
Patterning of supported lipid bilayers and proteins using material selective nitrodopamine-mPEG†‡
Philipp R.
Spycher
a,
Heike
Hall
a,
Viola
Vogel
a and
Erik
Reimhult
*bc
aLaboratory of Applied Mechanobiology, Department of Health Sciences and Technology, ETH Zurich, Switzerland
bLaboratory for Surface Science and Technology, Department of Materials, ETH Zurich, Switzerland
cInstitute for Biologically Inspired Materials, Department of Nanobiotechnology, University of Natural Resources and Life Sciences Vienna, Vienna, Austria. E-mail: erik.reimhult@boku.ac.at; Fax: +43 1 47891 12 00; Tel: +43 1 47654 22 30
First published on 8th September 2014
Abstract
We present a generic patterning process by which biomolecules in a passivated background are patterned directly from physiological buffer to microfabricated surfaces without the need for further processing. First, nitrodopamine-mPEG is self-assembled to selectively render TiO2 patterns non-fouling to biomolecule adsorption on hydrophilic and adhesive glass surfaces. After the controlled TiO2 passivation, the biomolecules can be directly adsorbed from solution in a single step creating large scale micropatterned and highly homogeneous arrays of biomolecules with very high pattern definition. We demonstrate the formation of fluid supported lipid bilayers (SLBs) down to the single μm-level limited only by the photolithographic process. Non-specific adsorption of lipid vesicles to the TiO2 background was found to be almost completely suppressed. The SLB patterns can be further selectively functionalized with retained mobility, which we demonstrate through biotin–streptavidin coupling. We envision this single step patterning approach to be very beneficial for membrane-based biosensors and for pattering of cells on a passivated background with complex, sub-cellular geometries; in each application the adherent areas have a tunable mobility of interaction sites controlled by the fluidity of the membrane.
Introduction
Surfaces with biomolecular micro- and nanopatterns are nowadays broadly applied for the study of fundamental biological phenomena.1–6 For the patterning of biomolecules and cells, different patterning techniques have been developed to meet the increasing demands in this field including microcontact printing (μCP),7–9 microfluidic devices,10–12 dip-pen lithography,13,14 or e-beam lithography15 with demonstrated pattern resolution down to a couple of nm.16,17 Selective molecular assembly patterning (SMAP)18 is another method to pattern high-quality biomolecular contrasts; it was originally based on the binding selectivity of alkanephosphates towards certain metal oxides. The phosphate anchor group binds to TiO2, but not to SiO2 where it instead shows a cleaning effect. Thus, after creating a pattern of TiO2/SiO2 by e.g. a photolithographic process the sample is first incubated with alkanephosphates which form a self-assembled monolayer on the TiO2 pattern and subsequently is backfilled with PLL-g-PEG; the PLL-g-PEG coats the SiO2 background through charge interactions but can be rinsed from the alkanephosphate covered TiO2 surface. If the surface now is exposed to a protein solution, the protein will easily and selectively adsorb to the hydrophobic SAM on the TiO2, while being repelled on the PEG-coated SiO2 areas, making it functional for cell studies.18 However, the protein adsorbs to the alkane SAM in a random fashion through non-specific hydrophobic interactions that also can lead to denaturation and loss of function.Nitrocatechols were recently introduced as high affinity surface coating anchor groups for nanoparticles,19 inspired by earlier work on catechol anchors derived from mussel adhesive proteins.20,21 They have since been shown to bind with high affinity to various oxides including Fe3O4,19 TiO2 and Nb2O5, while they display extremely weak if any binding to SiO2 surfaces.22 The catechol anchor group can be attached to non-adhesive (e.g. mPEG) or functional (e.g. PEG-biotin) polymers.23 Nitrocatechol-PEGs could therefore be a very useful tool to selectively passivate or alternatively functionalize TiO2 patterns on a SiO2 (glass) substrate. The resulting pattern could be made to display specific binding sites on a pure, clean and hydrophilic oxide transparent substrate suitable for a multitude of non-denaturing biomolecular assembly approaches. Importantly, it avoids the drawback of one part of the pattern having non-specific, denaturing, hydrophobic interactions, as in the original SMAP approach.
In this study, we exploit the material selectivity of the nitrocatechols and demonstrate direct patterning of functional biomolecules like avidin directly from physiological buffer. We further show patterning of liposomes and supported lipid bilayers (SLBs), which in recent years gained considerable interest for the development of biosensors.24–27 Patterning of liposomes with incorporated membrane proteins is envisioned to be an attractive tool for the design of biosensors on which a large library of molecules rapidly can be screened and potential drugs identified. One of the most critical aspects in liposome array development is to suppress non-specific liposome and protein adsorption in order to generate a high signal-to-noise ratio; we investigated the suitability of our method to produce patterned biotinylated liposome arrays by area-specific pre-patterning of avidin on a nitrodopamine-mPEG brush background. Patterned SLBs have also proved to be very useful for patterning cell attachment ligands4,6,28 in order to retain and restrict their lateral rearrangement possibilities by the attached cell; this, for example, helped unravelling the minimal amount of antigen ligands necessary to stimulate a T-cell.29 We address the non-fouling property of the PEG-brush which is key to design liposome and SLB arrays and demonstrate supported lipid bilayer patterning to feature sizes as small as 1 μm. For SLBs, this is not straightforward to achieve with standard patterning methods due to the delicate nature of the self-assembled structure held together by weak entropic and van der Waals interactions. It excludes, e.g., approaches involving processing steps of e.g. photoresist removal with organic solvents. The introduced method thus meets a significant and difficult patterning challenge not met by previous photolithographic or SMAP approaches; this is particularly valuable for applications involving membrane-based microarrays or for the fabrication of membrane-mimetic surfaces functionalized with mobile cell attachment ligands that can be rearranged during interactions with cells.4–6
Experimental
Chemicals and buffers
All solvents used are commercially available and had HPLC purity grade. All buffers described were 0.2 μm filtered before use. High salt buffer to dissolve nitrodopamine-mPEG (Susos, Switzerland) consisted of 0.1 M 3-(N-morpholino)propanesulfonic acid (MOPS) (Sigma Aldrich, Switzerland) at pH 6 with 0.6 M NaCl and 0.6 M K2SO4 re-adjusted to pH 6. Hepes buffer consisting of 10 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (Sigma Aldrich, Switzerland) and 150 mM NaCl at pH 7.4 was used for 24 h rinsing and removal of unbound PEG-polymer.Avidin and streptavidin labelled with Alexa488 and Alexa633, resp. (Life Technologies, USA) were dissolved in tris buffered saline (TBS): 10 mM Tris(hydroxymethyl)aminomethane (Sigma Aldrich, Switzerland), 150 mM NaCl, pH 7.4. Bovine serum albumin (BSA) (Sigma Aldrich, Switzerland) was labelled with Alexa546-N-hydroxysuccinimide (Life Technologies, USA) according to manufacturer's protocol and finally dialyzed against TBS to remove excess dye and adjust the buffer conditions.
