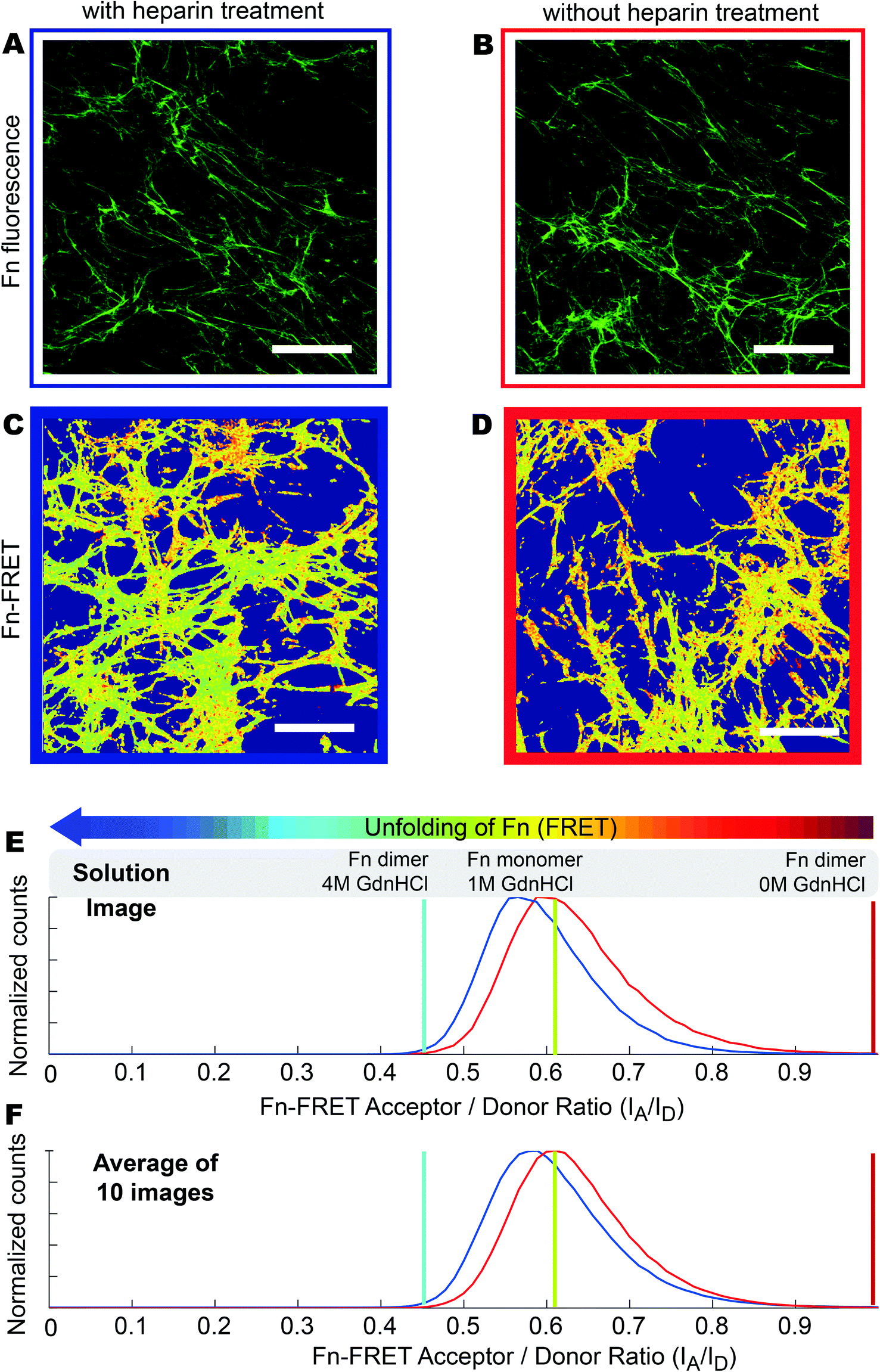 | ||
| Fig. 2 Heparin treatment induces a more extended Fn conformational distribution within cell-derived, decellularized ECM scaffolds. (A and B) Decellularized HFF derived ECM scaffolds with (A) or without (B) heparin treatment were incubated with sheep anti-human fibronectin antibody, and then stained with FITC labeled donkey anti-sheep secondary antibody. The samples were checked under Confocal microscope. (C and D) FRET false color images of decellularized HFF derived ECM scaffolds, which contained trace amounts of FRET-labeled Fn, with (C) or without heparin treatment for 12 hours at 4 °C (D). The FRET false color scheme represents the relative conformational changes of Fn fibrils with a color range of red to blue indicating compact to completely unfolded states, respectively. (E) Histograms of Fn-FRET IA/ID ratios of decellularized ECM with (blue curve, C) and without heparin treatment (red curve, D) representing the ECM shown in image C and D respectively. (F) Histograms of average Fn FRET IA/ID ratios (average of 10 images of different samples) of decellularized ECM scaffolds with (blue curve) or without heparin treatment (red curve). The red, green and blue vertical lines represent IA/ID ratios of native Fn-FRET in 0 M GdnHCl, monomeric Fn-FRET denatured in 1 M GdnHCl and dimeric Fn-FRET denatured in 4 M GdnHCl respectively. Scale bars: 50 μm. | ||
Fibronectin conformational differences between heparin-treated and native ECM scaffolds are eliminated after reseeding the scaffolds with hMSCs
In addition to heparin treatment, most of the quaternary structure that defines Fn's conformation in solution is broken open during Fn fibrillogenesis (Fig. 2E) and cell-generated forces are sufficient to further stretch ECM fibrils, thereby shifting the conformations of fibrillar Fn to even more extended conformations.26,27,59 To monitor the Fn conformational changes of ECM under cell-generated forces, 3 × 103 cells cm−2 hMSCs were seeded on heparin-treated or native decellularized ECM scaffolds, and cultured for 24 hours. Intramolecular FRET was reduced after cell seeding. The average FRET ratios decreased from 0.63 ± 0.03 (Fig. 2F) to 0.57 ± 0.02 (Fig. 3F) in native ECM scaffolds and from 0.59 ± 0.02 (Fig. 2F) to 0.57 ± 0.03 (Fig. 3F) in heparin-treated ECM, and the FRET histograms of native and heparin-treated ECM were shifted to similar low levels. This suggests that cell-generated forces are high enough to stretch the Fn ECM fibrils far beyond the conformational alterations induced by heparin treatment.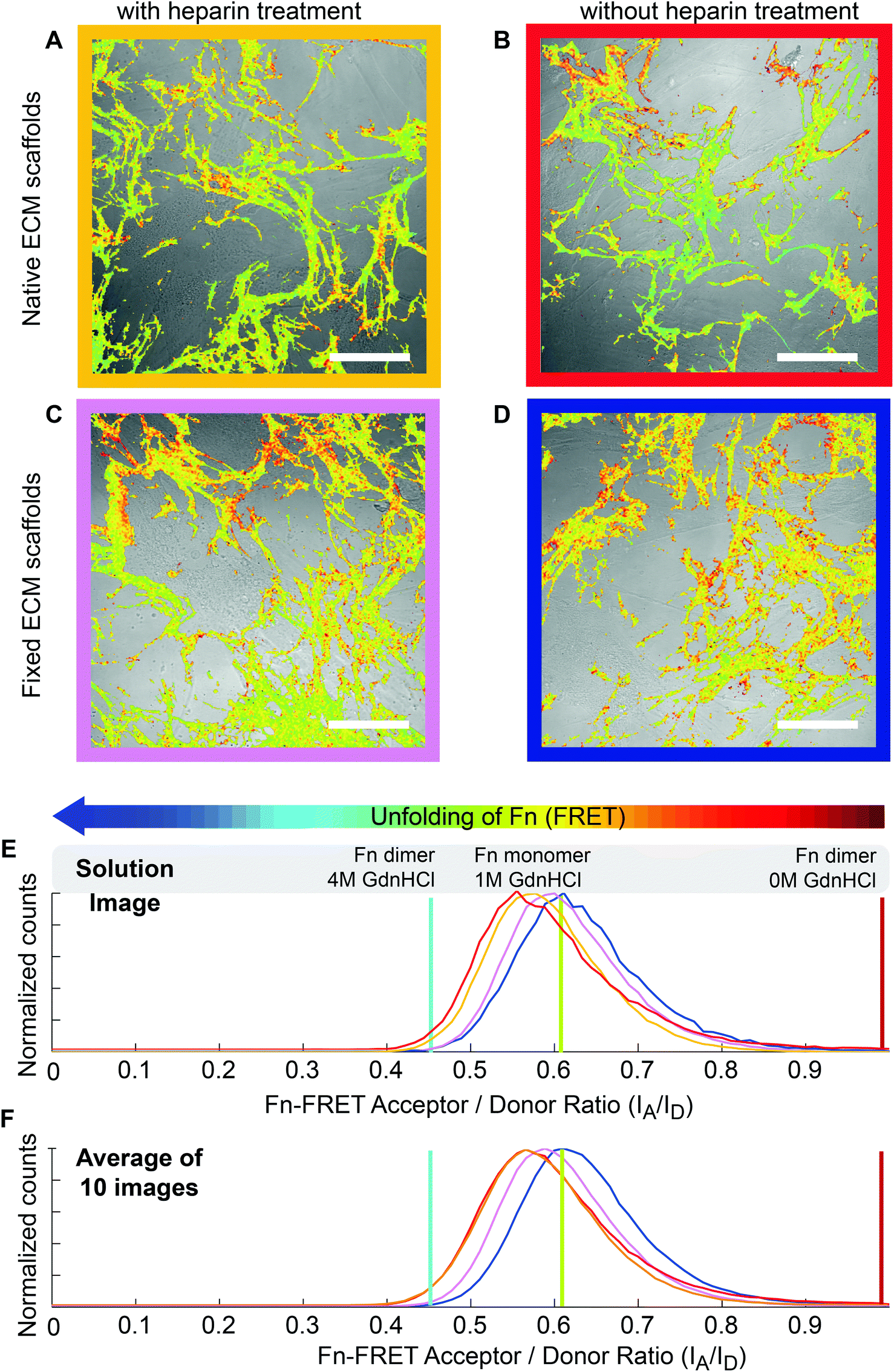 | ||
| Fig. 3 ECM scaffolds 24 hours after reseeding with hMSCs. After chemical fixation, the heparin-induced changes of the Fn conformational distributions within ECM scaffolds remained stable upon reseeding with hMSCs. (A, B, C and D) Merged images of reseeded HFF-derived ECM scaffolds (FRET false colors) with Confocal brightfield images of hMSCs (3 × 103 cells cm−2). hMSCs were cultured in mixed induction medium for 24 hours on heparin-treated (A), native (B), fixed heparin-treated (C) or fixed native ECM scaffolds (D). Scale bars: 50 μm. (E) Histograms of Fn-FRET IA/ID ratios of HFF assembled Fn ECM following hMSCs attachment of the images shown in A–D. hMSCs cultured on native (red curve, B), heparin-treated (yellow curve, A), fixed heparin-treated (purple curve, C) and fixed native ECM (blue curve, D). (F) Histograms of average IA/ID ratios (average of 10 images of different samples) of HFF assembled Fn ECM following hMSCs attachment: Red: native scaffolds; Yellow: heparin-treated scaffolds; purple: heparin-treated and fixed scaffolds; blue: native and fixed scaffolds. | ||