Production of lipid vesicles
Lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-(12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl)-sn-glycero-3-phosphocholine (NBD-PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (Avanti Polar Lipids, USA) were mixed in the desired ratio and the solvent was evaporated with N2 for 1 h to obtain a dried lipid film. Rehydration of the lipid film was done by adding TBS such that a final lipid concentration of 1 mg ml−1 was reached. The liposome suspension was then extruded 31 times through two polycarbonate filters (Avestin, Canada) with a pore size of 100 nm using a Lipofast extruder (Avestin, Canada). Liposome size was confirmed by dynamic light scattering (DLS) using a Nano ZS (Malvern Instruments Ltd, UK).Creation of patterns by photolithography, magnetron sputtering and lift-off
Micropatterns were created on 24 mm × 24 mm × 0.17 mm borosilicate cover slip glass slides #1 (Menzel Gläser, Germany) by standard photolithographic process. Briefly, glass slides were first cleaned by ultrasonication for 5 min in acetone, followed by isopropanol and subsequently, rinsed with ultrapure water. After drying with N2, the glass slides were spin coated with maN-1405 (Microresist, Germany), prebaked 2 min at 130 °C and illuminated with 350 mJ cm−2 at 365 nm. After developing in ma-D 533S (Microresist, Germany) for 80s, the glass slides were rinsed with ultrapure water and dried with N2. Glass slides were then coated with TiO2 using reactive ion magnetron sputtering under an oxygen atmosphere (PVD Products, USA). Lift-off was performed by 1-methyl-2-pyrrolidinone (NMP, Sigma Aldrich, Switzerland) by applying heating and ultrasonication until no residual photoresist was visible anymore by microscopy. The thickness of the sputtered TiO2 thickness was determined by ellipsometry (Sentech, Germany) to 22–26 nm. Different types of micropatterns consisting of simple lines or more complex patterns such as star-shaped were created having channel lengths of 30–600 μm and widths of 1–5 μm.Passivation of patterned surfaces by material selective PEGylation
Prior to use, patterned glass slides were cleaned by 20 min ultrasonication in toluene and 20 min in isopropanol, rinsed with ultrapure water, blow dried with N2 and then by a pre-heated UV ozone cleaner for 30 min (UV/Ozone Procleaner, Bioforce Nanosciences, USA). Cleaned glass slides were then incubated with 0.1 mg ml−1 nitrodopamine-mPEG for 4 h under cloud point conditions, i.e. at 80 °C using high salt buffer. After incubation, the samples were rinsed with ultrapure water and then further rinsed in a 24 h Hepes buffer incubation step while continuous gentle stirring was applied to remove unbound polymer. Afterwards, the samples were rinsed with ultrapure water, dried with N2 and mounted into a home-built microscopy cell. The PEGylated surfaces were always freshly prepared prior to the experiment and therefore were not stored in order to prevent PEG degradation by oxidation.Patterning of proteins, liposomes, supported lipid bilayers and BSA anti-fouling test
For patterning avidin-Alexa488, PEGylated surfaces were incubated first with 40 μg ml−1 avidin in TBS buffer for 1 h at room temperature and subsequently rinsed 10 times with 1 ml TBS. Liposomes (1 mol% rhodamine B lipids, 5 mol% biotinylated lipids, 94 mol% POPC) in TBS were afterwards added at a concentration of 0.2 mg ml−1 and incubated for 1 h, followed by washing 10 times with 1 ml TBS. Patterned biotinylated supported lipid bilayers (bSLBs) were created by exposing 0.1 mg ml−1 liposomes (1 mol% NBD-PC, 0.1 mol% biotinylated lipids, 98.9 mol% POPC) in TBS to PEGylated surfaces for 1 h at room temperature. The bSLBs were then either exposed to BSA546 (50 μg ml−1) for 1 h or to streptavidin-Alexa633 (40 μg ml−1) for 20 min in TBS, whereby after each step it was rinsed 10 times with 1 ml TBS. Each patterning experiment was repeated at least three times.Fluorescence photobleaching
Images were acquired with a Zeiss LSM 510 (Carl Zeiss AG, Germany) using 20 × 0.8 NA and 63 × 1.4 NA objectives. Photobleaching and fluorescence recovery after photobleaching (FRAP), resp. were done by applying a short high laser power pulse using the 63 × 1.4 NA objective and a 25 mW Argon laser. To bleach the green dyes, only the Argon laser was used and for the red dyes a Helium-Neon laser at 633 nm was additionally engaged. After the bleaching process of bSLB and streptavidin bound to bSLB, images were acquired until the fluorescence was estimated to be homogenous again.Calculation of non-specific protein and liposome adsorption to the PEG-brush
Fluorescence image analysis was done by ImageJ software (National Institutes of Health, Bethesda, USA). Non-specific adsorption was calculated by first photobleaching both the pattern and the background within one bleach spot. The fluorescence intensities of the bleached part of each respective area were averaged. The fluorescence intensity of the designated non-bleached pattern and background were then area averaged and the corresponding bleached background average was subtracted. The ratio of the background subtracted values of the pattern and of the background was calculated to give a quantitative estimation of the suppression of non-specific molecule adsorption to the PEG-brush.Results and discussion
In previous work, it was shown that nitrodopamine-mPEG brushes self-assembled under cloud point conditions on TiO2 surfaces are highly efficient to suppress biomolecule adsorption.22 We therefore investigated the application of nitrodopamine-mPEG for the controlled surface passivation of TiO2 patterns on hydrophilic glass. Subsequently, we investigated the use of the created pattern of passivated and adhesive surface areas to pattern biomolecules directly via adsorption from bulk solution in a single step. The aim was to create high pattern fidelity with low non-specific adsorption to the PEGylated-background. The TiO2/glass patterns were incubated under cloud point conditions (Fig. 1), i.e. high salt concentration and high temperature, in order to achieve a high PEG-grafting density on the TiO2 surfaces; grafting under these conditions has been shown sufficient for non-fouling by ellipsometry, since the PEG is strongly bound to the surface in a highly collapsed state.22,30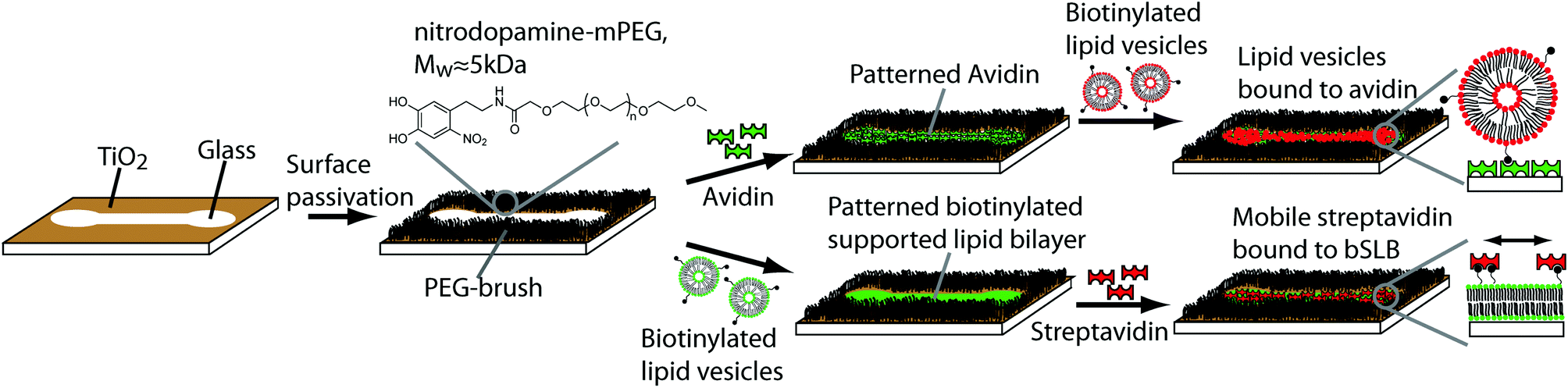 | ||
| Fig. 1 Schematic representation of a TiO2/glass pattern, the passivation process of the surface with nitrodopamine-mPEG and the patterning procedure. Upon sample exposure to the nitrodopamine-mPEG solution under cloud point conditions the nitrodopamine-mPEG binds to the TiO2 background forming a PEG-brush in physiological buffer. Subsequently, the passivated surface is exposed to either avidin or biotinylated lipid vesicles forming a biotinylated supported lipid bilayer (bSLB) through lipid vesicle fusion. The surface is then further functionalized by adding biotinylated lipid vesicles to the patterned avidin (upper part) forming an array of surface-bound liposomes or streptavidin to the patterned, biotinylated SLB (lower part). The latter remains laterally mobile after binding to the fluid bSLB. | ||
Direct patterning of avidin from solution
First, we investigated whether it is possible to adsorb a functional protein, avidin, directly from solution exclusively onto the exposed hydrophilic glass patterns. With a patterned functional protein on the surface, a second biomolecule can be introduced and patterned; for example, biotinylated lipid vesicles coupled to avidin have been demonstrated for liposome based biosensing arrays.24 We expected that avidin due to its positive charge at pH 7.4 adsorbs to the negatively charged glass surface. The patterned substrates were thus exposed to fluorescently labelled avidin (Alexa488) and subsequently rinsed with TBS buffer (Fig. 1, upper part). Large scale, defect free and very homogeneous fluorescence limited to the patterns was found using highly sensitive confocal microscopy (Fig. 2A). Only some minor fluorescence from the background was observed which indicated the presence of a PEG-brush protecting the background surface from fluorescent avidin adsorption. Closer inspection by confocal microscopy revealed a high contrast between the non-passivated glass and the PEGylated TiO2 (Fig. 2B). Furthermore, the fluorescence intensity within the patterns was highly homogeneous indicating a defect free glass surface. This shows that fast, large scale and highly homogenous patterns with well-defined contrasts can be achieved with this method. Compared to other patterning approaches, the direct assembly from physiological bulk solution by this method offers the advantage that dense and homogenous layers with high definition can be formed in one step. If one compares to other one-step methods such as microcontact printing, optimization of wetting and biomolecule transfer to the surface is always a challenge to produce similar patterns as demonstrated here; the boundary between pattern and background typically is diffuse and smeared.8,9 Due to the direct material-selective binding of the nitrodopamine-mPEG to TiO2, no post-processing steps are required which is a key advantage of our approach; it obviates the need for potentially denaturing chemicals necessary for pattern formation.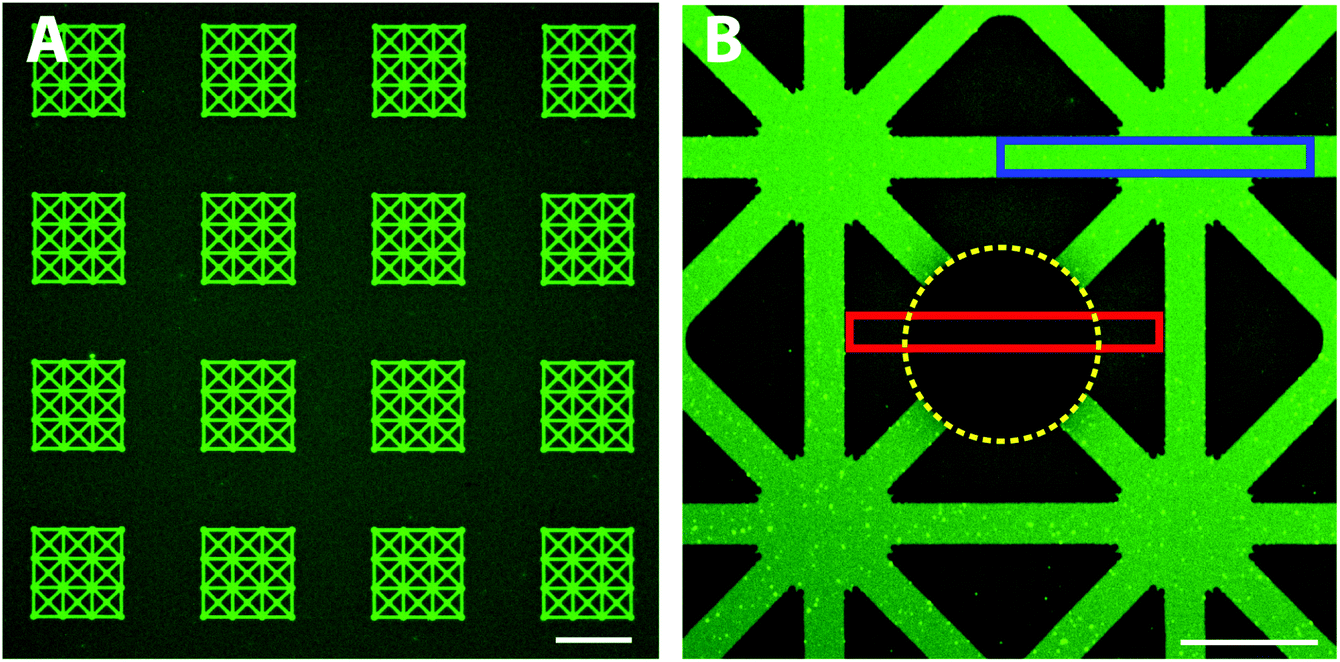 | ||
| Fig. 2 Representative fluorescence micrographs of nitrodopamine-mPEG/glass patterns incubated with fluorescently labeled avidin (Alexa488) and a high resolution view of a bleached pattern. (A) Strong fluorescence is found to be restricted to the patterns with only minor fluorescence from the background indicating the presence of a PEG-layer which suppresses non-specific protein adsorption. (B) A close up view shows a sharp transition between the pattern and the background. Photobleaching (indicated by the yellow dotted circle) was done to provide a reference level for quantitative fluorescence microscopy; the blue box indicates the area from which the fluorescence intensity of the pattern was averaged; the non-bleached parts of the red box indicate the area of the background that was averaged. A reduced avidin adsorption of more than 4 times to the PEG-brush was calculated compared to the glass pattern. Scale bar (A) 100 μm, (B) 15 μm. | ||
To quantify the avidin adsorption to the hydrophilic glass pattern relative to the PEG-brush covered background area, an area covering both the pattern and the background was photobleached (see Experimental for details). The fluorescence intensities in the pattern (blue box, Fig. 2B) and the PEG-brush modified background (non-bleached areas within the red box, Fig. 2B) were averaged with the bleach spot (yellow line, Fig. 2B) as reference. The more than 4-times reduced avidin adsorption to the PEG-brush covered area compared to the glass demonstrates the presence of a PEG-brush. However, protein adsorption was only partially suppressed, since we also see some limited non-specific avidin adsorption to the background. A possible reason could be that the PEG chains during the grafting process under cloud point conditions were not fully collapsed to achieve a higher grafting density on the surface and therefore the PEG-brush was likely not dense enough to completely block non-specific adsorption. Alternatively, the cloud point buffer did not produce an optimal environment for strong nitrocatechol binding to the TiO2 substrate. Thus, by increasing the grafting density or alternatively, increasing the chain length30,31 the protein resistance of the PEG-brush could be improved.