Chemical fixation can preserve the heparin-induced fibronectin conformations, even in the presence of cell-generated forces
To clarify whether the heparin-induced Fn conformational changes could impact hMSC differentiation, hMSCs were seeded and differentiated on scaffolds treated with or without heparin. However, since cell generated forces are sufficient to overwrite the heparin-induced effect on Fn conformation as shown above, we locked-in the Fn conformational distribution prior to hMSC seeding by chemical fixation of the heparin-treated ECM scaffolds with 4% formaldehyde. For this fixation protocol, we reported before that cell-generated forces are not high enough to significantly stretch fixed ECM fibrils and cause a detectable Fn conformational change as probed by FRET.59 hMSCs were then seeded on the fixed heparin-treated ECM (Fig. 3C) or fixed native ECM (Fig. 3D) at sufficiently low seeding densities (3 × 103 cells cm−2) to prevent cell–cell contacts. In contrast to native ECM scaffolds, cell attachment did not decrease the Fn-FRET ratios of fixed ECM. The mean IA/ID values before cell attachment were 0.58 ± 0.03 and 0.62 ± 0.03 for heparin-treated and native ECM samples respectively, whereas mean IA/ID values after cell attachment were 0.59 ± 0.02 and 0.63 ± 0.03 for heparin-treated and native ECM samples respectively. This suggests that our fixation protocol was able to lock-in the Fn conformations of heparin-treated ECM scaffolds and protected its conformational display against destruction by cell-generated forces.Heparin treatment of ECM followed by chemical fixation significantly increased the osteogenic differentiation of hMSCs, but rigidification by fixation alone did not
Using the fixed ECM scaffolds with the Fn conformations locked-in, we tested whether the heparin-induced Fn conformations have an effect on hMSC differentiation. Briefly, hMSCs (∼3000 cm−2) were seeded on scaffolds that were either heparin-treated (Fig. 4C) or native (Fig. 4D) and fixed with formaldehyde. As a control, hMSCs were also seeded on heparin-treated or native scaffolds (Fig. 4A and 4B), but without fixation.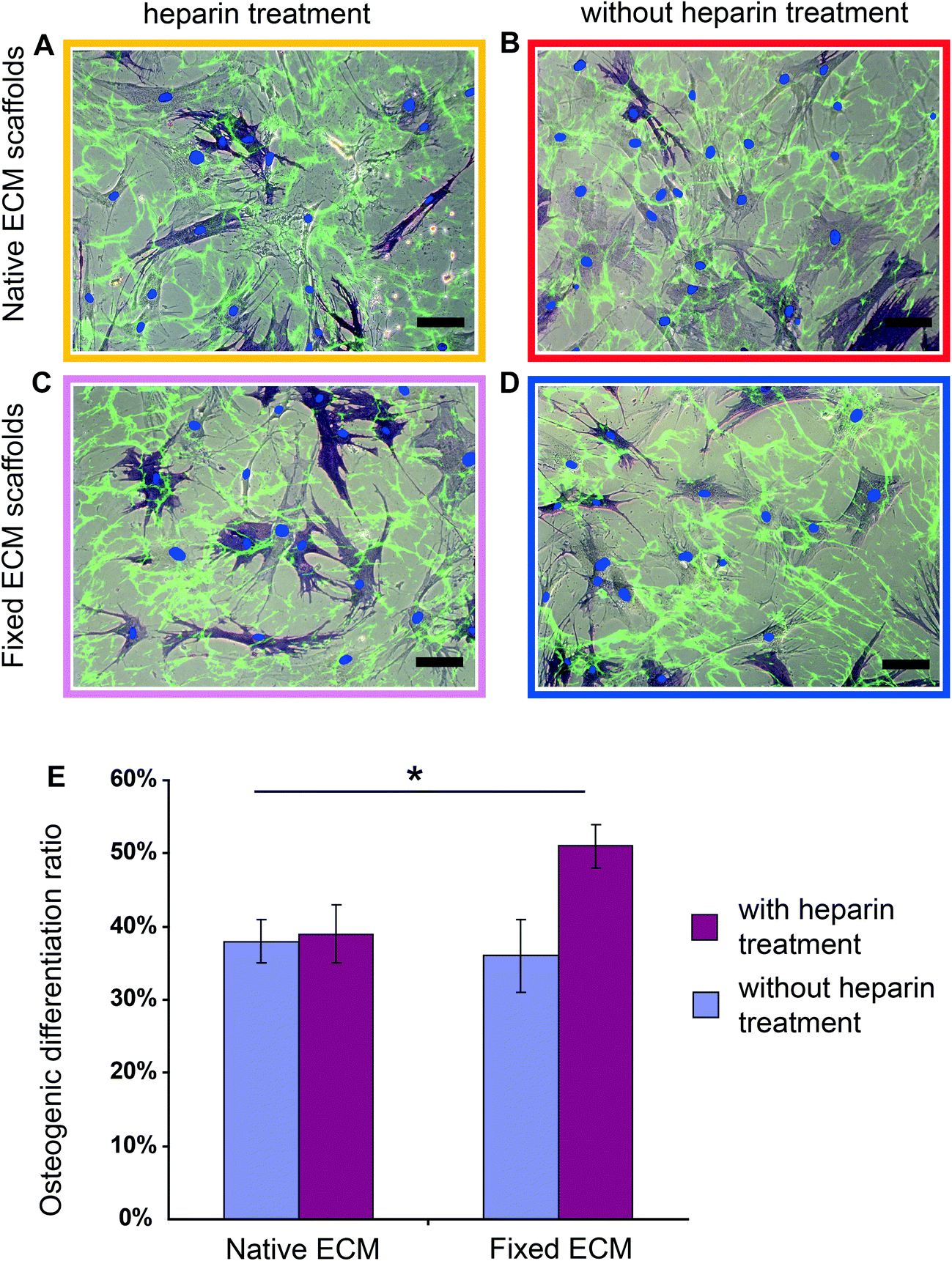 | ||
| Fig. 4 Heparin treatment followed by chemical fixation of the ECM scaffolds significantly increased hMSC osteogenic differentiation. (A, B, C and D) Merged brightfield and fluorescence images of hMSCs cultured for 7 days in mixed induction medium on heparin-treated (A), native (B), fixed heparin-treated (C) and fixed native ECM scaffolds (D). Decellularized ECM scaffolds were labeled with Fn-FRET (green); cell nuclei were stained with DAPI (blue dot) and histochemical staining was performed for ALP (dark blue). Scale bars: 100 μm. (E) Percentage of ALP positive cells when hMSCs were cultured for 7 days on heparin-treated (red bar) or native (blue bar) ECM scaffolds, with or without fixation, in mixed induction medium (50% adipogenic plus 50% osteogenic). Data are shown for ALP positive cells and represent mean ± s.d. (n = 5). Asterisk p < 0.05. | ||
After culturing hMSCs for 7 days in mixed induction medium (50/50 vol% adipogenic/osteogenic induction medium), osteogenic and adipogenic differentiation was examined by histochemical staining for alkaline phosphatase (ALP) and lipid droplets (Oil Red O), respectively.60 Heparin treatment of ECM followed by chemical fixation increased significantly the osteogenic differentiation of hMSCs (Fig. 4E): in native ECM scaffolds, 36 ± 6% hMSCs showed positive ALP staining, while 52 ± 4% stained positive in the conformationally locked ECM scaffolds. This observation was confirmed in 5 independent experiments and a total of more than 1000 cells were counted for each sample.
Since chemical fixation has complex effects on cell-derived ECM scaffolds in addition to the locking of Fn conformation,59 such as increasing the rigidity of ECM fibrils,59,61 changing the molecular composition of cell adhesion sites62 and increasing the force necessary to detach cells from ECM,61 we asked whether chemical fixation alone has an effect on hMSC osteogenic differentiation. Importantly, as shown in Fig. 4E, chemical fixation alone did not significantly influence hMSC osteogenic differentiation (38 ± 4% hMSCs on native ECM and 36 ± 6% hMSCs on chemically fixed ECM showed positive ALP staining) compared with native ECM. Rigidifying Fn ECM fibrils to an extent that the cells cannot stretch them any further thus does not upregulate osteogenesis. Also the osteogenic differentiation ratios of hMSCs on native and heparin treated ECM scaffolds that have not been fixed are similar (38 ± 4% on native and 39 ± 5% on heparin treated scaffolds). Therefore, tight preservation of the heparin-induced Fn conformation within the ECM scaffolds is required to upregulate the osteogenic differentiation of hMSCs.
To exclude that cell proliferation rates differ for hMSCs reseeded on untreated or treated ECM scaffolds, we checked the cell densities on all tested ECM scaffolds after 7 days in cell culture and found no significant differences in cell densities (Fig. S2†).
Osteogenic differentiation is not upregulated by heparin retained in the ECM scaffolds, but due to heparin-induced conformational alterations of fibrillar fibronectin
Since it was reported that heparin-functionalized PEG hydrogels, where heparin was covalently bound to PEG gels could promote hMSC osteogenesis in 2D or 3D culture,51,52 we tested whether the observed effect on hMSC differentiation is mediated by heparin retained within the scaffolds. ECM scaffolds were thus treated with fluorescently labeled heparin. The Fn ECM scaffolds were treated with 100 µg ml−1 Alexa633-labeled heparin for 12 hours. After extensive washing with PBS, microscopic observations did not reveal detectable fluorescence signal (Fig. S3†), suggesting that most of the heparin was removed. This is consistent with a previous report, which showed that after heparin treatment of ECM only about 1% of the added heparin was retained.30Importantly and further supporting the notion that residual heparin does not cause the effect, removal of any remaining heparin from fixed heparin-treated ECM by active degradation with heparinase-I did not significantly impact hMSC osteogenic differentiation (Fig. S4†). Following heparinase-I treatment of fixed heparin-treated samples, 53% ± 4% hMSCs were positively stained with ALP, a ratio similar to that observed without heparinase treatment. As an additional evidence to support our hypothesis that the observed effect on osteogenic differentiation are not mediated by remaining heparin, the non-fixed ECM scaffolds that were treated with heparin and would retain similar amounts of heparin did not impact hMSC osteogenesis (Fig. 4E).