While selective molecular assembly patterning (SMAP) has previously been shown to pattern proteins like streptavidin,18 alkanephosphates are hydrophobic molecules that potentially denature adsorbed proteins; this disturbs secondary functionalization and makes creation of complex biomimetic surface functionalization and further application challenging. Nitrodopamine has a much higher binding affinity to TiO2 than phosphate32 and thus long term stability of the coating is expected to be increased.33,34 Indeed, phosphates can only be used for assembly of SAMs with high mutual attraction and low solubility. Sterically repulsive layers such as PEG are therefore excluded. Long-term stability is also difficult for thiols attached to PEG, as the thiols are susceptible to oxidation.35,36 Another advantage of nitrocatechols compared to previously used surface-anchoring groups for pattern formation, is that they can be used with several different metal oxides surfaces or oxidized metal surfaces,19,22 but show no affinity to silicon oxide. PEG-thiols, on the other hand, are restricted to a few metal surfaces such as Au or Pt surfaces which have the severe disadvantages for pattern functionalization for cell studies that they are opaque and quench fluorescent dyes in their proximity. Our approach therefore opens up new opportunities for cell biology applications over longer time periods due to stable patterning of substrates suitable for fluorescence microscopy. An additional advantage is that after surface passivation the proteins can directly be backfilled to the surface from aqueous solutions, because in every process step chemicals are used that are non-denaturing and non-toxic. This is in contrast to the rather toxic chemicals used for PEG-surface passivation based on silanes. Additionally, in case of chlorosilanes the assembly process requires careful control of the environmental conditions as they are sensitive to moisture. Badly controlled immobilization additionally might either lead to an incomplete monolayer resulting in defects or to multilayer formation induced by uncontrolled head-group cross-linking. Most importantly, silanes bind indiscriminately to most oxides, making them unsuitable for a SMAP approach. The use of high-affinity, but specifically binding, nitrocatechol anchors thus has several important advantages over silanes and other competing anchor strategies.
Functionalization with biotinylated lipid vesicles
After patterning of avidin, the samples were exposed to biotinylated liposomes that were fluorescently labelled with rhodamine lipids (1 mol%) (Fig. 1, upper part) in order to demonstrate the feasibility of creating patterned lipid vesicle arrays for biosensing. Biotinylated liposomes are expected to bind only to the pre-patterned avidin and not to the nitrodopamine-mPEG passivated TiO2 areas. The result should be a strong, slightly speckled fluorescence on the glass/avidin areas that does not recover after photobleaching.37 Phosphatidylcholine liposomes adsorbed on a bare glass surface would instead rupture into a planar supported lipid bilayer for which the homogenously distributed fluorescence recovers after photobleaching (see next section).38 The fluorescence was found to be restricted to the patterns, i.e. to the avidin covered surface, while the background only showed minimal lipid vesicle adsorption (Fig. 3A); thus, the sharp contrast between pattern and background was reproduced also after the liposome adsorption step. Bleaching the rhodamine (Fig. 3B, bleached spot indicated by the yellow dotted line) quantitatively demonstrated very high quality patterns (Fig. 3C). The stable bleach spot also demonstrates that the fluorescence emanates from intact liposomes.39 Ratiometric fluorescence intensity measurements from the blue and red boxes (Fig. 3B and C) yielded a ∼30 times decreased liposome adsorption to the PEG-brush compared to the glass patterns. In fact, the liposome adsorption to the PEGylated surface is almost completely suppressed by comparing to the bleach spot as the background signal difference is within the noise level (see Fig. 3C). This implies that the observed non-specific binding of avidin as described in the previous section (Fig. 2B) occurs via penetration into the PEG-brush restricting accessibility for interaction with at least large biomolecules or supramolecular assemblies such as liposomes. Patterned cells should thus not be able to bind to or interact with proteins buried in the PEG-brush. Cells are thus also expected to be confined to the patterns.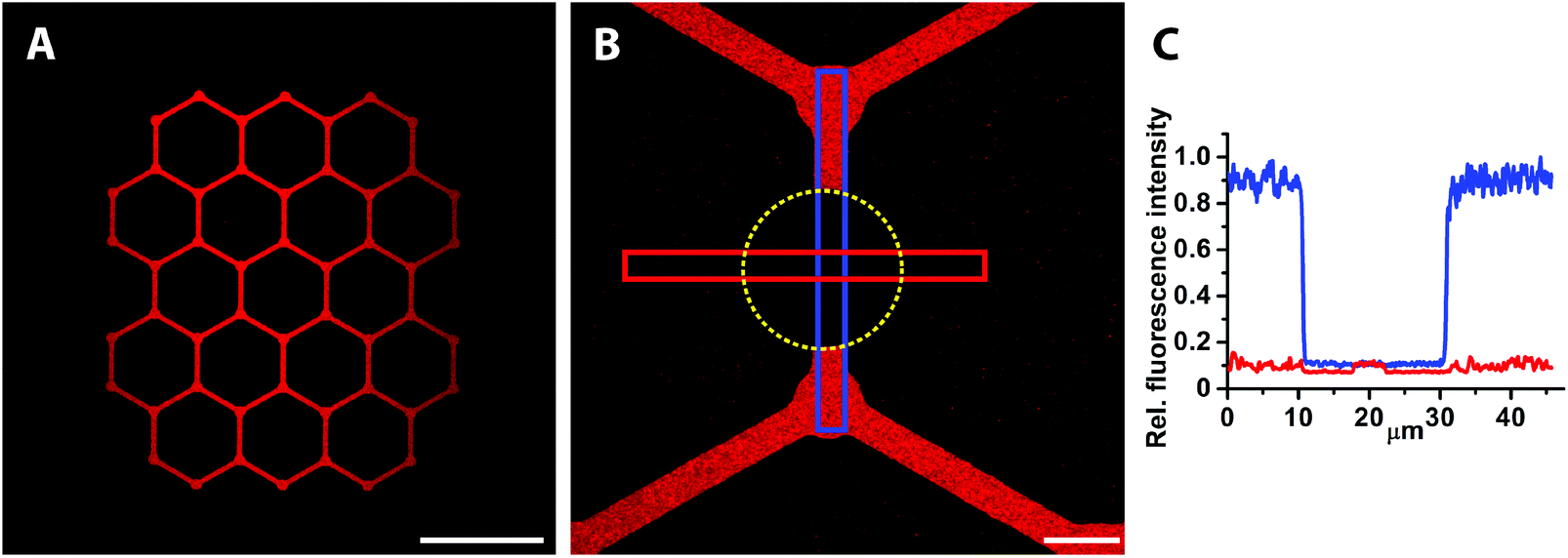 | ||
| Fig. 3 Representative images of patterned biotinylated lipid vesicles with incorporated rhodamine labelled lipids serving as fluorescent tags and photobleaching to quantitatively determine lipid vesicle adsorption to the PEG-background. (A) High fluorescence intensity that is limited to the glass pattern implies the presence of a PEG-brush that prevents non-specific lipid vesicle adsorption. (B) Photobleaching of the pattern and the background was done to address non-specific liposome adsorption (yellow dotted circle delineates the bleached spot) with the blue box serving to average the fluorescence intensity from the pattern and the red box from the background. (C) Plotting the averaged fluorescence intensities from the boxes shown in (B) and with the bleached spot as reference, demonstrates a near complete reduction in lipid vesicle adsorption to the PEG-background. Scale bar (A) 100 μm, (B) 10 μm. | ||
Near-zero, non-specific liposome adsorption and sharply defined pattern boundaries make this approach very interesting for the development of chemical and biological sensors since this allows for generating high signal-to-noise ratio and to suppress unspecific bleeding of non-desired interactions in the background.