Heparin treatment of cell-derived ECM has no effect on hMSC adipogenesis
After hMSC attachment and differentiation on the cell-derived ECM scaffolds, we observed only few cells (less than 1/1000 cells) that stained with Oil Red O, an indicator of adipogenic differentiation (Fig. 4). As shown in Fig. 4, most of the hMSCs on ECM were spread well and showed a similar dendritic shape, while hMSC adipogenesis was strongly inhibited.60 It was reported that addition of heparin in adipogenic induction medium could promote the adipogenic differentiation of immortalized MSCs.53 Our results thus show that the heparin treatment of cell-derived ECM had no effect on hMSC adipogenesis.Heparin treatment of Fn functionalized polyacrylamide gels or Fn coated glass coverslip has no effect on hMSC differentiation
Finally, we tested whether adsorbed Fn could affect hMSC differentiation in similar ways as fibrillar Fn. Fn was therefore either crosslinked to soft polyacrylamide gels (0.1 kPa),63 or adsorbed to glass and then treated with heparin as described before, but heparin treatment did not affect hMSC differentiation in both cases (Fig. S5†). This suggests that the fibrillar organization of Fn within the ECM scaffolds is a necessary factor for the heparin-mediated changes in Fn conformation to upregulate the osteogenic differentiation potential of hMSCs.The Fn conformations in hMSC newly assembled ECM are similar on all tested decellularized ECM scaffolds
So far, we have studied the effect of Fn conformation within the ECM scaffolds on which hMSCs were seeded. However, hMSCs not only respond to and tune the conformational display of the ECM scaffold fibers by pulling on them, but they also deposit new ECM fibrils that can have a distinctly different conformational distribution.59 According to a previous report hMSCs could harvest plasma Fn and assemble ECM within 24 hours after reseeding on biomaterials, and the Fn conformations in this newly assembled ECM could further guide hMSC differentiation potential.23 In order to assess the effect of hMSC newly assembled ECM on hMSC differentiation, the Fn conformations in newly assembled ECM were observed by adding FRET-labeled Fn after 24 hours of hMSC reseeding (3 × 103 cells cm−2) on decellularized HFF derived ECM scaffolds.59 Prior to hMSC reseeding, the scaffolds were treated with or without heparin and then were either fixed with formaldehyde, or were left native. On all tested scaffolds and within the first 24 hours after reseeding, no significant FRET differences are seen within the ECM newly assembled by hMSCs (Fig. S6†), and this newly assembled ECM had a conformational distribution similar to the more relaxed conformations seen in our crosslinked ECM scaffolds (Fig. 2E). This suggests that this newly assembled ECM within the first 24 hours after seeding provides similar Fn conformational signals for hMSCs, despite the conformational differences and rigidities of the initial ECM scaffolds (Fig. 3). For the heparin-treated ECM, our data suggest that heparin-treatment changes the Fn conformation of the ECM and thus regulates growth factor binding to the scaffold Fn fibers.30 Though the Fn conformations in newly assembled ECM are similar in different types of scaffolds, the fixed heparin-inducted Fn conformations in the heparin-treated scaffolds remains available and constitutes an integral part of ECM functionalities displayed to the cells.Discussion
By exploiting trace amounts of FRET-labeled Fn which the cells can incorporate into their own ECM during ECM assembly,27 we could show here that heparin treatment can change the conformational distribution of fibrillar Fn within fibroblast-derived ECM scaffolds to more extended conformations (Fig. 2). However, upon reseeding of such scaffolds with hMSCs, the heparin-induced conformational changes of the ECM fibrils as observed by FRET were abolished (Fig. 3), suggesting that hMSCs could further tune the Fn conformations by mechanical stretching of the ECM fibrils. In contrast, when the heparin-activated conformation of fibrillar Fn was locked-in by chemical fixation, osteogenic differentiation of hMSCs was significantly increased in mixed induction medium (Fig. 4), whereas heparin treatment or chemical fixation of the ECM scaffold alone had no effect (Fig. 4). In the context of the ongoing debate whether and how rigidity directs stem cell differentiation,23,63,64 it is important to note that the effect of chemical fixation we observed here was due to the prevention of further stretching of ECM fibrils by cell-generated forces as suggested by FRET (Fig. 3), and was not due to ECM rigidification since chemical fixation alone had no effect (Fig. 4).We hypothesize that the observed effect on osteogenic differentiation is due to the effect of heparin on Fn conformation and not due to heparin remaining in the chemically-fixed, heparin treated ECM. Indeed, by utilizing fluorescently labeled heparin, we did not detect any remaining heparin in the ECM (Fig. S3†). Even though we cannot fully exclude the possibility that small amounts of heparin below the detection limit by fluorescence remain in the ECM, our observation that degradation of such remaining heparin by heparinase-I did not impact hMSC osteogenic differentiation (Fig. S4†), supports our hypothesis that the heparin-induced increase of hMSC osteogenic differentiation is not mediated directly by heparin acting on hMSCs, but through alterations of Fn's conformation in the ECM scaffold. The capacity of heparinase to access and degrade immobilized heparin and heparin sulfate chains has been demonstrated and utilized in many studies,65–67 supporting the notion that heparin molecules fixed in the matrix can be degraded by heparinase-I. Heparinase treatment will generate mainly disaccharides (http://www.ibex.ca/ENZglyco_hepI.htm), and since it has been already shown that heparin fragments smaller than 22 disaccharide units are unable to elicit any effect on the conformation of Fn,29 it is unlikely that any heparin fragments still remaining in the matrix would cause the observed effect. Additionally, in contrast to the effect on fibrillar Fn within ECM, heparin treatment of surface adsorbed Fn had no effect on the osteogenic differentiation of hMSCs (Fig. S5†). In this situation then, any remaining amount of heparin in the substrate could not promote osteogenic differentiation, suggesting that the presence of heparin in the ECM cannot be the main cause for the effect. Our study thus suggests that heparin induced Fn conformational changes, when preserved within fibrillar ECM scaffolds, can promote hMSC osteogenesis, although we cannot exclude the possibility that some heparin may remain in the ECM and contribute to the observed effect.
While several studies have indicated that heparin could change Fn's conformation,29,30,68–71 the underpinning mechanisms remain unknown. Since heparin is a highly negatively charged molecule,32 and Fn contains segments of positively charged residues including FnIII13,72,73 heparin might interfere with some residual quaternary structure that still exists in relaxed Fn fibers.27 The presence of heparin might break apart the intramolecular salt bridges by which the FnIII13 repeat might interact with other Fn-repeats thereby breaking apart any remaining backfolding of Fn within fully relaxed Fn-fibrils. Based on our FRET data (Fig. 3) and results of a previous study using manually pulled Fn fibers,74 this residual quaternary structure can be eliminated in the early events of fiber stretching. At higher fiber strains, spatial distances between the FnIII modules that contain the growth factor binding sites could start to mechanically unfold, thereby destroying the binding motif. This could indeed explain why cell generated forces can ultimately eliminate the heparin-induced effect on osteogenic differentiation. With our FRET labeling scheme of Fn, we are particularly sensitive to conformational changes that happen in the surroundings of FnIII7 and FnIII15 whereby the latter just follows the FnIII12–14 fragment that displays one heparin binding site (also called Heparin II binding site) and serves as binding site for many growth factors,46 such as basic fibroblast growth factor-2 (FGF-2),75 platelet-derived growth factor (PDGF),46 VEGF76 and bone morphogenetic protein-2 (BMP-2).46 Several of these growth factors, including FGF-2, BMP-2 and VEGF, have positive effect on hMSC osteogenesis.54,55,77,78 As mentioned before, the osteogenic promoting effect of heparin is not mediated directly by any heparin retained on the matrix. This implies that the effects seen here do not result from heparin itself serving as bridge to attract growth factors, but that the heparin-induced conformational alterations of Fn are primarily responsible alterations in Fn signaling. Taken together, our study suggests that the heparin-induced Fn conformational changes upregulate the binding of growth factors to Fn, and that the tethered growth factors subsequently impact hMSC osteogenic differentiation.
In summary, our data suggest that heparin might interfere with residual quaternary structure in relaxed Fn fibers thereby opening up buried sites which might include growth factor binding sites. In a biphasic manner, these buried sites might then become accessible, but our data also suggest that increasing mechanical strain might ultimately start to unfold the FnIII modules thereby again destroying these binding site motifs.
The finding that heparin induced Fn conformational changes of ECM scaffolds can promote hMSC osteogenesis is also significant to the biomaterials community in another context. Since tissue-derived scaffolds have found widespread clinical applications, this new method opens new possibilities to treat ECM scaffold or tissues after decellularization, thereby tuning their biological activity. In the context of the clinical challenges to find better methods of promoting bone healing and regeneration, or to improve the efficiencies of stem cell therapies,79 our data suggest a way to modify the Fn conformation within ECM scaffolds by heparin treatment and chemical fixation. Since heparin treatment of surface adsorbed or fixed Fn alone cannot influence hMSC differentiation (Fig. S5†), it seems that this method might be particularly powerful when treating cell-derived or tissue derived ECM scaffolds with heparin followed by chemical fixation. Though some studies suggested that the chemical fixation process changes the host tissue response to a pro-inflammatory and foreign body response,80,81 chemically crosslinked biological scaffold derived from ECM of intact mammalian tissues have been successfully used in at least some clinical applications.82 For instance, Vascu-Guard® (Synovis Surgical, USA) which is prepared by chemical crosslinking of bovine pericardium with glutaraldehyde has been successfully used in peripheral vascular reconstruction including the carotid, renal, iliac, femoral, profunda and tibial blood vessels. In summary, our findings suggest that cell-derived extracellular matrix combined with heparin treatment and fixation can provide novel ways of modifying biomaterials used for bone graft.
Materials and methods
Cell culture
hMSCs were purchased from Lonza and cultured in growth medium (DMEM, 10% FBS, 0.3 mg ml−1 glutamine, 100 units ml−1 penicillin and 100 µg ml−1 streptomycin). Only early passage hMSCs (up to passage 5) were used in our experiments. Osteogenic and adipogenic induction media were purchased from Lonza. Mixed induction medium was composed of 50% adipogenic induction medium and 50% osteogenic induction medium (by volume). To test the hMSC differentiation on matrices, 3 × 103 cells cm−2 hMSCs were seeded on cell-derived ECM scaffolds and incubated for 1.5 hour before changing to mixed induction medium. Medium was changed every two days, and the differentiation of hMSCs was examined by histochemical staining after 7 days in culture. Human Foreskin Fibroblast (HFF) cells were cultured in serum-free medium (NHDF with supplements, Promocell).Cell staining
Alkaline phosphatase (ALP) was stained using the Sigma kit #85 according to the manufacturer's protocol. For the staining of lipids, cells were fixed with 10% formaldehyde and rinsed with 60% isopropanol. Cells were then stained with 30 mg ml−1 Oil Red O (Sigma) in 60% isopropanol. Cells were stained with 3 μg ml−1 DAPI (Invitrogen) to visualize cell nuclei. Cells were photographed and counted using an Axiovert 200M inverted microscope (Carl Zeiss).Fn labeling and chemical denaturation curves
Fn was isolated and labeled according to a previously described protocol.27 The Fn-FRET molecules used for the present study had an average of 7 donors and 3.5 acceptors per molecule. Fn-FRET was stored as 10 μl aliquots in PBS at −20 °C and used within 5 days upon thawing. The same batch of Fn-FRET was used for all FRET data shown in this paper. FRET analysis was performed according to the method described in a previous paper.27 FRET IA/ID ratios were calibrated to different Fn conformations in GdnHCl solution. Dimeric and fully folded Fn in PBS showed strong energy transfer (IA/ID = 0.99), while monomeric Fn-FRET in 4 M GdnHCl, where the Fn molecule is significantly unfolded, showed dramatically decreased energy transfer (IA/ID = 0.45). Monomeric Fn-FRET (generated from dimeric FN-FRET by DTT reduction) in 1 M GdnHCl which is partially unfolded, showed a medium energy transfer (IA/ID = 0.61). According to previous studies on Fn conformations in solution, the IA/ID value of monomeric Fn-FRET in 1 M GdnHCl (IA/ID = 0.61) was used to indicate the very first onset of loss of secondary structure.27FRET analysis