Patterning of biotinylated supported lipid bilayers (bSLBs)
Traditional SMAP with alkanephosphates prevents the formation of SLBs due to the hydrophobic and denaturing interface presented by the self-assembled monolayers on TiO2. A hydrophilic glass surface pattern as presented here should be an ideal substrate for micro- or nanopatterned supported lipid bilayers. Hence, we investigated whether fluid SLBs, very sensitive to surface contaminations and organic solvents, could be formed when exposing the mPEG-nitrodopamine-TiO2/glass patterned surfaces to lipid vesicles (Fig. 1, lower part). A successful demonstration would prove the suitability of our patterning approach for e.g. cell adhesion and membrane interaction studies.4,6,28,29 The lipid fluorescence (1 mol% NBD-PC) was found to be limited to the patterned areas with a sharp definition of the edge (Fig. 4A). Within the patterns the fluorescence was found to be homogenously distributed all over the visually inspected surface (1 cm × 1 cm). Almost no fluorescence due to defects within the brush were observed. The smallest line width of 1 μm wide lines realized here was limited by the photolithographic process used to fabricate the glass/TiO2 patterns (Fig. 4B, white arrows). All patterns were found to exhibit near homogenous fluorescence thereby indicating essentially defect-free patterns. The best indication that a homogenous SLB has formed is whether fluorescence recovery within a photobleached area (FRAP) is observed. After photobleaching, the fluorescence quickly recovered throughout the bleached pattern areas, including within the 1 μm-wide lines (Fig. 5A, a–d, purple circle indicates bleached spot and Fig. S1‡), which demonstrates the presence of a fluid supported lipid bilayer. It therefore seems plausible that the pattern resolution was only limited by the resolution of the photolithographic process used to produce the underlying TiO2/glass pattern. In contrast, for other patterning techniques like μCP the stamped boundaries are often poorly defined with poor material transfer leading to inhomogeneous patterns. Feature sizes of a few micrometers are difficult to achieve due to the elastomeric nature of the material. Smaller feature sizes over large areas can be achieved by, e.g., colloidal lithography40 with which SiO2 patterns on a TiO2 matrix has already been demonstrated.41 Other SLB patterning methods to create sub-μm features have been accomplished by, e.g., dip-pen nanolithography in which the SLBs are directly written using an AFM-based tip.42 In another AFM-based patterning method, stripes on a BSA coated surface were removed with an AFM tip and subsequently backfilled with liposomes resulting in localized sub-100 nm SLB patterns with mobile lipids as demonstrated by FRAP.43 However, these approaches are time and work intensive. They have to be applied to every single sample individually and do not allow for making large SLB patterns in a parallel manner and on multiple substrates as shown here. With our new SMAP method, SLBs can fast and easily be patterned over arbitrary pattern geometries and large surface areas, with high contrast and minimal defects to a hydrophilic glass surface with a PEG-brush background.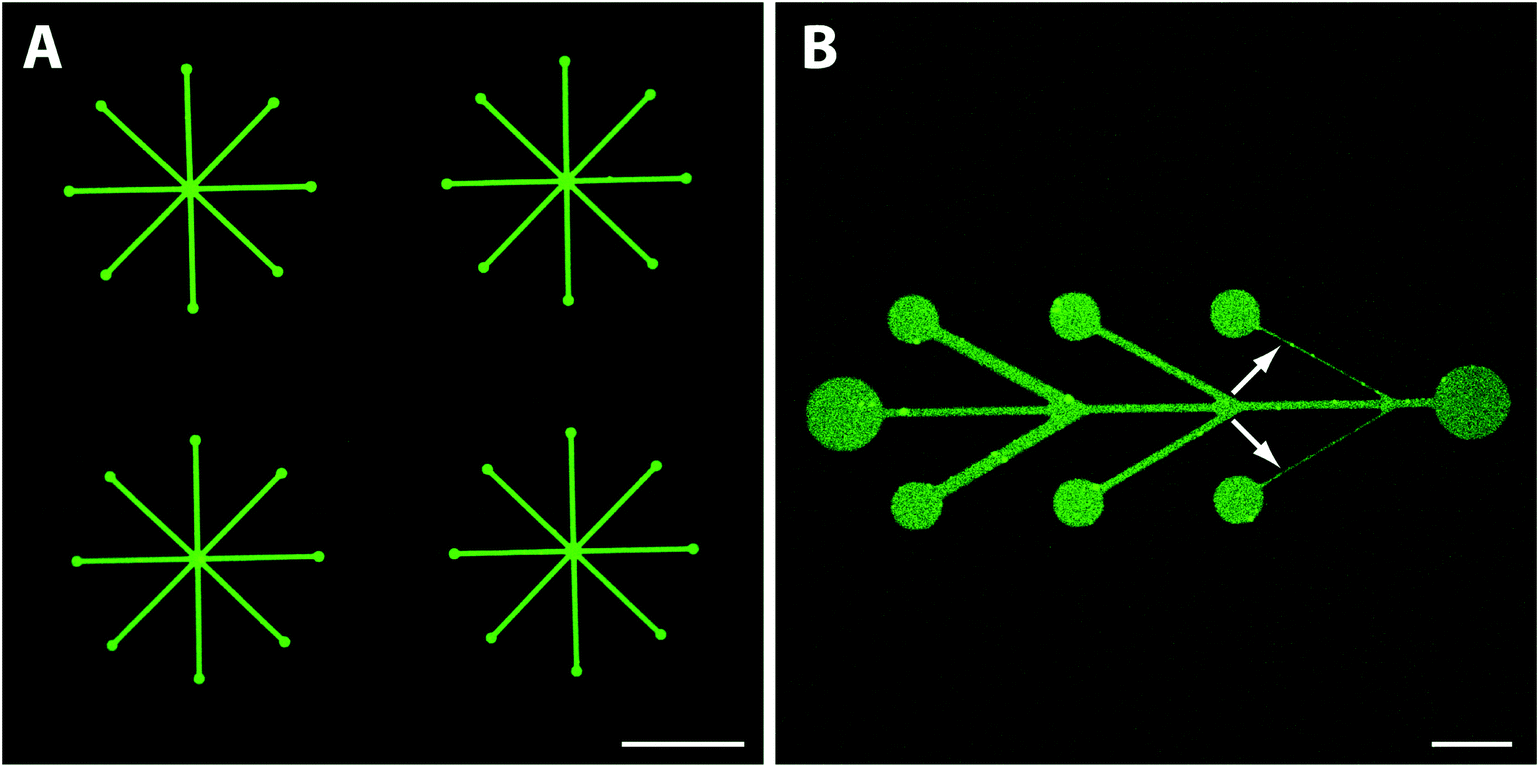 | ||
| Fig. 4 Fluorescence images representatively showing patterned biotinylated supported lipid bilayers (bSLBs) containing 1 mol% NBD-PC. (A) High fluorescence intensity was found to be limited to the patterns. (B) Close up view of a single pattern showed hardly any non-specific liposome adsorption to the PEG-background. The smallest investigated pattern feature size was limited to 1 μm wide lines (white arrows) due to the photolithographic process; it exhibited homogeneous fluorescence. Scale bar (A) 100 μm, (B) 15 μm. | ||
 | ||
| Fig. 5 Fluorescence recovery after photobleaching (FRAP) to determine lateral lipid mobility of the bSLB within the patterns and non-specific liposome adsorption. (A) A short high-laser power pulse (purple circle delineates the bleached spot) was used to photobleach the fluorescently labelled lipids. 225s after bleaching the fluorescence has recovered indicating the presence of a supported lipid bilayer. (B) After photobleaching (bleached spot indicated by the purple dotted circle) non-specific liposome adsorption to the background was analyzed by area averaging the fluorescence intensity of the non-bleached background (left part of yellow box) and of the pattern (dark purple box) with the bleached background subtracted (right part of yellow box). Calculating the ratio, a >100 times reduced liposome adsorption to the background compared to the glass pattern was found. This indicates complete suppression of non-specific liposome adsorption. The red box served to area average the fluorescence intensity of the bleached part and the blue box for the non-bleached part in order to estimate the fluorescence recovery within the 1 μm structure. Image was acquired 8 min after bleaching. (C) Plotted averaged fluorescence intensity profiles from the red and blue boxes depicted in (B). From this ratio and after subtracting the bleached background (right part of yellow box) the fluorescence recovery in the 1 μm structure was estimated to ≥70%. This indicates the presence of a highly fluid and connected SLB. Scale bar (A) and (B), 15 μm. | ||
The quality of SLB formation can be quantified by the percentage of the recovered fluorescence; a high percentage of fluorescence recovery is indicative of a fluid SLB while from the recovery time the lipid mobility (diffusion coefficient) can be estimated. To quantitatively estimate the recovered fluorescence fraction and lipid mobility within the 1 μm structure, the fluorescence intensity was averaged in the bleached and non-bleached areas respectively (Fig. 5B and C, red and blue box, resp.), with the bleached background subtracted (part of yellow box, bleached spot indicated by the purple dotted circle, Fig. 5B). This yielded ≥70% fluorescence recovery and thereby mobile lipid fraction even on the smallest features of the pattern. For bigger channel structures, 100% fluorescence recovery was calculated (Fig. S1‡). Due to the line geometry the lipid mobility could not be determined with standard models which require a circular bleach profile,44 but the recovery time on the order of a few minutes indicates similar mobility as on unpatterned glass, i.e. 1–2 μm2 s−1. The formation of fluid supported membranes is known to be strongly hindered already by traces of organic or inorganic residues on silica surfaces, particularly for small feature sizes and close to pattern edges where resist residues and other materials gradients can occur. Thus, methods that include a material removal step for patterning of e.g. photoresists such as imprint lithography require complete material removal. Otherwise a low or patchy SLB coverage is obtained. Additionally, the resist removal often requires treatment with solvents or other chemicals before the second step of molecular assembly; this might damage and disassemble delicate SLB structures as well as other biomolecules pre-patterned on the surface. Our success in this respect demonstrates that the patterning with nitrodopamine-mPEG onto oxidatively cleaned hydrophilic substrates leaves the glass areas essentially free of contaminants; it allows for straightforward backfilling of and creation of large scale patterning of biomolecules down to sub-cellular features sizes. Only one type of liposomes (in terms of composition) that self-assemble into SLBs on SiO2 can be patterned with our SMAP approach. Techniques such as microfluidics45,46 or robotic spotting47 can be used to pattern multiple lipid mixtures onto the same substrate sequentially. Conversely, these approaches are more work intensive and in case of robotic spotting only allow creating feature sizes of hundreds of micrometers. Most importantly, these methods do not allow for arbitrary pattern creation, but they could be combined by our type of SMAP substrates to create more detailed structured patterns of varied composition.
Finally, the non-specific liposome adsorption was quantitatively assessed by area averaging the fluorescence intensities of the pattern and the non-bleached background (Fig. 5B, dark purple and non-bleached part of the yellow box, respectively) with the bleached part of the background subtracted. From the ratio of the two values a reduction of liposome adsorption to the background of >100 times compared to the glass was calculated; this is consistent with complete suppression of liposome adsorption on the PEGylated TiO2 areas. This again demonstrates that our approach could be useful for patterning biosensors with liposomes and supported lipid membranes for which suppressed background adsorption is key for a high signal-to-noise ratio.