All images were acquired using an Olympus (http://www.olympus-global.com/) FV-1000 scanning laser Confocal microscope with a 1.35NA 60× oil immersion objective. Alexa Fluor 488 donors of the Fn-FRET were excited with a 488 nm laser. Emitted light was split using a 50/50 beam splitter and detected in two separate photomultiplier tubes (PMTs). Emission detection windows were set at 514–526 nm (donor channel) and 566–578 nm (acceptor channel) to capture peak emissions. Images were acquired at a resolution of 512 × 512 pixels for a 212 × 212 µm field of view with a pinhole diameter of 200 µm. The images were analyzed using Matlab (http://www.mathworks.com/) according to a previous script.27 First, images were averaged with 2 × 2 pixel sliding blocks, and the dark current background was subtracted from donor and acceptor images (previously acquired for each experiment). Donor images were corrected for light attenuation from the 50/50 beam splitter with a multiplication factor of 1.09. A threshold mask of 100 relative intensity units was applied to both images and the acceptor image was divided pixel by pixel by the donor image for all pixels above threshold intensity values to yield Fn-FRET IA/ID ratios. Decreasing Fn-FRET IA/ID ratios indicated more extended Fn conformations. Histograms were computed from all data pixels within each field of view and Fn-FRET IA/ID ratios were color-coded within the range of 0.05 to 1.0 to produce FRET images. For each sample, histograms were also collected from 10 randomly chosen images showing in all cases that the histograms given in Fig. 2–4 for single images are representative. Brightfield images were background subtracted using a polynomial fit (degree of 32) with the ImageJ software (http://rsbweb.nih.gov/ij/).Preparation of HFF derived 3D ECM scaffolds
Following a previous protocol,59 12.5 cm2 tissue culture flasks were coated with Fn (20 μg ml−1 in PBS), and 35 mm glass-bottom dishes (MatTek, Ashland, MA) were covalently functionalized with Fn to prevent Fn ECM detachment during decellularization. Briefly glass surfaces were plasma cleaned for 30 seconds (0.36 mbar, 200 W load coil power) and silanized with aminopropyltriethoxysilane (Sigma) molecules. Silanized surfaces were treated with glutaraldehyde, followed by incubation with Fn (20 μg ml−1 in PBS) for 1 hour. Before seeding of cells, the Fn-functionalized glass surfaces were rinsed briefly with serum-containing (10% by volume) medium (DMEM, 10% FBS). HFF cells were seeded on the Fn-adsorbed flasks and Fn-functionalized dishes at 45 × 103 cells cm−2 and cultured for a total of 4 days in serum-free medium (NHDF with supplements, Promocell) containing 45 μg ml−1 unlabeled Fn and 5 μg ml−1 Fn-FRET. The cultures were decellularized to generate the 3D ECM scaffolds according to a previous protocol,83 Briefly, the cultures were incubated with extraction solution (20 mM NH4OH solution in PBS (pH = 9.95) with 0.5% (v/v) Triton X) for 5 minutes at room temperature, and then up to 10 additional minutes at 37 °C. The resulting scaffolds were washed with deionized water and PBS.Heparin treatment of HFF derived 3D ECM scaffolds
Heparin (sodium salt from bovine intestinal mucosa) was purchased from Sigma-Aldrich. The decellularized ECM scaffolds were treated with 100 µg ml−1 heparin in PBS for 12 hours at 4 °C,29 and then extensively washed with PBS for 5 times. The samples were chemically fixed with 4% formaldehyde in PBS for 30 min and then washed with PBS. Degradation of heparin retained on fixed heparin-treated scaffolds was conducted by incubating the ECM scaffolds with 0.025 milliunits ml−1 heparinase I (IBEX) for 12 h at 37 °C, and followed by extensive washing with PBS.Immunostaining of HFF derived ECM scaffolds
The HFF derived ECM scaffolds were fixed in 4% formaldehyde for 30 min, and washed with PBS. The fixed samples were blocked with 5% donkey serum and 2% BSA in PBS for 1 hour at room temperature. After washing with PBS, the samples were incubated with 5 µg ml−1 sheep anti-human fibronectin polyclonal antibody (AbD Serotec) for 1 hour at room temperature. Then the samples were washed with PBS and incubated with 20 µg ml−1 FITC labeled donkey anti-sheep secondary antibody (Abcam) for 1 hour at room temperature. Finally the samples were washed with PBS, and observed under Olympus FV-1000 scanning laser Confocal microscope.Labeling heparin with Alexa Fluor 633 succinimidyl ester
Heparin was labeled with Alexa Fluor 633 succinimidyl ester (Molecular Probes) according to the manufacturer's protocols. Briefly, 300 µg heparin were dissolved in 300 µl of 0.1 M sodium bicarbonate buffer (pH = 8.5), and incubated with 1 mg Alexa Fluor 633 succinimidyl ester on ice for 2.5 hours. The reaction was quenched by incubating with 0.1 ml of freshly prepared 1.5 M hydroxylamine (pH 8.5) on ice for 1 hour. The labeled heparin was separated from free dye in a PD-10 Sephadex G-25 column (GE Healthcare life sciences). The concentration of labeled heparin was determined by the dimethylmethylene blue assay.84hMSC newly assembled ECM on HFF derived ECM scaffolds
hMSCs (3 × 103 cells ml−1) were allowed to attach for 24 hour on decellularized HFF derived ECM scaffolds in growth medium supplemented with 45 μg ml−1 unlabeled Fn and 5 μg ml−1 Fn-FRET. After extensive wash with PBS the new assembled ECM scaffolds were checked under Olympus FV-1000 scanning laser Confocal microscope.Preparation of 2D polyacrylamide substrates
As previously described,85 35 mm glass-bottom dishes were plasma cleaned, silanized using aminopropyltriethoxysilane and treated with glutaraldehyde. The surfaces were coated with 10 μl droplets of 3% polyacrylamide/0.05% bisacrylamide for the ∼0.1 kPa soft substrate (0.13 kPa ± 0.005 kPa) and covered with 12 mm diameter coverslips. Coverslips were removed and the polyacrylamide surfaces covalently functionalized with Fn using sulfosuccinimidyl-6 (4′-azido-2′-nitrophenylamino) hexanoate (sulfo-SANPAH, Pierce) to allow cell attachment. Briefly polyacrylamide gels were placed in a 24-well plate and 500 µl of a 0.2 mg ml−1 solution of sulfo-SANPAH in milli-Q H2O were added to each well. The PDMS surface was irradiated for 5 minutes using the 365 nm UV LED array. The solution was removed and the procedure was repeated once. After washing with 50 mM HEPES in PBS (twice), the substrates were coated with 20 µg ml−1 Fn (purified by ourselves) in PBS. The Young's moduli of the polyacrylamide gels were determined by atomic force microscopy (AFM) using a silicon nitride tip with an attached polystyrene bead (Novascan, 4.5 μm bead diameter, 10 pN nm−1 spring constant) and a modified Hertz model as previously described.86 The AFM-derived Young's moduli were in good agreement with recent literature values of comparable polyacrylamide gel compositions.87Author contributions
BL and VV designed the research and BL, ZL, YZ and MM performed it. BL and VV analyzed the data. BL, MM and VV wrote the paper.Acknowledgements
We wish to thank the following group members for helpful discussions, comments and technical advice: Dr Michael Smith, Dr William Little, Dr Jens Möller and Susanna Früh. Financial support from the CCMX Matlife program, an ERC Advanced Grant (Mechanochemical Switches, 223157, VV), as well as funding from the Swiss National Science Foundation (SNF 3103A-116236, VV) and from ETH Zurich are gratefully acknowledged.Notes and references
- J. R. Mauney, V. Volloch and D. L. Kaplan, Tissue Eng., 2005, 11, 787–802 CrossRef CAS PubMed.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- R. M. Salasznyk, W. A. Williams, A. Boskey, A. Batorsky and G. E. Plopper, J. Biomed. Biotechnol., 2004, 2004, 24–34 CrossRef PubMed.
- H. Petite, V. Viateau, W. Bensaid, A. Meunier, C. de Pollak, M. Bourguignon, K. Oudina, L. Sedel and G. Guillemin, Nat. Biotechnol., 2000, 18, 959–963 CrossRef CAS PubMed.
- R. Quarto, M. Mastrogiacomo, R. Cancedda, S. M. Kutepov, V. Mukhachev, A. Lavroukov, E. Kon and M. Marcacci, N. Engl. J. Med., 2001, 344, 385–386 CrossRef CAS PubMed.