Functionalization of the bSLB using streptavidin and exposure to serum protein albumin
To make SLBs functional for cell studies, they need to be functionalized with cell adhesive proteins. Anchoring of such proteins to the membrane can be done covalently or non-covalently,48 with streptavidin-biotin being a commonly used method. We therefore investigated if fluorescently labelled streptavidin could be coupled to the biotinylated SLB while still being fluid to show the applicability of this platform to pattern functionalized lipid membranes for cell studies. Adsorption of streptavidin was found to be homogenously distributed within the patterns (Fig. 6A), however, with some streptavidin adsorption also in the PEGylated background areas, possibly caused by an insufficiently dense PEG-brush.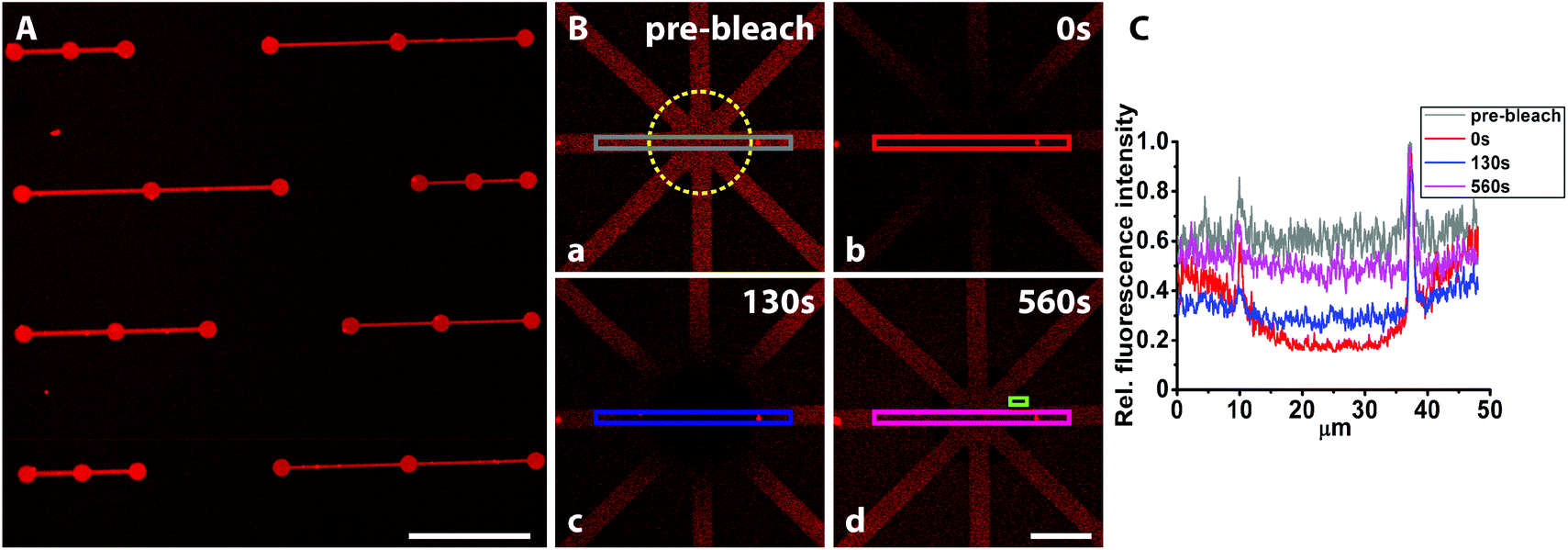 | ||
| Fig. 6 Representative fluorescence images of patterned fluorescently labelled streptavidin (Alexa633) coupled to bSLB and photobleaching to estimate the recovery fraction. (A) The fluorescence intensity increase was mainly confined within the patterns, indicating that the streptavidin was coupled to the bSLB. (B) Mobility of the streptavidin was demonstrated by FRAP (yellow circle corresponds to the bleached spot). To quantitatively estimate the adsorbed mobile streptavidin fraction, the fluorescence intensity was averaged after apparent full recovery (d) (indicated by the purple box) and before bleaching (a) (indicated by the grey box). From this ratio and after subtraction of the averaged background (green box in (d)) a recovery rate of ∼76% was calculated. This shows that the streptavidin was laterally mobile through binding to mobile biotin lipids in the lipid bilayer. The remaining bleached spot in the PEG-background indicates some non-specific streptavidin adsorption to the PEGylated areas. (C) Fluorescence recovery traces obtained by averaging the fluorescence intensity from the boxes (a)–(d) depicted in (B). Scale bar (A) 100 μm, (B) 15 μm. | ||
In contrast, exposure of fluorescently labelled BSA (Alexa546) did not result in any microscopically detectable fluorescence signal on the bSLB patterns or on the PEGylated surface (Fig. S2‡). Adding BSA to the passivated surface was done because BSA is the most common protein in serum and therefore often serves as a standard protein to test the non-fouling properties of passivated surfaces. Our results demonstrate that both the PEGylated surface and the lipid membrane were sufficiently defect free to repel BSA adsorption. These findings are in agreement with previous experiments in which the non-fouling of nitrodopamine-mPEG functionalized TiO2 for non-patterned substrates were studied by ellipsometry before and after exposure to human serum.22 Why albumin in contrast to the avidin/streptavidin was completely repelled by the PEG-brush is not clear, but its affinity to TiO2 is lower. Lateral lipid mobility is an important parameter for cells attaching to functionalized SLBs as recently demonstrated.49–51 Thus, we investigated the lateral mobility of fluorescently labelled streptavidin (Alexa633) by FRAP. The fluorescence recovered within several minutes inside the pattern after photobleaching with an estimated recovery of ∼76% (Fig. 6B and C). This demonstrates that the streptavidin was laterally mobile due to binding to mobile biotin lipids in the supported lipid bilayer. The presented platform to pattern functionalized SLBs is a very attractive tool to analyze ligand mobility and rearrangement in cell adhesion studies, particularly when complex patterns are required. Functionalized, e.g. biotinylated,23 PEG can be used to create the TiO2-assembled PEG-brush, which then allows controlled functionalization also of these areas. Mobile and stationary attachment zones can therefore simultaneously be presented using the same, easy method.
Conclusions
To address the limitations that are associated with the SMAP approach relying on hydrophobic alkanephosphates for patterning of biomolecules and supported lipid bilayers we introduced an improved method to structurally array biomolecules to transparent (glass) patterns in a passivated PEG background. The simple process steps carried out under physiological conditions allow for patterning of delicate protein and even lipid membrane structures. Lipid mobility in patterned lipid membranes could be demonstrated down to sub-cellular (1 μm) features and transferred to membrane linked ligands. We envision this method to be very useful for patterning of cells and in studying complex cell recognition processes requiring a native-like presentation of ligands with reconfigurable spacings. In fact, only dependent on the solid-state TiO2/glass contrast patterning step, arbitrary shapes and sizes of mobile or non-mobile recognition zones can be implemented where the microscope substrate acts as an artificial cell. With the growing realization that dimensionality and mechanical properties of cell adhesion sites strongly influence cell development and cell response to drugs,52,53 we anticipate that our simple to fabricate platform provides an important new tool for cell studies, biosensors and drug screening.Acknowledgements
This work was supported by the Novartis Foundation (grant 8B27) and the National Competence Center for Biomedical Imaging (NCCBI) in Lausanne, Switzerland. We also thank ETH Zurich and BM.W-F Austria for funding.Notes and references
- T. Blattler, C. Huwiler, M. Ochsner, B. Stadler, H. Solak, J. Voros and H. M. Grandin, J. Nanosci. Nanotechnol., 2006, 6, 2237–2264 CrossRef CAS PubMed.
- A. B. Faia-Torres, T. Goren, M. Textor and M. Pla-Roca, in Comprehensive Biomaterials, ed. Editor-in-Chief: D. Paul, Elsevier, Oxford, 2011, pp. 181–201 Search PubMed.
- D. B. Weibel, W. R. DiLuzio and G. M. Whitesides, Nat. Rev. Microbiol., 2007, 5, 209–218 CrossRef CAS PubMed.
- B. N. Manz and J. T. Groves, Nat. Rev. Mol. Cell Biol., 2010, 11, 342–352 CrossRef CAS PubMed.
- C. H. Yu, J. B. K. Law, M. Suryana, H. Y. Low and M. P. Sheetz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20585–20590 CrossRef CAS PubMed.
- M. L. Dustin and J. T. Groves, Annu. Rev. Biophys., 2012, 41, 543–556 CrossRef CAS PubMed.
- S. A. Ruiz and C. S. Chen, Soft Matter, 2007, 3, 168–177 RSC.
- Y. N. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- O. Akbulut, A. A. Yu and F. Stellacci, Chem. Soc. Rev., 2010, 39, 30–37 RSC.
- A. C. Siegel, S. K. Y. Tang, C. A. Nijhuis, M. Hashimoto, S. T. Phillips, M. D. Dickey and G. M. Whitesides, Acc. Chem. Res., 2010, 43, 518–528 CrossRef CAS PubMed.
- M. Hitzbleck and E. Delamarche, Chem. Soc. Rev., 2013, 42, 8494–8516 RSC.
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7 CrossRef PubMed , 041501.
- K. Salaita, Y. H. Wang and C. A. Mirkin, Nat. Nanotechnol., 2007, 2, 145–155 CrossRef CAS PubMed.
- C. C. Wu, D. N. Reinhoudt, C. Otto, V. Subramaniam and A. H. Velders, Small, 2011, 7, 989–1002 CrossRef CAS PubMed.
- C. M. Kolodziej and H. D. Maynard, Chem. Mater., 2012, 24, 774–780 CrossRef CAS.
- H. Tran, K. L. Killops and L. M. Campos, Soft Matter, 2013, 9, 6578–6586 RSC.
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed.
- R. Michel, J. W. Lussi, G. Csucs, I. Reviakine, G. Danuser, B. Ketterer, J. A. Hubbell, M. Textor and N. D. Spencer, Langmuir, 2002, 18, 3281–3287 CrossRef CAS.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
- J. L. Dalsin, B. H. Hu, B. P. Lee and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 4253–4258 CrossRef CAS PubMed.
- X. W. Fan, L. J. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS PubMed.
- B. Malisova, S. Tosatti, M. Textor, K. Gademann and S. Zurcher, Langmuir, 2010, 26, 4018–4026 CrossRef CAS PubMed.
- E. Amstad, S. Zurcher, A. Mashaghi, J. Y. Wong, M. Textor and E. Reimhult, Small, 2009, 5, 1334–1342 CrossRef CAS PubMed.
- M. Bally, K. Bailey, K. Sugihara, D. Grieshaber, J. Voros and B. Stadler, Small, 2010, 6, 2481–2497 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS PubMed.
- L. Tiefenauer and S. Demarche, Materials, 2012, 5, 2205–2242 CrossRef CAS PubMed.
- E. Reimhult and K. Kumar, Trends Biotechnol., 2008, 26, 82–89 CrossRef CAS PubMed.
- C. H. Yu, W. W. Luo and M. P. Sheetz, Cell Adhes. Migr., 2012, 6, 280–284 CrossRef PubMed.
- B. N. Manz, B. L. Jackson, R. S. Petit, M. L. Dustin and J. Groves, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9089–9094 CrossRef CAS PubMed.
- P. Kingshott, H. Thissen and H. J. Griesser, Biomaterials, 2002, 23, 2043–2056 CrossRef CAS.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. Degennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- M. Rodenstein, Ph.D. Thesis, ETH Zurich, 2010.
- S. Saxer, C. Portmann, S. Tosatti, K. Gademann, S. Zurcher and M. Textor, Macromolecules, 2010, 43, 1050–1060 CrossRef CAS.
- Q. Ye, F. Zhou and W. M. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- M. H. Schoenfisch and J. E. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502–4513 CrossRef CAS.
- M. J. Tarlov and J. G. Newman, Langmuir, 1992, 8, 1398–1405 CrossRef CAS.
- P. Nollert, H. Kiefer and F. Jahnig, Biophys. J., 1995, 69, 1447–1455 CrossRef CAS.
- E. Reimhult, M. Zach, F. Hook and B. Kasemo, Langmuir, 2006, 22, 3313–3319 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- X. Ye and L. Qi, Nano Today, 2011, 6, 608–631 CrossRef CAS PubMed.
- T. M. Blattler, A. Binkert, M. Zimmermann, M. Textor, J. Voros and E. Reimhult, Nanotechnology, 2008, 19 CrossRef CAS PubMed , 075301.
- S. Lenhert, P. Sun, Y. H. Wang, H. Fuchs and C. A. Mirkin, Small, 2007, 3, 71–75 CrossRef CAS PubMed.
- J. J. Shi, J. X. Chen and P. S. Cremer, J. Am. Chem. Soc., 2008, 130, 2718–2719 CrossRef CAS PubMed.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS.
- M. R. Dusseiller, B. Niederberger, B. Stadler, D. Falconnet, M. Textor and J. Voros, Lab Chip, 2005, 5, 1387–1392 RSC.
- N. J. Wittenberg, H. Im, T. W. Johnson, X. H. Xu, A. E. Warrington, M. Rodriguez and S. H. Oh, ACS Nano, 2011, 5, 7555–7564 CrossRef CAS PubMed.
- B. Stadler, M. Bally, D. Grieshaber, J. Voros, A. Brisson and H. M. Grandin, Biointerphases, 2006, 1, 142–145 CrossRef CAS PubMed.
- P. M. Nair, K. Salaita, R. S. Petit and J. T. Groves, Nat. Protocols, 2011, 6, 523–539 CAS.
- K. Spendier, A. Carroll-Portillo, K. A. Lidke, B. S. Wilson, J. A. Timlin and J. L. Thomas, Biophys. J., 2010, 99, 388–397 CrossRef CAS PubMed.
- A. Carroll-Portillo, K. Spendier, J. Pfeiffer, G. Griffiths, H. T. Li, K. A. Lidke, J. M. Oliver, D. S. Lidke, J. L. Thomas, B. S. Wilson and J. A. Timlin, J. Immunol., 2010, 184, 1328–1338 CrossRef CAS PubMed.
- C. J. Hsu, W. T. Hsieh, A. Waldman, F. Clarke, E. S. Huseby, J. K. Burkhardt and T. Baumgart, PLoS One, 2012, 7, e32398 CAS.
- E. Cukierman and D. E. Bassi, Semin. Cancer Biol., 2010, 20, 139–145 CrossRef CAS PubMed.
- M. B. Meads, R. A. Gatenby and W. S. Dalton, Nat. Rev. Cancer, 2009, 9, 665–A674 CrossRef CAS PubMed.
Footnotes |
| † We dedicate this work to Dr Heike Hall who sadly passed away far too early. We sorely miss her as a colleague, supervisor and person who meant a lot to us all. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00090k |
| This journal is © The Royal Society of Chemistry 2015 |