- A. S. Rowlands, P. A. George and J. J. Cooper-White, Am. J. Phys., 2008, 295, C1037–C1044 CrossRef CAS PubMed.
- A. Ode, G. N. Duda, J. D. Glaeser, G. Matziolis, S. Frauenschuh, C. Perka, C. J. Wilson and G. Kasper, J. Biomed. Mater. Res., 2010, 95, 1114–1124 CrossRef PubMed.
- S. R. Chastain, A. K. Kundu, S. Dhar, J. W. Calvert and A. J. Putnam, J. Biomed. Mater. Res., 2006, 78, 73–85 CrossRef PubMed.
- U. Hempel, V. Hintze, S. Moller, M. Schnabelrauch, D. Scharnweber and P. Dieter, Acta Biomater., 2012, 8, 659–666 CrossRef CAS PubMed.
- S. Kliemt, C. Lange, W. Otto, V. Hintze, S. Moller, M. von Bergen, U. Hempel and S. Kalkhof, J. Proteome Res., 2013, 12, 378–389 CrossRef CAS PubMed.
- T. Velling, J. Risteli, K. Wennerberg, D. F. Mosher and S. Johansson, J. Biol. Chem., 2002, 277, 37377–37381 CrossRef CAS PubMed.
- J. Sottile and D. C. Hocking, Mol. Biol. Cell, 2002, 13, 3546–3559 CrossRef CAS PubMed.
- J. A. Buckwalter, M. J. Glimcher, R. R. Cooper and R. Recker, Instr. Course Lect., 1996, 45, 371–386 CAS.
- P. G. Robey and A. L. Boskey, in Osteoporosis, Academic Press Ltd., 14 Belgrave Square, 24–28 Oval Road, London NW1 70X, England, UK, 1250 Sixth Ave., San Diego, California 92101, USA, 1996, pp. 95–183 Search PubMed.
- R. E. Weiss and A. H. Reddi, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 2074–2078 CrossRef CAS.
- A. M. Moursi, C. H. Damsky, J. Lull, D. Zimmerman, S. B. Doty, S. Aota and R. K. Globus, J. Cell Sci., 1996, 109(Pt 6), 1369–1380 CAS.
- N. Ogura, M. Kawada, W. J. Chang, Q. Zhang, S. Y. Lee, T. Kondoh and Y. Abiko, J. Oral Sci., 2004, 46, 207–213 CrossRef CAS.
- Y. Sogo, A. Ito, T. Matsuno, A. Oyane, G. Tamazawa, T. Satoh, A. Yamazaki, E. Uchimura and T. Ohno, Biomed. Mater., 2007, 2, 116–123 CrossRef CAS PubMed.
- A. J. Garcia, M. D. Vega and D. Boettiger, Mol. Biol. Cell, 1999, 10, 785–798 CrossRef CAS.
- S. N. Stephansson, B. A. Byers and A. J. Garcia, Biomaterials, 2002, 23, 2527–2534 CrossRef CAS.
- B. G. Keselowsky, D. M. Collard and A. J. Garcia, J. Biomed. Mater. Res., 2003, 66, 247–259 CrossRef PubMed.
- B. G. Keselowsky, D. M. Collard and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5953–5957 CrossRef CAS PubMed.
- B. Li, C. Moshfegh, Z. Lin, J. Albuschies and V. Vogel, Sci. Rep., 2013, 3 CAS.
- H. P. Erickson, J. Muscle Res. Cell Motil., 2002, 23, 575–580 CrossRef.
- C. A. Lemmon, T. Ohashi and H. P. Erickson, J. Biol. Chem., 2011, 286, 26375–26382 CrossRef CAS PubMed.
- G. Baneyx, L. Baugh and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5139–5143 CrossRef CAS PubMed.
- M. L. Smith, D. Gourdon, W. C. Little, K. E. Kubow, R. A. Eguiluz, S. Luna-Morris and V. Vogel, PLoS Biol., 2007, 5, e268 Search PubMed.
- V. Vogel, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 459–488 CrossRef CAS PubMed.
- M. Mitsi, Z. Hong, C. E. Costello and M. A. Nugent, Biochemistry, 2006, 45, 10319–10328 CrossRef CAS PubMed.
- M. Mitsi, K. Forsten-Williams, M. Gopalakrishnan and M. A. Nugent, J. Biol. Chem., 2008, 283, 34796–34807 CrossRef CAS PubMed.
- J. L. Dreyfuss, C. V. Regatieri, T. R. Jarrouge, R. P. Cavalheiro, L. O. Sampaio and H. B. Nader, An. Acad. Bras. Cienc., 2009, 81, 409–429 CrossRef CAS PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, Freeman, p. 1100, 2005 Search PubMed.
- R. Sasisekharan and G. Venkataraman, Curr. Opin. Chem. Biol., 2000, 4, 626–631 CrossRef CAS.
- D. D. Metcalfe, D. Baram and Y. A. Mekori, Phys. Rev., 1997, 77, 1033–1079 CAS.
- D. E. Humphries, G. W. Wong, D. S. Friend, M. F. Gurish, W. T. Qiu, C. Huang, A. H. Sharpe and R. L. Stevens, Nature, 1999, 400, 769–772 CrossRef CAS PubMed.
- C. Page, ISRN Pharmacol., 2013, 910743 Search PubMed.
- R. Lever, B. Mulloy, C. P. Page and T. W. Barrowcliffe, in Heparin – A Century of Progress, Springer, Berlin Heidelberg, 2012, vol. 207, pp. 3–22 Search PubMed.
- D. Xu and J. D. Esko, Annu. Rev. Biochem., 2014, 83, 129–157 CrossRef CAS PubMed.
- M. Ishihara, M. Sato, H. Hattori, Y. Saito, H. Yura, K. Ono, K. Masuoka, M. Kikuchi, K. Fujikawa and A. Kurita, J. Biomed. Mater. Res., 2001, 56, 536–544 CrossRef CAS.
- C. L. Casper, N. Yamaguchi, K. L. Kiick and J. F. Rabolt, Biomacromolecules, 2005, 6, 1998–2007 CrossRef CAS PubMed.
- S. S. Cai, Y. C. Liu, X. Z. Shu and G. D. Prestwich, Biomaterials, 2005, 26, 6054–6067 CrossRef CAS PubMed.
- K. M. Yamada, D. W. Kennedy, K. Kimata and R. M. Pratt, J. Biol. Chem., 1980, 255, 6055–6063 CAS.
- M. J. Benecky, C. G. Kolvenbach, D. L. Amrani and M. W. Mosesson, Biochemistry, 1988, 27, 7565–7571 CrossRef CAS.
- K. C. Ingham, S. A. Brew and M. Migliorini, Arch. Biochem. Biophys., 1994, 314, 242–246 CrossRef CAS PubMed.
- K. Sekiguchi, S. Hakomori, M. Funahashi, I. Matsumoto and N. Seno, J. Biol. Chem., 1983, 258, 14359–14365 CAS.
- M. M. Martino and J. A. Hubbell, FASEB J., 2010, 24, 4711–4721 CrossRef CAS PubMed.
- K. L. Bentley, R. J. Klebe, R. E. Hurst and P. M. Horowitz, J. Biol. Chem., 1985, 260, 7250–7256 CAS.
- P. P. Francois, K. T. Preissner, M. Herrmann, R. P. Haugland, P. Vaudaux, D. P. Lew and K. H. Krause, J. Biol. Chem., 1999, 274, 37611–37619 CrossRef CAS PubMed.
- T. P. Richardson, V. Trinkaus-Randall and M. A. Nugent, J. Biol. Chem., 1999, 274, 13534–13540 CrossRef CAS PubMed.
- R. Ruppert, E. Hoffmann and W. Sebald, Eur. J. Biochem., 1996, 237, 295–302 CAS.
- D. S. W. Benoit, A. R. Durney and K. S. Anseth, Biomaterials, 2007, 28, 66–77 CrossRef CAS PubMed.
- D. S. W. Benoit and K. S. Anseth, Acta Biomater., 2005, 1, 461–470 CrossRef PubMed.
- W. J. Luo, H. Shitaye, M. Friedman, C. N. Bennett, J. Miller, O. A. MacDougald and K. D. Hankenson, Exp. Cell Res., 2008, 314, 3382–3391 CrossRef CAS PubMed.
- H. Mayer, H. Bertram, W. Lindenmaier, T. Korff, H. Weber and H. Weich, J. Cell. Biochem., 2005, 95, 827–839 CrossRef CAS PubMed.
- N. Casap, N. B. Venezia, A. Wilensky and Y. Samuni, Tissue Eng., Part A, 2008, 14, 247–253 CrossRef CAS PubMed.
- E. Cukierman, Methods Mol. Biol., 2005, 294, 79–93 CAS.
- G. Baneyx, L. Baugh and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14464–14468 CrossRef CAS PubMed.
- I. Kii, N. Amizuka, J. Shimomura, Y. Saga and A. Kudo, J. Bone Miner. Res., 2004, 19, 1840–1849 CrossRef CAS PubMed.
- K. E. Kubow, E. Klotzsch, M. L. Smith, D. Gourdon, W. C. Little and V. Vogel, Integr. Biol., 2009, 1, 635–648 RSC.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- A. J. Engler, M. Chan, D. Boettiger and J. E. Schwarzbauer, J. Cell Sci., 2009, 122, 1647–1653 CrossRef CAS PubMed.
- E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science, 2001, 294, 1708–1712 CrossRef CAS PubMed.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. Strange, Y. Li, M. L. Oyen, M. A. Cohen Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. Huck, Nat. Mater., 2012, 11, 642–649 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- V. D. Nadkarni and R. J. Linhardt, BioTechniques, 1997, 23, 382–385 CAS.
- S. Murugesan, T. J. Park, H. Yang, S. Mousa and R. J. Linhardt, Langmuir, 2006, 22, 3461–3463 CrossRef CAS PubMed.
- D. Berry, D. M. Lynn, E. Berry, R. Sasisekharan and R. Langer, Biochem. Biophys. Res. Commun., 2006, 348, 850–856 CrossRef CAS PubMed.
- Sachchidanand, O. Lequin, D. Staunton, B. Mulloy, M. J. Forster, K. Yoshida and I. D. Campbell, J. Biol. Chem., 2002, 277, 50629–50635 CrossRef CAS PubMed.
- L. Vuillard, D. J. Hulmes, I. F. Purdom and A. Miller, Int. J. Biol. Macromol., 1994, 16, 21–26 CrossRef CAS.
- E. Osterlund, I. Eronen, K. Osterlund and M. Vuento, Biochemistry, 1985, 24, 2661–2667 CrossRef CAS.
- H. Richter, C. Wendt and H. Hormann, Bio. Chem. Hoppe-Seyler, 1985, 366, 509–514 CrossRef CAS.
- F. Citarella, H. te Velthuis, M. Helmer-Citterich and C. E. Hack, Thromb. Haemostasis, 2000, 84, 1057–1065 CAS.
- A. Sharma, J. A. Askari, M. J. Humphries, E. Y. Jones and D. I. Stuart, EMBO J., 1999, 18, 1468–1479 CrossRef CAS PubMed.
- E. Klotzsch, M. L. Smith, K. E. Kubow, S. Muntwyler, W. C. Little, F. Beyeler, D. Gourdon, B. J. Nelson and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18267–18272 CrossRef CAS PubMed.
- C. Bossard, L. Van den Berghe, H. Laurell, C. Castano, M. Cerutti, A. C. Prats and H. Prats, Cancer Res., 2004, 64, 7507–7512 CrossRef CAS PubMed.
- E. S. Wijelath, S. Rahman, M. Namekata, J. Murray, T. Nishimura, Z. Mostafavi-Pour, Y. Patel, Y. Suda, M. J. Humphries and M. Sobel, Circ. Res., 2006, 99, 853–860 CrossRef CAS PubMed.
- T. Ito, R. Sawada, Y. Fujiwara and T. Tsuchiya, Cytotechnology, 2008, 56, 1–7 CrossRef CAS PubMed.
- V. Karageorgiou, M. Tomkins, R. Fajardo, L. Meinel, B. Snyder, K. Wade, J. Chen, G. Vunjak-Novakovic and D. L. Kaplan, J. Biomed. Mater. Res., Part A, 2006, 78A, 324–334 CrossRef CAS PubMed.
- B. Parekkadan and J. M. Milwid, Annu. Rev. Biomed. Eng., 2010, 12, 87–117 CrossRef CAS PubMed.
- J. E. Valentin, J. S. Badylak, G. P. McCabe and S. F. Badylak, J. Bone Jt. Surg., 2006, 88, 2673–2686 CrossRef PubMed.
- S. F. Badylak and T. W. Gilbert, Semin. Immunol., 2008, 20, 109–116 CrossRef CAS PubMed.
- S. F. Badylak, D. O. Freytes and T. W. Gilbert, Acta Biomater., 2009, 5, 1–13 CrossRef CAS PubMed.
- E. Cukierman, in Current Protocols in Cell Biology, John Wiley & Sons, Inc., 2002 Search PubMed.
- R. W. Farndale, D. J. Buttle and A. J. Barrett, Biochim. Biophys. Acta, 1986, 883, 173–177 CrossRef CAS.
- M. Antia, G. Baneyx, K. E. Kubow and V. Vogel, Faraday Discuss., 2008, 139, 229–249 RSC ; discussion 309–225, 419–220.
- R. E. Mahaffy, C. K. Shih, F. C. MacKintosh and J. Käs, Phys. Rev. Lett., 2000, 85, 880–883 CrossRef CAS.
- J. R. Tse and A. J. Engler, Curr. Protoc. Cell Biol., 2010 Search PubMed , ch. 10, Unit 10.16, 10.16.1–10.16.16.
- M. D. Pierschbacher and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5985–5988 CrossRef CAS.
- S. D. Redick, D. L. Settles, G. Briscoe and H. P. Erickson, J. Cell Biol., 2000, 149, 521–527 CrossRef CAS.
- J. Takagi, K. Strokovich, T. A. Springer and T. Walz, EMBO J., 2003, 22, 4607–4615 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60326a |
| This journal is © The Royal Society of Chemistry 2015 |